
Removal of per- and polyfluoroalkyl substances (PFASs) from contaminated groundwater using granular activated carbon: a pilot-scale study with breakthrough modeling†

Abstract
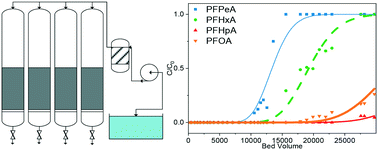
Granular activated carbon (GAC) is a commonly used technology for removal of per- and polyfluoroalkyl substances (PFAS). However, most studies characterizing PFAS absorption to GAC are performed in small bench-scale tests with synthetic groundwater, which poorly approximates conditions encountered in full-scale treatment systems. Pilot-scale studies, although somewhat rare, better predict full-scale performance due to realistic operating conditions. This study presents breakthrough results from a pilot-scale GAC system operated for seven months treating a continuous source of PFAS contaminated groundwater with four activated carbons, Calgon F400 and F600, and Norit GAC400 and GCN1240. Chain length dependent breakthrough was generally observed for perfluorocarboxylates (PFCAs) and perfluorosulfonates (PFSAs) where shorter chain PFASs broke through faster than longer chain PFASs. All tested GACs performed similarly for weakly adsorbing shorter chain PFASs, suggesting that GAC properties may not affect breakthrough for these PFASs. However, F400 and GAC400 performed 40–50% better than F600 and GCN1240 for strongly adsorbing long chain PFASs, which may be due to a higher volume of transport pores within F400 and GAC400. Breakthrough curves were fit by equilibrium and intraparticle diffusion models using the solid liquid partition coefficient (Kd). The equilibrium model was found to be a better fit for the data and a more practical model. Model Kd outputs from the pilot study were compared against a separate batch study. Comparisons showed that batch study Kd values were larger for longer chain compounds and smaller for shorter chain compounds.
- This article is part of the themed collection: PFAS
 Please wait while we load your content...
Please wait while we load your content...

