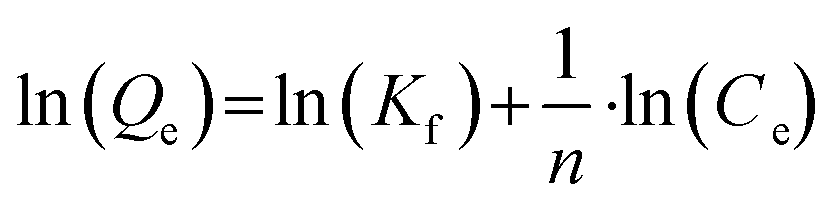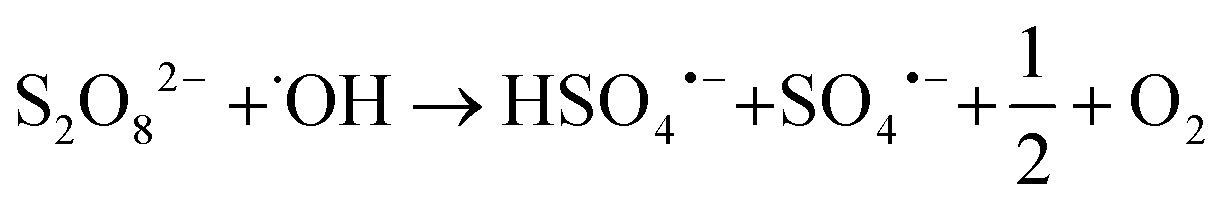Removal of per- and polyfluoroalkyl substances (PFASs) from tap water using heterogeneously catalyzed ozonation†
Received
23rd April 2019
, Accepted 5th September 2019
First published on 2nd October 2019
Abstract
Drinking water is one of the most important human exposure pathways of per- and polyfluoroalkyl substances (PFASs). As conventional water treatment techniques are unable to remove PFASs efficiently, novel treatment methods for the removal of PFASs in water are urgently needed. In the present study advanced oxidation processes (AOPs) based on heterogeneously catalyzed ozonation were evaluated on laboratory- and pilot-scales for their efficiency in removing PFASs from water. Laboratory-scale ozonation experiments were conducted with different combinations of ozone, a catalyst and persulfate and showed the highest efficiency for the treatment combining all three parameters. The method was further evaluated for the treatment of spiked drinking water on the pilot-scale. The concentrations of all 18 analyzed PFASs decreased significantly within three hours of treatment in the pilot-scale set-up. The perfluorocarbon chain length had a dominant influence on the removal efficiency, where CF7 − CF11 PFASs were removed with more than 98% removal efficiency, independent of the functional group, CF12 − CF17 PFASs with 64%, and CF4 − CF6 with 55% on average. As the evaluated ozonation treatment is already commercially available for large scale applications today, it could easily be applied in existing water treatment trains; however, ozonation can create potentially toxic transformation products which needs to be investigated in future research.
Water impact
Human exposure to per- and polyfluoroalkyl substances (PFASs) through contaminated drinking water is of great concern. Once contamination is discovered, impacted water needs to be treated on-site, which represents logistical and economic challenges. In this study, an available treatment technique based on heterogeneously catalyzed ozonation was evaluated for the removal of PFASs from tap water at environmentally relevant concentrations. PFASs were removed with up to 98% removal efficiency in a pilot-scale set-up and the removal depended on the perfluorocarbon chain length rather than the functional group. Laboratory scale trials showed that the treatment can be improved by the addition of persulfate. This treatment gives water providers not only a tool to treat acutely impacted water but potentially also a long-term solution as a step in the treatment train.
|
1 Introduction
Rising concern exists over human exposure to per- and polyfluoroalkyl substances (PFASs). Their ubiquitous use in various commercial and industrial products has led to unintended human exposure incidents in the past when local drinking water was contaminated.1–4 Conventional drinking water treatment does generally not decrease PFAS concentrations.5–8 Municipalities in which raw water is contaminated therefore often apply granulated activated carbon (GAC) filters, which are capable of removing PFASs if freshly regenerated or newly replaced.8 In order to meet current drinking water guidelines, these filters however need to be changed often and are therefore a costly interim solution. Due to the substitution of fluorine for hydrogen in the alkyl carbon chain of PFASs, they are recalcitrant towards conventionally applied oxidation treatment.9,10 Fluorine has a reduction potential of E0 = 3.6 (ref. 11) and is the strongest inorganic oxidant. Therefore it is thermodynamically unfavorable to oxidize fluorine in the oxidation state (−1) to its elemental state of F2(0) with any other one-electron oxidant.10 Nonetheless, the use of advanced oxidation processes (AOPs) has been discussed as a promising approach for the removal of PFASs on both laboratory and full-scales.12,13 AOPs describe a group of treatment techniques that are based on in situ generation of radicals. AOPs are widely applied in diverse combinations in water treatment to degrade a large number of different organic pollutants with the help of ozone (O3), hydrogen peroxide (H2O2), ultraviolet radiation (UV), persulfate (S2O82−) and other oxidizing agents.14–19 Different reaction conditions lead to generation of highly reactive radicals like hydroxyl radicals (OH˙), sulfate radicals (SO4˙−), hyperoxide radicals (O2˙−) or carbonate radicals (CO3˙−).8 Molecular ozone, which has a redox potential of E0 = 2.07 V, and OH˙ (E0 = 2.8 V)20 have shown to be insufficient for the degradation of PFASs.9,10,21 Other oxygen containing radical species created by means of catalytic oxidation processes (e.g. Fenton's reagent chemistry or catalyzed hydrogen peroxide) have on the other hand shown to degrade PFASs with various functional groups.22 Furthermore, several studies found that the application of sulfate radicals leads to efficient degradation of various organic micropollutants including perfluoroalkyl carboxylic acids (PFCAs; CnF(2n+1) COOH).8,23–25 Hori et al.23,26 found a substantial decrease of PFCA concentrations when treating contaminated water with photocatalytically activated persulfate while Wang et al.27 showed that F−, CO2 and small quantities of shorter chain PFCAs were the main reaction products in a similar experiment. Abu Amr et al.28 found an enhanced PFAS removal in landfill leachate for the combination of ozone and persulfate compared to a treatment with ozone only. Schröder et al.29 investigated PFAS destruction via a combination of ozone treatment and heterogeneous catalysis on celite and removal efficiencies of about 14% (PFOA) and 53% (PFOS) were reported. It should be noted that Schröder et al. hypothesized that various types of strong oxidative agents including hydroxyl radicals led to perfluoroalkyl chain shortening, while later research showed that hydroxyl radicals do not transform perfluoroalkyl substances, as mentioned above. Until now, there is however a lack of understanding and knowledge about the use of ozonation in combination with a catalyst and persulfate for the removal of PFASs.
In this study research on heterogeneously catalyzed ozonation for the removal of PFASs from water was assessed in laboratory- and pilot-scale treatment. Ozone was applied in situ in combination with a heterogeneous iron-oxide based catalyst using ultrapure water on a laboratory scale as well as spiked drinking water on a pilot scale. The specific objectives of this study were i) to test the combinations of ozone, a catalyst and persulfate in terms of removal efficiency of PFASs in order to draw conclusions about the potential to improve the existing, commercially available treatment, ii) to investigate the treatment efficiency in pilot-scale treatment of contaminated drinking water, and finally iii) to evaluate treatment specific thermodynamic and kinetic aspects regarding PFAS removal and possible reaction mechanisms.
