
Surface water treatment by UV/H2O2 with subsequent soil aquifer treatment: impact on micropollutants, dissolved organic matter and biological activity†
 ab
Julia
Plattner,
c
ab
Julia
Plattner,
c
Abstract
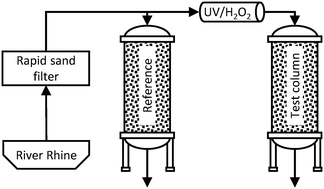
Because organic micropollutants (MP) are frequently detected in river waters that are used as drinking water sources, combining a relatively cost-efficient natural treatment with upstream advanced oxidation processes (AOP) appears promising for their efficient abatement. Such a multi-barrier system can be integrated in drinking water production schemes to minimize risks from potentially hazardous MPs. This study investigates the impact of an UV/H2O2 AOP before soil aquifer treatment (SAT) on the abatement of selected MPs (EDTA, acesulfame, iopamidol, iomeprol, metformin, 1H-benzotriazole, iopromide), dissolved organic matter (DOM) (apparent molecular size distribution, specific UV absorbance at 254 nm – SUVA) and microbial parameters (intact cell count, cell-bound ATP). A pilot plant consisting of an AOP (0.5 m3 h−1, 4 mg L−1 H2O2, 6000 J m−2) and two parallel soil columns (filtration velocity: 1 m d−1, column height: 1 m) was continuously operated over a period of 15 months with Rhine river water pre-treated with rapid sand filtration. The investigations revealed a shift towards longer retention times of the humic substances peak in LC analysis of DOM, lower SUVA and higher biodegradability of DOM after UV/H2O2 treatment. In addition, an overall higher abatement of all investigated MPs by the combined treatment was observed (AOP with subsequent SAT) compared to either process alone. This observation could be explained by an addition of the single treatment effects. The strong primary disinfection effect of the AOP was detectable along the first meter of infiltration, but did not lead to any change in the column performance (i.e., similar abatement of dissolved organic matter).
- This article is part of the themed collection: Best Papers 2019 – Environmental Science: Water Research & Technology
 Please wait while we load your content...
Please wait while we load your content...

