
Absorption of short-chain to long-chain perfluoroalkyl substances using swellable organically modified silica†

Abstract
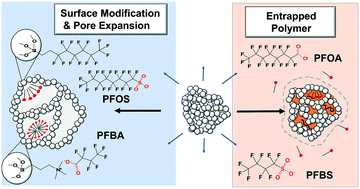
Organosilica synthesis chemistry via the sol–gel method was used to create porous organosilica adsorbents designed to remove PFAS compounds from water. A set of adsorbents was created by incorporating a fluorophilic amide or fluoroalkyl function group along with quaternary groups for ion exchange within a continuous porous matrix. A distinguishing aspect of the adsorbents was the ability to volumetrically swell 2.5× (>6 mL g−1) in the presence of organic liquids. Swelling results from a flexible pore architecture and was used to create expanded mesopores which were hypothesized to yield greater adsorption capacity for PFAS compounds. Adsorption isotherms and adsorption kinetics experiments were conducted for a set of 12 PFAS analytes in deionized water and 50 mM NaCl in order to understand the effect of adsorbate characteristics and ionic strength on adsorption. PFAS of all chain lengths ranging from C4–C10 were bound to adsorbents possessing both fluorophilic and cationic groups. In comparison, only long-chain PFAS >C6 are bound when the adsorbents are exclusively hydrophobic or fluorophilic. An adsorbent was created where a cationic imidazolium containing polymer was entrapped within the swellable pores. The polymer-infused organosilica demonstrated the highest capacity across the suite of 12 short-chain and long-chain PFAS. In column experiments, the polymer organosilica yielded 47 mg g−1 capacity for perfluorooctanoic acid (PFOA) prior to breakthrough with an influent concentration of 200 μg L−1, approximately an order a magnitude greater than GAC. The high adsorption capacity is attributed to expandable pores which can accommodate polymer and PFAS adsorbates while maintaining sufficient access for mass transport.
- This article is part of the themed collections: PFAS and Best Papers 2019 – Environmental Science: Water Research & Technology
 Please wait while we load your content...
Please wait while we load your content...