2 Materials and methods
2.1 Chemicals
In total 18 target analytes were analyzed which included CF3 − CF11, CF13, CF15, and CF17 PFCAs (CFn describing the amount of perfluorinated carbon atoms; PFBA, PFPeA, PFHxA, PFHpA, PFOA, PFNA, PFDA, PFUnDA, PFDoDA, PFTeDA, PFHxDA, and PFOcDA), CF4, CF6, and CF8 perfluoroalkyl sulfonic acids, PFSAs (CnFn+1SO2OH; PFBS, PFHxS, and PFOS), CF6 and CF8n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 fluorotelomer sulfonic acids, (CnF2n+1C2H4SO3H) and perfluorooctanesulfonamide, FOSA (C8F17SO2NH2). For quantification, 10 mass labeled PFASs were applied as internal standards (ISs); see Table A1 in the ESI.† All native and mass-labeled standard solutions used for analysis were purchased from Wellington Laboratories Inc. (Guelph, Ontario, Canada). For the applied spiking solution, native PFASs were purchased from Sigma Aldrich Sweden AB (Stockholm, Sweden), Alfa Aesar GmBH (Thermo Fisher (Kandel) GmbH, Karlsruhe, Germany) and Apollo Scientific Ltd (Manchester, United Kingdom); see Table A2 in the ESI.†
:2 fluorotelomer sulfonic acids, (CnF2n+1C2H4SO3H) and perfluorooctanesulfonamide, FOSA (C8F17SO2NH2). For quantification, 10 mass labeled PFASs were applied as internal standards (ISs); see Table A1 in the ESI.† All native and mass-labeled standard solutions used for analysis were purchased from Wellington Laboratories Inc. (Guelph, Ontario, Canada). For the applied spiking solution, native PFASs were purchased from Sigma Aldrich Sweden AB (Stockholm, Sweden), Alfa Aesar GmBH (Thermo Fisher (Kandel) GmbH, Karlsruhe, Germany) and Apollo Scientific Ltd (Manchester, United Kingdom); see Table A2 in the ESI.†
2.2 Experimental procedures
2.2.1 Kinetic experiments.
For the kinetic adsorption experiments 0.1 g of the catalyst material and 40 mL tap water were added into a 50 mL PP-tube. After addition of 20 μL PFAS stock solution containing the 18 target analytes (20000 μg mL−1 per compound, in methanol), the tubes were put on a horizontal shaking machine at 150 rpm. Single 100 μL samples were taken after 1, 2, 5, 10, 15, 30, 75 and 100 min. The experiment was conducted in duplicate and the samples were analyzed the same day. Further, a positive blank test (n = 2) was conducted without a catalyst to evaluate PFAS adsorption to the tube material.
2.2.2 Adsorption isotherms.
Adsorption isotherms for PFAS adsorption to the catalytic material were obtained for MilliQ water, tap water and dissolved organic carbon (DOC; 10.0 mg L−1) containing water. DOC containing water was prepared by shaking 1.33 g uncontaminated soil with 150 mL MilliQ water (Millipak© Express 20, 0.22 μm filter, Merck Millipore) for 48 h at 50 rpm and subsequently centrifuging for 30 min at 3000 rpm. The supernatant was removed with 10 mL HSW NORM-JECT© syringes, pre-filtered through quantitative filter paper (Munktell Ahlstrom, Grade 00H, pore size: 0.45 μm) and filtered in a second step with syringe filters (VWR International, 0.45 μm pore size, nylon membrane). After diluting with MilliQ water, it had a DOC concentration of 10.0 mg L−1. For each trial, 40 mL of the respective water type, 0.1 g catalyst material and 20μL−1 of a respective PFAS mix (200, 1000, 2000, 10000 or 20000 μg L−1) containing the 18 target analytes (in methanol) were added to 50 m PP-tubes. The PP-tubes were shook horizontally for 100 h at 150 rpm. Experiments were conducted in duplicate for MilliQ, tap-, and DOC water each and the mean value was taken for further calculations. Experimental data obtained from the 100 h shaking experiment were evaluated regarding the fit to different adsorption isotherm models (i.e. Freundlich and Langmuir). Freundlich sorption parameters were calculated from eqn (1):| | 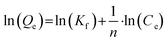 | (1) |
where Qe represents the equilibrium adsorbate amount [mg g−1] and Ce corresponds to the equilibrium concentration of the analyzed compounds [mg L−1]. Kf [(μg g−1)(μg L−1)−n] describes the adsorption capacity of the catalytic material and n [unitless] is the adsorption intensity for analyzed PFASs. Since positive blank experiments performed during the kinetic trials showed no significant change in concentrations after the first 10–20 min, these positive blank experiments were further used to account for losses during adsorption isotherm experiments.
2.2.3 Laboratory-scale trials.
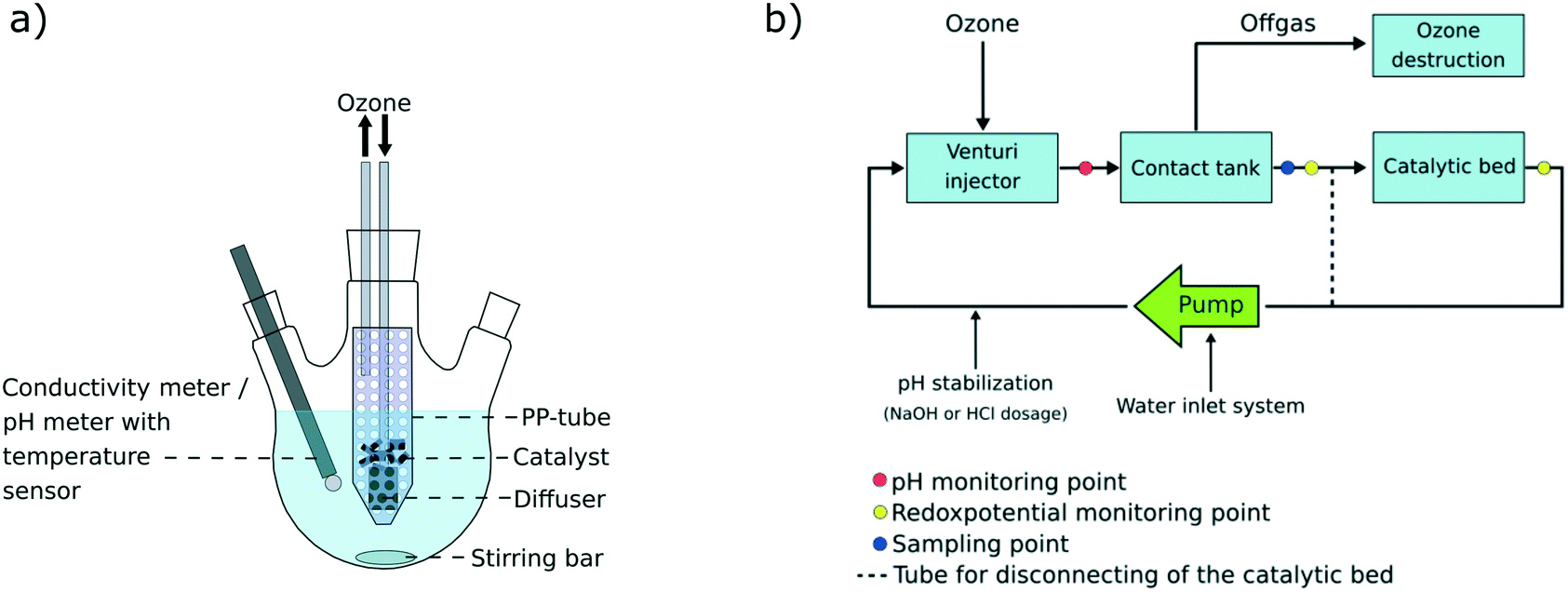
Batch experiments were conducted at room temperature (22 °C) in 500 mL three necked flasks under constant stirring at 500 rpm in a system schematically shown in Fig. 1a. A perforated 50 mL polypropylene (PP)-tube was placed in the flask and depending on the experiment, filled with 5 g catalyst material and/or an O3 gas diffuser. 500 mL MilliQ water (Millipak® Express 20, 0.22 μm filter, Merck Millipore) was spiked with 50 μL of a PFAS solution (PFBA, PFHxA, PFOA, PFDA, PFDoDA, PFTeDA, PFHxDA, PFOcDA, PFBS, PFHxS, PFOS, 6:2 FTSA, 8:2 FTSA, and FOSA; containing 10 μg mL−1 per compound). After 10 min of balance adjustment, the first set of samples was taken and the ozone flow (300 mg ozone per h) was turned on and/or 187 mg ammonium persulfate (99.5%, Peroxy Chem, USA) (mole ratio PFASs/ammonium persulfate 1:50) was added, depending on the respective trial. Further 1 mL samples were taken after 5, 15, 30, 60, 90 and 120 min and immediately purged with nitrogen to remove dissolved ozone gas. All the samples were taken in duplicate. An overview over the conducted experiments on the laboratory scale is listed in Table 1. Both positive and negative blank experiments were conducted in the same manner as the actual trials with the same sampling intervals. Duplicate experiments were performed for all experiments and the mean value was taken for the discussion of results. Changes in PFAS concentrations for the positive blank tests (see Table 1) were within 0–15% and were considered in the calculations for deriving relative removal percentages in the laboratory-scale trials. Positive blank 2a was used for degradation trials using the catalyst, while positive blank 2b was used for trials not applying the catalyst. A correction factor was derived for each PFAS compound, where the deviation of the initial concentration in the positive blank samples was divided by the mean of the concentrations measured during the remaining 120 min of the positive blank experiment.
