
Enhanced adsorption of perfluoro alkyl substances for in situ remediation†
 ac
Matt F.
Simcik
*ab
ac
Matt F.
Simcik
*ab
Abstract
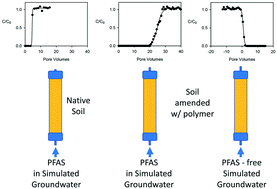
Numerous groundwater sites around the globe have been contaminated by aqueous film forming foam (AFFF) as a result of firefighting, fire training activities and the storage and accidental spillage of AFFF. AFFF contains numerous per- and polyfluoroalkyl substances (PFAS), including perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) for which the U.S. Environmental Protection Agency has set a combined health advisory level of 70 ng L−1 in drinking water. One approach for in situ remediation of PFAS-impacted groundwater is to enhance the adsorption of PFAS by introducing adsorbents, which serve to increase the total adsorption capacity of aquifer solids. This paper describes a method for increasing the sorption of PFAS to aquifer solids by adding commercially available drinking water coagulants. These coagulants are the cationic polymers polydiallyldimethyl ammonium chloride (polyDADMAC) and polyamine (a co-polymer of epichlorohydrine and dimethyl amine). The six PFAS on the Unregulated Contaminant Monitoring Rule 3 (UCMR3) list were studied including perfluorobutane sulfonate (PFBS), perfluorohexane sulfonate (PFHxS), PFOS, perfluoorbutanoic acid (PFBA), PFOA and perfluorononanoic acid (PFNA). Treatability tests were conducted with natural soil excavated from an Air Force Base located in the south central United States. In completely mixed batch reactors studies, the coagulants increased the sorption capacity of the soil for PFAS by a factor of 2.0–6.1. One-dimensional columns pre-loaded with coagulant delayed the breakthrough of PFAS by as much as 20 pore volumes with an applied PFAS concentration of 100 μg L−1. Following loading with PFAS and coagulant the columns were then flushed with PFAS-free simulated groundwater to assess desorption behavior. For all PFAS the retention on the column showed hysteresis where only 1 to 20% of the PFAS was recovered from the column after flushing with 30 pore volumes of simulated groundwater. The observed increase in PFAS adsorption could not be accounted by solely by the increase in organic carbon content resulting from the addition of adsorption enhancers, suggesting that exchange interactions contributed to PFAS retention. These results indicate that amending soils and aquifer solids with cationic polymers acting as sorption enhancers holds promise as a viable method for in situ PFAS sequestration.
- This article is part of the themed collection: PFAS
 Please wait while we load your content...
Please wait while we load your content...