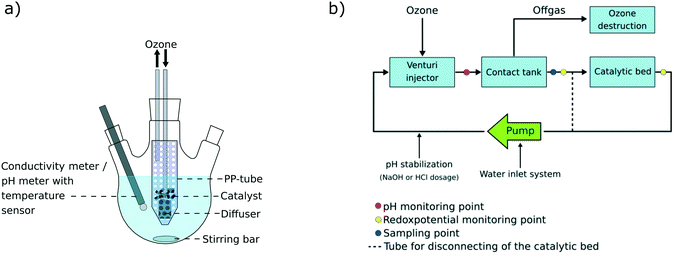 |
| | Fig. 1 a) Experimental set up of the 500 mL laboratory-scale system and b) scheme of the 50 L pilot-scale system. | |
Table 1 Overview over the conducted laboratory-scale trials with the applied combinations of ozone (O3), the catalyst (cat) and persulfate (PS); all trials were conducted in duplicate
| Sample ID |
Persulfate |
Ozone |
Catalyst |
Spikea |
Description |
|
The native PFAS stock solution contained 10 μg mL−1 per compound resulting in a concentration of 1 μg L−1 per compound in the flask.
|
| Negative blank |
| 1 |
— |
— |
— |
— |
Contamination check |
| Positive blanks |
Adsorption to: |
| 2a |
— |
— |
Yes |
Yes |
Laboratory materials and catalyst |
| 2b |
— |
— |
|
Yes |
Laboratory materials |
| Degradation trials |
Treatment of PFASs through: |
| T1: O3 |
— |
Yes |
— |
Yes |
Ozone |
| T2: O3/cat |
— |
Yes |
Yes |
Yes |
Ozone and catalyst |
| T3: PS |
Yes |
— |
— |
Yes |
Persulfate |
| T4: O3/PS |
Yes |
Yes |
— |
Yes |
Ozone and persulfate |
| T5: cat/PS |
Yes |
— |
Yes |
Yes |
Persulfate and catalyst |
| T6: O3/cat/PS |
Yes |
Yes |
Yes |
Yes |
Ozone, catalyst and persulfate |
2.2.4 Pilot-scale trials.
The experiment on the pilot-scale was separated into three parts: A) adsorption to the system walls (day 1), B) adsorption to the catalyst (days 2–5) and C) degradation experiment (day 6), as summarized in Table 2. The pilot-scale system (Fig. 1b) was filled with 50 L tap water and spiked with 0.1 mL, followed by 1 mL, respectively, of a PFAS mix containing the 18 target analytes (50000 ng mL−1 per compound, in methanol), equal to a final concentration of 1.1 μg L−1 per compound. During the first test phase (A), the catalytic bed was excluded and the water circulated through the pipes of the system only, at a flow of 850 L h−1. The water was spiked with 0.1 mL of the PFAS mix in order to observe possible adsorption to the walls of the system. Samples were taken after 0.5, 1, 4, 9.5 and 23.5 h. On the next day, prior to the second spike, the catalytic bed was opened and further 10 L tap water was introduced into the system since the catalytic bed did not contain water. A second period of the water circulating through the system was awaited during days 2–5 and several samples were taken in order to monitor equilibrium adjustment during this period (see Table 2). Diverse ad- and desorption processes to and from the catalyst material were observed during the second test period (part B; see Table 2). The degradation trials (part C) investigated the effectiveness of the ozonation treatment in terms of PFAS removal and samples were collected at 0.5, 1, 2, 3, 4, 5, 6, 7 and 8 h during which ozone was applied to the system. Samples were taken in duplicate and the mean value was taken for calculations. For data analysis and better data comparability, each calculated concentration per analyzed compound and sampling time was normalized to the sample taken directly prior to turning on the ozone flow (S14 (0 h); Table 2). During the whole time of the experiment, the pH was kept constant at pH = 7.5 by automated sodium hydroxide (NaOH) or hydrochloric acid (HCl) addition. Additionally, the redox potential was constantly monitored.
Table 2 Chronological procedure of the pilot-scale trials
| Part |
Experiment |
Modifications on the system |
Timea |
Sampling |
| DD:HH:MM |
(Ozonation time) |
|
The time measurement was started with the spike of 0.1 mL of the standard mix and continued until the end of the trial.
|
| A |
Positive blank (investigate loss to system walls) |
|
00:00:00 |
S1 |
| +1st spike PFASs (0.1 mL of 50000 ng mL−1) |
00:00:00 |
| 00:00:30 |
S2 |
| 00:01:07 |
S3 |
| 00:04:00 |
S4 |
| 00:09:30 |
S5 |
| B |
Positive blank (investigate loss to catalyst) |
|
00:23:30 |
S6 |
| +5–10 L water catalytic bed was opened |
|
|
| +2nd spike PFASs (1.0 mL of 50000 ng mL−1) |
00:23:45 |
|
| 01:00:15 |
S7 |
| 01:00:46 |
S8 |
| 01:03:45 |
S9 |
| 01:07:45 |
S10 |
| 03:23:47 |
S11 |
| 04:01:47 |
S12 |
| 04:03:46 |
S13 |
| C |
Degradation trial |
|
04:23:25 |
S14 (0 h) |
| Ozone flow was turned on |
04:23:30 |
| 05:00:00 |
S15 (0.5 h) |
| 05:00:30 |
S16 (1 h) |
| 05:01:30 |
S17 (2 h) |
| 05:02:30 |
S18 (3 h) |
| 05:03:30 |
S19 (4 h) |
| 05:04:30 |
S20 (5 h) |
| 05:05:30 |
S21 (6 h) |
| 05:06:30 |
S22 (7 h) |
| 05:07:30 |
S23 (8 h) |
2.3 Instrumental analysis
Samples for the adsorption isotherm experiments, kinetic experiments and laboratory-scale experiments T1, T2 and T3 were analyzed via direct injection by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS; TSQ Quantiva, Thermo Fisher Scientific, San Jose, CA, USA) whilst on-line solid phase extraction (SPE) UHPLC-MS/MS was conducted for the pilot-scale experiment (LC/UPLC system; Thermo Scientific Dionex UltiMate 3000 Pumps; TSQ Quantiva, Thermo Fisher Scientific, San Jose, CA, USA). An Acquity UPLC BEH-C18 column (100 mm × 2.1 i.d., 1.7 μm particle size from Waters Corporation, Manchester, UK) was used as an analytical column. A Hypersil GOLD aQ column (20 mm × 2.1 mm i.d, 12 μm particles, from Thermo Fisher Scientific, San Jose, CA, USA) was used as an on-line SPE column. In order to distinguish signals due to mobile phase contamination from sample peaks, a trapping column was applied after the mixing chamber (ACQUITY UPLC column reversed-phase 1.7 μm spherical hybrid, 3 mm × 30 mm from Waters Corporation, Manchester, UK). Both the trapping column and the analytical column were equipped with an ACQUITY UPLC BEH C18 VanGuard pre-column (130 Å, 1.7 μm, 2.1 mm × 5 mm from Waters Corporation, Manchester, UK). The injection volumes were 10 μL for direct injection samples and 1.0 mL for on-line SPE samples. Data were evaluated using TraceFinder™ 3.3 software (Thermo Fisher). A detailed overview of instrumental parameters as well as single reaction monitoring transitions can be found in Tables A3 to A5 in the ESI,† respectively. Samples from laboratory-scale experiments T4, T5 and T6 were analyzed by direct injection HPLC-MS/MS (HPLC, Agilent Technologies 1200 Series, Palo Alto, CA, USA). The analysis was conducted according to a method described elsewhere.30
2.4 Quality control and quality assurance
Limits of detection (LODs) are defined as the concentrations found in the replicate negative blank samples plus three times the relative standard deviation (eqn A1 in the ESI†). Further, the limits of quantification (LOQs) are defined similarly, however by adding 10 times the relative standard deviation to blank concentrations (eqn A2 in the ESI†). In the case where no PFAS contamination was detected in the negative blank, the lowest calibration point at the same concentration with a signal to noise ratio of S/N > 3 and S/N > 10 was set as LOD and LOQ, respectively; see also Table A6 in the ESI.† Negative blank samples (n = 2) were retrieved by conducting experiments with unspiked water using the same procedure as for each respective experiment (Table A7 in the ESI†). For each experiment series performed in the laboratory positive blank experiments were conducted in duplicate. In the case where significant losses were observed, they were taken into consideration when calculating removal efficiencies or adsorption for the actual experiments. For this, a correction factor was calculated by dividing the concentration at time 0 min (sample size n = 2) by the mean concentrations across the full positive blank time series (n = 8 for kinetic and adsorption isotherm trials; n = 5 for laboratory-scale trials; for details see Table A8 in the ESI†). The recovery of the internal standards was on average 97% for the adsorption tests (n = 11), 81% for the pilot-scale test (n = 8) and 110% for the laboratory-scale test (n = 11); see also Table A9 in the ESI.†
3 Results and discussion
3.1 Sorption behaviour of PFASs to the catalyst
A kinetic study was performed in the laboratory to gain information on the adsorption velocity of PFASs to the catalytic material. All evaluated PFASs reached equilibrium concentrations within the first 10 min (data not shown). Small changes in concentration during the remaining time of the sorption experiment of 100 h were observed for PFHxS, PFOS, and FOSA, however not significant.
Adsorption isotherms showed a fit according to the Freundlich adsorption model for all the evaluated PFASs in all three water types (MilliQ, tap water and DOC water); see Table A10 in the ESI.† The adsorption intensity n was found to be ≥1 for all PFASs and water types suggesting that the adsorption is a physical process rather than a process involving chemical binding.31–33Kf describes the loading of the adsorbent with the adsorbate and was found to vary between 3.75 × 10−6 (μg g−1)(μg L−1)−n (PFOS, DOC water) and 1.36 × 10−3 (μg g−1)(μg L−1)−n (PFDoDA, tap water); see Table A10 in the ESI.† A linear relationship between ln(Kf) and the number of perfluorocarbon atoms was found for PFSAs and most PFCAs in MilliQ water (see Fig. A1 in the ESI†). This observation was also found in previous studies investigating the adsorption of PFOA and PFOS on diverse types of GAC, powdered activated carbon (PAC) and three activated carbon fibers34 and for various PFASs.35 According to Tang et al.,36 it can be assumed that the observed increase in ln(Kf) for longer chain PFCAs (Fig. A1 in ESI†) is an indication of increasing hydrophobic interactions between the catalyst surface and PFASs with increasing chain lengths. An increase in ln(Kf) for shorter chain PFASs (PFBA, PFBS) in tap water suggests increasing electrostatic interactions with decreasing chain lengths. The present study found lower ln(Kf) values for PFSAs compared to PFCAs with the same perfluorocarbon chain length in MilliQ and DOC water (Fig. A1 in the ESI†), suggesting that the PFSA adsorption strength to the catalyst material is slightly weaker than that of PFCAs with respect to the same perfluorocarbon chain length. Hansen et al.37 found that PFSAs adsorb more strongly to GAC and PAC compared to PFCAs, and the increase of the Kf is more pronounced with a higher perfluorocarbon chain length. In contrast to GAC, the catalyst material used in this study is an inorganic material. The inverted behavior of the functional groups could be due to the weak ligand properties of the slightly larger PFSA molecules compared to those of PFCAs.38 The adsorption isotherm data for tap water showed the independence of the PFAS functional group and the ln(Kf) values of all the compounds seemed to depend on the perfluorocarbon chain length only (Fig. A1 in the ESI†).
3.2 Laboratory-scale trials combining the catalyst, ozone and persulfate
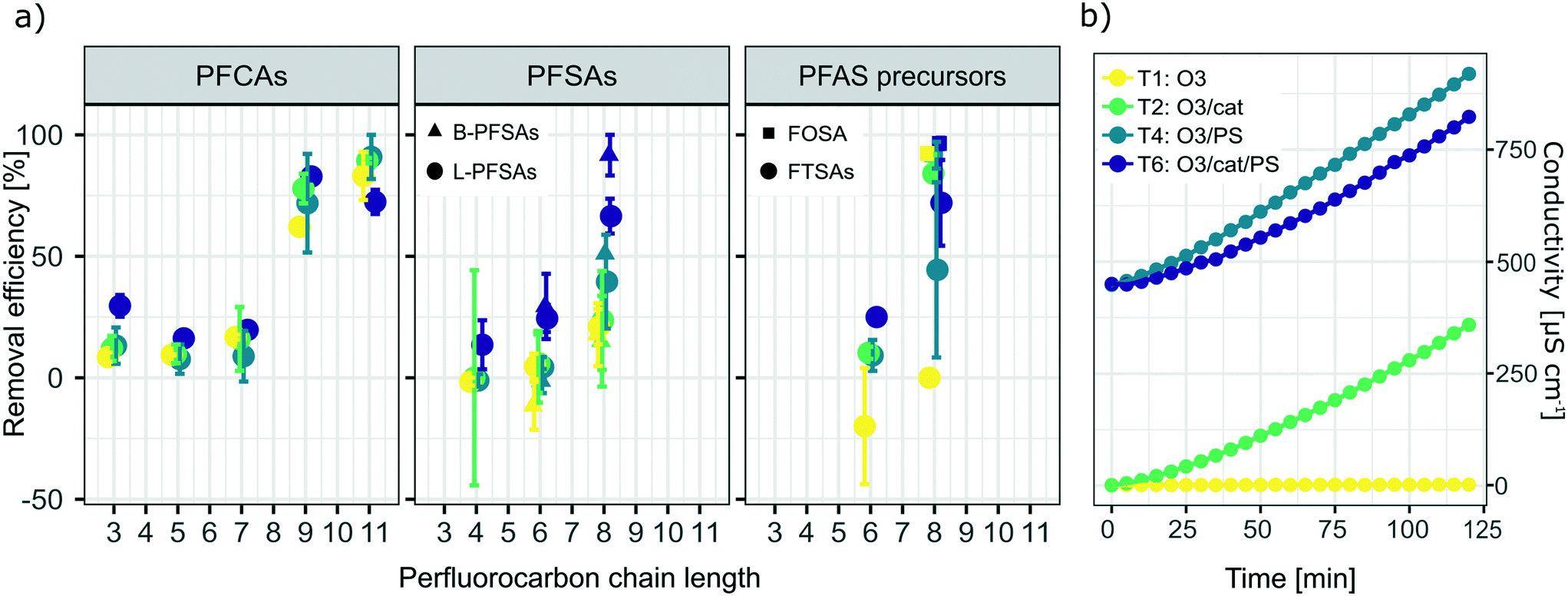
Fig. 2a) shows a summary of removal of all evaluated PFASs after 120 min of treatment on the laboratory-scale for the ozone applying trials as a function of perfluorocarbon chain length. Significant removal of several PFASs was observed for the experiments using ozone in combination with the catalyst (T2: O3/cat, which was also used on the pilot-scale, see section 3.3), ozone in combination with persulfate (T4: O3/PS), ozone in combination with the catalyst and persulfate (T6: O3/cat/PS) and applying ozone only (T1: O3); see also Table A11 in the ESI.† In contrast to that, for all other possible treatment combinations described in Table 1 (i.e. T3: PS and T5: cat/PS) no significant PFAS removal was found, indicating that the catalyst did not activate persulfate into sulfate radicals for transformation of PFASs. It was noted that i) the removal increased with increasing perfluorocarbon chain length and ii) the removal did not show a general trend regarding the PFAS functional group (Fig. 2). The observed trend of increasing removal with increasing perfluorocarbon chain length was consistent within a set of PFASs with the same functional group with the exception of PFBA in experiments T2 (O3/cat), T4 (O3/PS) and T6 (O3/cat/PS) for which the removal was larger than that for some longer chain PFCAs. No significant trend was found for the difference in removal between linear (L) and branched (B) isomers (p-value < 0.05, Fig. 2 and Table A11 in the ESI†). The treatment with ozone only (T1) showed the worst performance in comparison to the other three ozone applying treatment approaches (T2, T4, T6). This was expected, as other authors have reported that ozone alone does not have a significant impact on PFAS concentrations.8–10,39 The average removal efficiencies for the different experiments increased in the order T1 (O3): 22% average removal efficiency <T4 (O3/PS): 24% < T2 (O3/cat): 26% < T6 (O3/cat/PS): 47%. Compared to the combination used in the pilot-scale treatment (O3/cat, see section 3.3), the combination of all three parameters (O3/cat/PS) was found to improve the removal efficiency for many of the spiked PFASs significantly: +18% (PFBA), +14% (PFBS), +19% (linear PFHxS; L-PFHxS), +77% (branched PFHxS; B-PFHxS), +43% (L-PFOS), +15% (B-PFOS) and +15% (6:2 FTSA). An improved oxidizing ability for the combination of ozone and persulfate was previously reported by Abu Amr et al., 2014 (ref. 28) and Yang et al., 2016.19 It is argued that persulfate can be activated to form SO4˙− in the presence of OH˙:| | 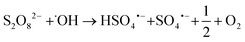 | (2) |
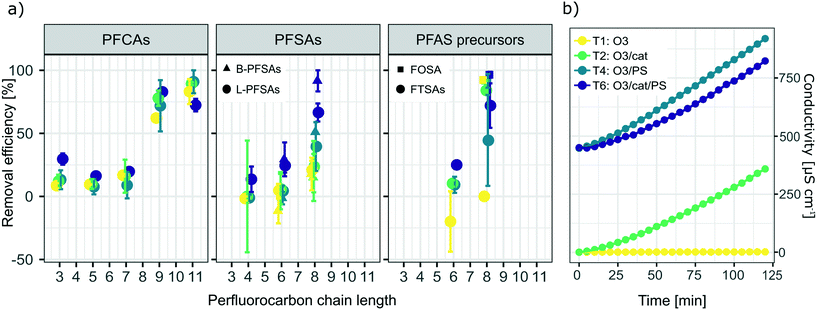 |
| | Fig. 2 a) Removal of all analyzed PFASs after 120 min treatment on the laboratory-scale for experiments applying ozone in combination with the catalyst (T2: O3/cat), ozone in combination with persulfate (T4: O3/PS), ozone in combination with the catalyst and persulfate (T6: O3/cat/PS) and applying ozone only (T1: O3). Error bars depict the variation between duplicate experiments. The significance of concentration changes is listed in Table A11 in the ESI.† b) Development of conductivity throughout the duration of the laboratory-scale experiments illustrated in a). | |
Other authors have assumed the presence of other reactive radical species like superoxide to play an important role in catalyzed persulfate systems, as reviewed by Waclawek et al.40 Therefore, it can be assumed that an additional application of the catalyst material further increases the amount of radicals and thereby leads to increased PFAS transformation as observed in T6 (O3/cat/PS). The conductivity increased for all ozone applying trials, with the exception of T1 (ozone only); see also Fig. 2b). This further indicates the formation of charged species, such as radical species described above and possible activation of persulfate. It should be noted that a decrease of the initial pH from pH = 6–7 to a final value of pH = 3 was observed for all trials in which ozone was applied (see Fig. A2 in the ESI†). During the treatment with no ozone, the observed changes in pH were within less than one pH unit (see Fig. A3 in the ESI†). Curiously, a similar decrease in pH was observed for the blank trial, applying only the catalyst without ozone. As repeated blank trials of the same kind did not show a decrease in pH (data not shown), the observed drop in pH might be due to a faulty pH meter, and thus cannot be evaluated further. The observed removal of the long-chain PFASs during the laboratory-scale trials could be a result of the existence of these PFASs at the gas–water interface of introduced ozone bubbles, leading to an underestimation of PFASs over time.41,42 As all ozone applying trials however were run under the very same conditions, the trials can still be considered comparable to each other and the reasoning for the discussion points given above applies.
3.3 Removal efficiency of ozone and catalyst treatment on pilot-scale
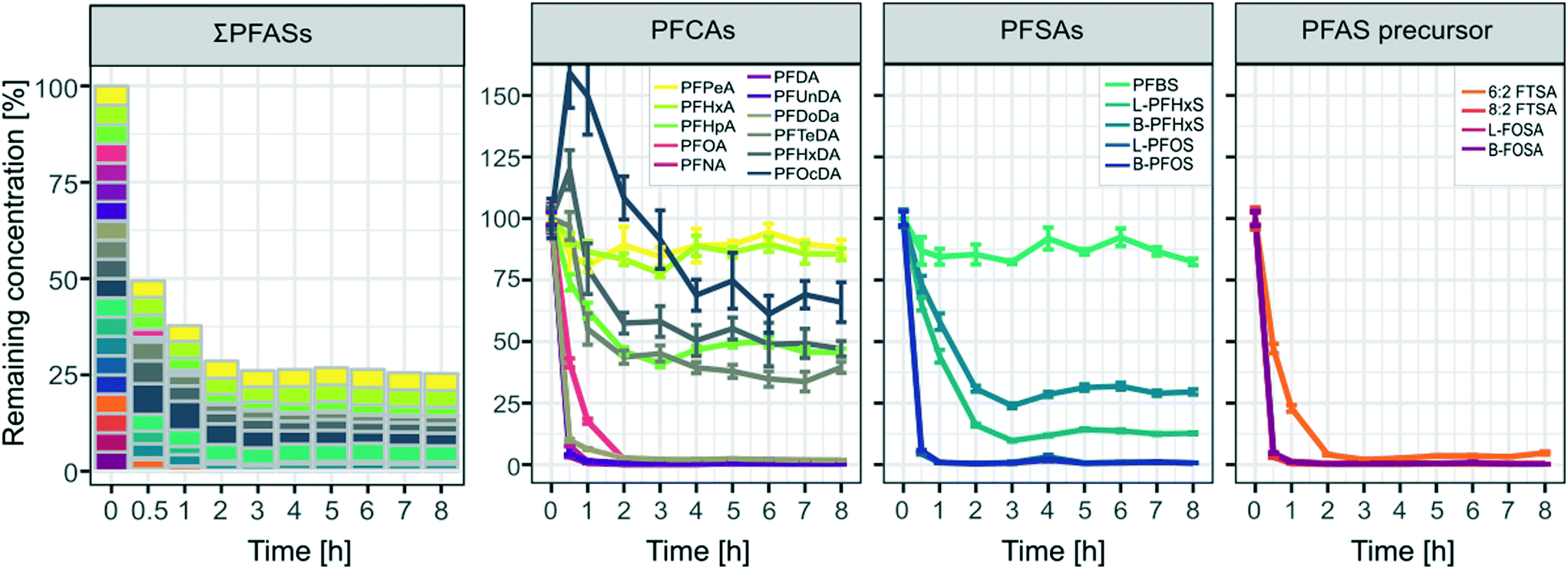
The concentrations after the first spike (part A, see also Table 2) remained almost constant indicating that adsorption to walls of the pilot-scale system was negligible even at low concentrations of 100 ng L−1 for individual PFASs (Fig. A4 in the ESI†). As can be seen in Fig. 3 a rapid decrease of >50% for ∑PFAS concentrations was observed during the first 30 min of the ozonation experiment. After 1 h and 2 h, the removal of PFASs slowed down and stagnated at ca. 70% after 3 h with remaining amounts of 38% (1 h), 29% (2 h) and 26% (3 h) of ∑PFASs, respectively. The decrease in concentration was significant (two sample t-test, one sided) with a p-value < 0.05 for PFPeA and p < 0.01 for all other analyzed PFASs from the moment when the ozonation started until the end of the trial. PFCAs with the longest evaluated perfluorocarbon chain length PFHxDA (CF15) and PFOcDA (CF17) showed an increase in concentrations in the first 30 min of the experiment. This can most likely be explained by desorption from the catalyst after disturbing the system by the introduction of ozone. Removal was observed for PFHxDA and PFOcDA after 30 min of ozone introduction.
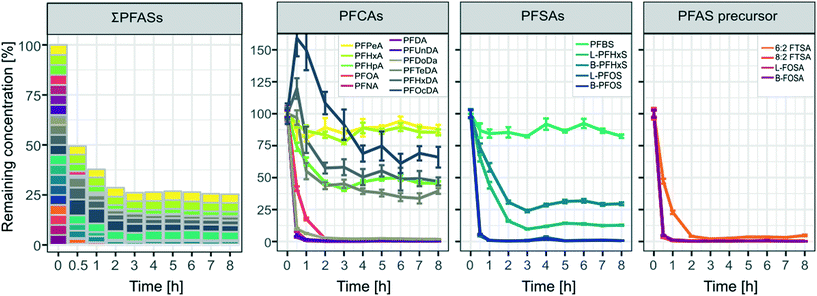 |
| | Fig. 3 Remaining concentrations of all spiked PFASs [%] over time for the pilot-scale ozonation trial. Values were derived from the mean concentrations from the duplicate sample taken at each respective time point. Error bars indicate the mean deviation of the duplicate samples from their mean. All calculations were normalized to concentrations found in the samples taken at 0 h, right before ozone was introduced into the system. | |
The pH in the pilot-scale system decreased as soon as ozone was introduced (see Fig. A5 in the ESI†) and needed to be stabilized by adding NaOH, as described in section 2.2.4. A drop in pH during ozonation can commonly be observed if organic matter containing double bonds is present in the treated water matrix, which can be the case for tap water. Ozonation leads to the formation of lower molecular weight organic acids, causing the pH to drop significantly.43,44 Next to a thesis published in 2016,45 the present study is one of the first to report removal of PFASs by a catalytic ozonation treatment on a pilot scale. Other studies evaluating pilot or full-scale ozone treatments found no significant PFAS removal.7,46–48 Ozonation studies reporting PFAS degradation are on the other hand limited to laboratory-scale experiments.9,21,49
3.4 Impact of the perfluorocarbon chain length and functional group
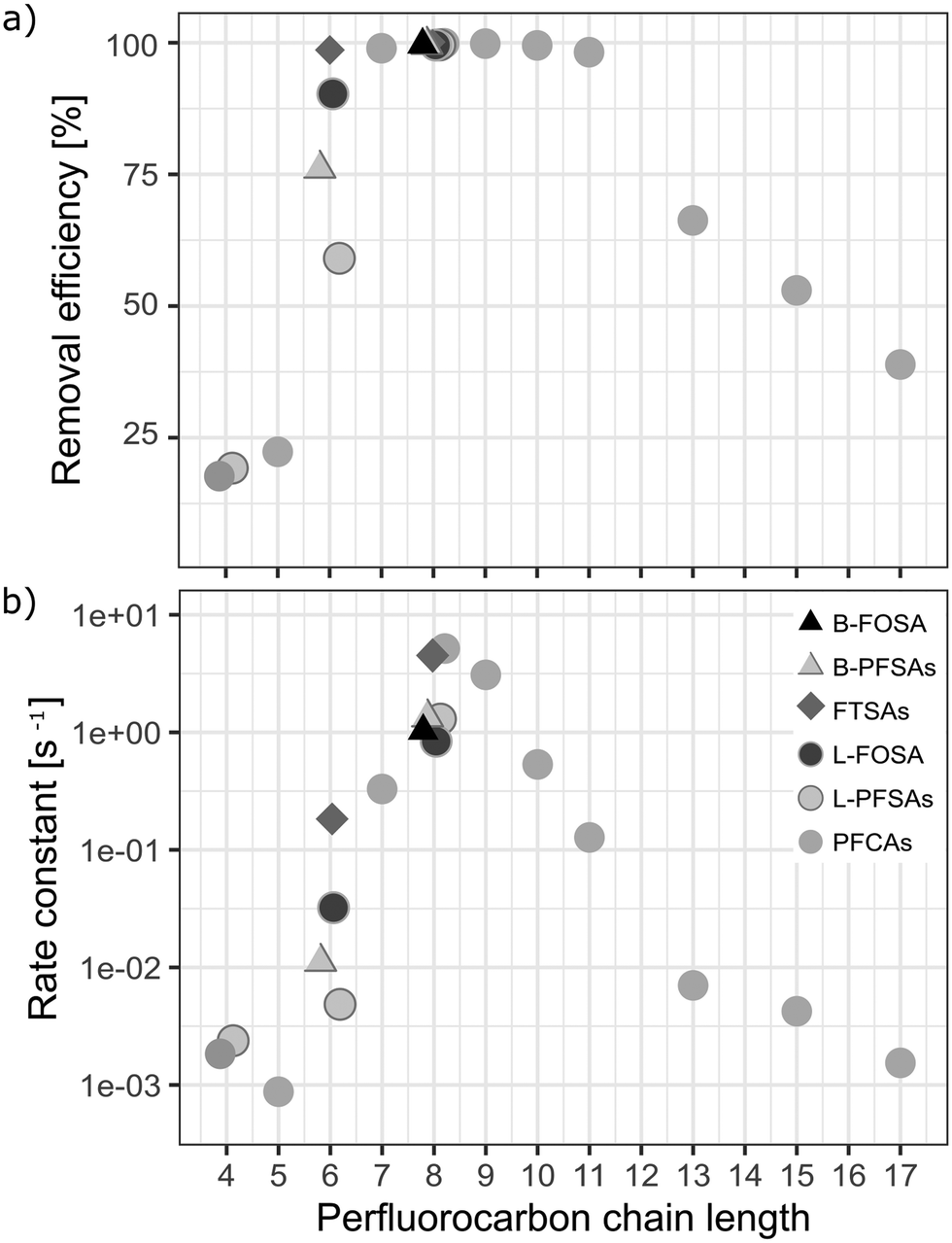
A removal efficiency of more than 98% was found for PFASs with a chain length of seven to eleven perfluorocarbon atoms (CF7 − CF11) independent of the functional group in the pilot-scale ozonation trial, as shown in Fig. 4a. PFASs with CF4 − CF6 showed the lowest average removal efficiency (55% on average) compared to CF12 − CF17 (64%) and CF7 − CF11 (99%). The outstanding removal efficiencies for CF7 − CF11 PFASs were observed for both partially (i.e. 6:2 FTSA and 8:2 FTSA) and fully fluorinated PFASs (i.e. PFOA, PFNA, PFDA, PFUnDA, PFOS, and FOSA). Short chain PFCAs, such as PFPeA, PFHxA and PFHpA, showed a slight increase of 11%, 21% and 25%, respectively (significant with p-value < 0.05, two sample t-test, one sided), in concentration after 3 h while longer chain congeners, CF12 − CF17 showed continued removal at that point. This may indicate step-wise unzipping of the perfluoroalkyl carbon chain through splitting-off of a –CF2 moiety followed by hydrolysis, as proposed previously for the reaction of PFASs with superoxide radicals that can be created in AOPs.22,51 The removal reactions for all analyzed PFASs followed second order reaction rates (Fig. 4b). Interestingly, these reaction rates showed a similar trend to the maximal removal efficiencies in terms of chain-length dependence observed during the 8 h treatment, see Fig. 4a. This indicates that the treatment is highly efficient (removal efficiency >99% in less than 3 h of treatment) for CF7 − CF11 PFASs independent of the functional group.
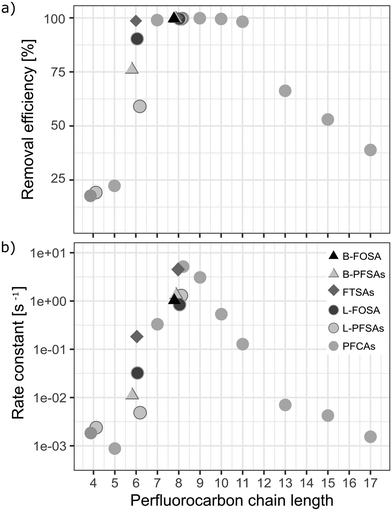 |
| | Fig. 4 a) Maximum removal efficiencies [%] after 8 h of ozonation and b) second order reaction rate constants [s−1] for all analyzed PFASs of varying perfluorocarbon chain lengths and functional groups in the pilot-scale ozonation treatment. Rate constants were calculated according to the second order rate law described by Atkins et al.50 | |
Overall, a correlation between the adsorption strength and PFAS removal efficiency was not found. This means that the removal mechanism cannot be predicted from the adsorption strength of PFASs to the catalyst. Previous studies found that the degradation efficiency of PFASs in water by means of uncatalyzed ozonation on a laboratory-scale as well as on an industrial scale is low.7,10,46 This was confirmed in the laboratory-scale experiments conducted in this study. For several AOP approaches, it is argued that hydroxyl radicals are not responsible for PFAS degradation.22 Mitchell et al. found that non-hydroxyl radical reactive oxygen species or low concentrations of other reducing species (superoxide radical anions, perhydroxyl radicals, and/or hydroperoxide anions) were responsible for PFOA degradation in a 2-propanol scavenged reaction.22,39
4 Conclusions
In this study a commercially available treatment based on heterogeneous catalysis was evaluated, where the PFAS adsorption behaviour to the catalyst material was investigated in detail. Laboratory-scale trials were performed in order to gain knowledge about potential improvement of the treatment method and degradation of PFASs in tap water was investigated on a pilot scale. An investigation of the catalyst adsorption capacity Kf showed that the binding strength increased for shorter as well as for longer chain PFASs compared to the other PFASs measured in this study suggesting both electrostatic and hydrophobic interactions of PFASs with the catalytic material. Results of the conducted laboratory-scale experiments indicate that the treatment can be improved by the use of persulfate, as the combination of the catalyst and ozone with persulfate showed increased removal efficiencies of up to 77% in comparison to the method of the catalyst combined with ozone. In the pilot-scale experiment, PFAS concentrations were successfully decreased in tap water with an overall maximum removal efficiency of ca. 70% and removal efficiencies of >98% for CF7 − CF11 PFASs, independent of the functional group. The high removal performance of CF7 − CF11 PFASs observed in the pilot-scale trial did not correspond to the adsorption behaviour found in the conducted adsorption studies. Therefore it can be suggested that the dominant factor for the removal is rather the type of interaction of PFASs with reactive species created during the process than the binding strength to the catalytic surface. The present study implies that the evaluated treatment method is a promising technique for the removal of PFASs from water, especially for PFASs with a perfluorocarbon chain length of six to eleven. Ozone can be created, applied and destroyed safely on-site. The evaluated system is a commercially available treatment method, and also available as a mobile system, and could therefore easily be applied in water treatment or in pump-and-treat processes for the treatment of contaminated water. Further research should investigate the production of degradation products and potentially unwanted by-products, monitor the occurrence of compounds in the gas-phase and prove the effectiveness of the treatment for other types of contaminants and application on different water types.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly funded by the Swedish Research Council Formas (PFAS-FREE, contract nr. 942-2015-1554). The authors would like to thank Ozone Tech Systems for the provided support and especially recognize the extensive help of Ramnath Lakshmanan. Further, the authors acknowledge the generous loan of an ozone generator for the laboratory scale trials from ACES at Stockholm University.
References
- K. Steenland, S. Tinker, S. Frisbee, A. Ducatman and V. Vaccarino, Association of Perfluorooctanoic Acid and Perfluorooctane Sulfonate With Serum Lipids Among Adults Living Near a Chemical Plant, Am. J. Epidemiol., 2009, 170, 1268–1278 CrossRef PubMed.
- A. Glynn, U. Berger, A. Bignert, S. Ullah, M. Aune, S. Lignell and P. O. Darnerud, Perfluorinated alkyl acids in blood serum from primiparous women in Sweden: serial sampling during pregnancy and nursing, and temporal trends 1996–2010, Environ. Sci. Technol., 2012, 46, 9071–9079 CrossRef CAS PubMed.
- I. Gyllenhammar, U. Berger, M. Sundström, P. McCleaf, K. Eurén, S. Eriksson, S. Ahlgren, S. Lignell, M. Aune, N. Kotova and A. Glynn, Influence of contaminated drinking water on perfluoroalkyl acid levels in human serum – A case study from Uppsala, Sweden, Environ. Res., 2015, 140, 673–683 CrossRef CAS PubMed.
- Z. Wang, J. C. DeWitt, C. P. Higgins and I. T. Cousins, A Never-Ending Story of Per- and Polyfluoroalkyl Substances (PFASs)?, Environ. Sci. Technol., 2017, 51, 2508–2518 CrossRef CAS PubMed.
- J. Thompson, G. Eaglesham, J. Reungoat, Y. Poussade, M. Bartkow, M. Lawrence and J. F. Mueller, Removal of PFOS, PFOA and other perfluoroalkyl acids at water reclamation plants in South East Queensland, Australia, Chemosphere, 2011, 82, 9–17 CrossRef CAS PubMed.
- T. D. Appleman, E. R. Dickenson, C. Bellona and C. P. Higgins, Nanofiltration and granular activated carbon treatment of perfluoroalkyl acids, J. Hazard. Mater., 2013, 260, 740–746 CrossRef CAS PubMed.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed.
- N. Merino, Y. Qu, R. A. Deeb, E. L. Hawley, M. R. Hoffmann and S. Mahendra, Degradation and Removal Methods for Perfluoroalkyl and Polyfluoroalkyl Substances in Water, Environ. Eng. Sci., 2016, 33, 615–649 CrossRef CAS.
- H. F. Schröder and R. J. Meesters, Stability of fluorinated surfactants in advanced oxidation processes—A follow up of degradation products using flow injection–mass spectrometry, liquid chromatography–mass spectrometry and liquid chromatography–multiple stage mass spectrometry, J. Chromatogr. A, 2005, 1082, 110–119 CrossRef PubMed.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA), Front. Environ. Sci. Eng. China, 2009, 3, 129–151 CrossRef CAS.
- P. Wardman, Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution, J. Phys. Chem. Ref. Data, 1989, 18, 1637 CrossRef CAS.
- V. A. A. Espana, M. Mallavarapu and R. Naidu, Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA): A critical review with an emphasis on field testing, Environmental Technology and Innovation, 2015, 4, 168–181 CrossRef.
- K. H. Kucharzyk, R. Darlington, M. Benotti, R. Deeb and E. Hawley, Novel treatment technologies for PFAS compounds A critical review, J. Environ. Manage., 2017, 204, 757–764 CrossRef CAS PubMed.
- H. Suty, C. De Traversay and M. Cost, Applications of advanced oxidation processes: present and future, Water Sci. Technol., 2004, 49, 227–233 CrossRef CAS PubMed.
- W. H. Glaze, J.-W. Kang and D. H. Chapin, The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation, Ozone Sci Eng, 1987, 9, 335–352 CrossRef CAS.
- R. Andreozzi, V. Caprio, A. Insola and R. Marotta, Advanced oxidation processes (AOP) for water purification and recovery, Catal. Today, 1999, 53, 51–59 CrossRef CAS.
- J. N. Barbara Kasprzyk-Hordern and M. Ziólek, Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment, Appl. Catal., B, 2003, 46, 63–669 Search PubMed.
- M. A. Oturan and J.-J. Aaron, Advanced Oxidation Processes in Water/Wastewater Treatment: Principles and Applications. A Review, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- Y. Yang, H. Guo, Y. Zhang, Q. Deng and J. Zhang, Degradation of bisphenol A using ozone/persulfate process: kinetics and mechanism, Water, Air, Soil Pollut., 2016, 227, 53 CrossRef.
-
D. R. Brink, B. Langlais and D. A. Reckhow, Ozone in Water Treatment: Application and Engineering: Cooperative Research Report, Lewis Publishers, 1991 Search PubMed.
- X. Yang, J. Huang, K. Zhang, G. Yu, S. Deng and B. Wang, Stability of 6: 2 fluorotelomer sulfonate in advanced oxidation processes: degradation kinetics and pathway, Environ. Sci. Pollut. Res., 2014, 21, 4634–4642 CrossRef CAS PubMed.
- P. M. Dombrowski, P. Kakarla, W. Caldicott, Y. Chin, V. Sadeghi, D. Bogdan, F. Barajas-Rodriguez and S.-Y. D. Chiang, Technology review and evaluation of different chemical oxidation conditions on treatability of PFAS, Remediation, 2017, 28, 135–150 CrossRef.
- H. Hori, A. Yamamoto, E. Hayakawa, S. Taniyasu, N. Yamashita, S. Kutsuna, H. Kiatagawa and R. Arakawa, Efficient Decomposition of Environmentally Persistent Perfluorocarboxylic Acids by Use of Persulfate as a Photochemical Oxidant, Environ. Sci. Technol., 2005, 39, 2383–2388 CrossRef CAS PubMed.
-
S. G. Huling and B. E. Pivetz, In-situ chemical oxidation, Environmental protection agency washington dc office of water technical report, 2006 Search PubMed.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbæk, R. Siegrist and P. Bjerg, In Situ Chemical Oxidation of Contaminated Soil and Groundwater Using Persulfate: A Review, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- H. Hori, Y. Nagaoka, T. Sano and S. Kutsuna, Iron-induced decomposition of perfluorohexanesulfonate in sub- and supercritical water, Chemosphere, 2008, 70, 800–806 CrossRef CAS PubMed.
- B. Wang, M. Cao, Z. Tan, L. Wang, S. Yuan and J. Chen, Photochemical decomposition of perfluorodecanoic acid in aqueous solution with VUV light irradiation, J. Hazard. Mater., 2010, 181, 187–192 CrossRef CAS PubMed.
- S. S. Abu Amr, H. Abdul Aziz, M. N. Adlan and J. M. A. Alkasseh, Effect of Ozone and Ozone/Persulfate Processes on Biodegradable and Soluble Characteristics of Semiaerobic Stabilized Leachate, Environ. Prog. Sustainable Energy, 2014, 33, 184–191 CrossRef CAS.
- H. F. Schröder, H. José, W. Gebhardt, R. Moreira and J. Pinnekamp, Biological wastewater treatment followed by physicochemical treatment for the removal of fluorinated surfactants, Water Sci. Technol., 2010, 61, 3208 CrossRef PubMed.
- L. Ahrens, H. Gashaw, M. Sjöholm, S. G. Gebrehiwot, A. Getahun, E. Derbe, K. Bishop and S. Åkerblom, Poly-and perfluoroalkylated substances (PFASs) in water, sediment and fish muscle tissue from Lake Tana, Ethiopia and implications for human exposure, Chemosphere, 2016, 165, 352–357 CrossRef CAS PubMed.
- V. Ochoa-Herrera and R. Sierra-Alvarez, Removal of perfluorinated surfactants by sorption onto granular activated carbon, zeolite and sludge, Chemosphere, 2008, 72, 1588–1593 CrossRef CAS PubMed.
- S. Senevirathna, S. Tanaka, S. Fujii, C. Kunacheva, H. Harada, B. Shivakoti and R. Okamoto, A comparative study of adsorption of perfluorooctane sulfonate (PFOS) onto granular activated carbon, ion-exchange polymers and non-ion-exchange polymers, Chemosphere, 2010, 80, 647–651 CrossRef CAS PubMed.
- X. Yang, J. Huang, K. Zhang, G. Yu, S. Deng and B. Wang, Batch Sorption Experiments: Langmuir and Freundlich Isotherm Studies for the Adsorption of Textile Metal Ions onto Teff Straw (Eragrostis tef) AgriculturalWaste, J. Thermodyn., 2013, 1, 1–6 Search PubMed.
- Y. Zhi and J. Liu, Adsorption of perfluoroalkyl acids by carbonaceous adsorbents: Effect of carbon surface chemistry, Environ. Pollut., 2015, 202, 168–176 CrossRef CAS PubMed.
-
Y. Qiu, Study on treatment technologies for perfluorochemicals in wastewater, PhD thesis, Kyoto University, 2007 Search PubMed.
- C. Y. Tang, Q. S. Fu, D. Gao, C. S. Criddle and J. O. Leckie, Effect of solution chemistry on the adsorption of perfluorooctane sulfonate onto mineral surfaces, Water Res., 2010, 44, 2654–2662 CrossRef CAS PubMed.
- M. C. Hansen, M. H. Børresen, M. Schlabach and G. Cornelissen, Sorption of perfluorinated compounds from contaminated water to activated carbon, J. Soils Sediments, 2010, 10, 179–185 CrossRef CAS.
- C. P. Higgins and R. G. Luthy, Modeling Sorption of Anionic Surfactants onto Sediment Materials:âĂL’ An a priori Approach for Perfluoroalkyl Surfactants and Linear Alkylbenzene Sulfonates, Environ. Sci. Technol., 2007, 41, 3254–3261 CrossRef CAS PubMed.
- S. M. Mitchell, M. Ahmad, A. L. Teel and R. J. Watts, Degradation of Perfluorooctanoic Acid by Reactive Species Generated through Catalyzed H2O2Propagation Reactions, Environ. Sci. Technol. Lett., 2014, 1, 117–121 CrossRef CAS.
- S. Waclawek, H. V. Lutze, K. Grubel, V. V. Padil, M. Cernik and D. D. Dionysiou, Chemistry of persulfates in water and wastewater treatment: A review, Chem. Eng. J., 2017, 330, 44–62 CrossRef CAS.
- M. Reth, U. Berger, D. Broman, I. Cousins, E. D. Nilsson and M. S. McLachlan, Water-to-air transfer of perfluorinated carboxylates and sulfonates in a sea spray simulator, Environ. Chem., 2011, 8, 381–388 CrossRef CAS.
- L. Vierke, L. Ahrens, M. Shoeib, W.-U. Palm, E. M. Webster, D. A. Ellis, R. Ebinghaus and T. Harner, In situ airâĂSwater and particleâĂSwater partitioning of perfluorocarboxylic acids, perfluorosulfonic acids and perfluorooctyl sulfonamide at a wastewater treatment plant, Chemosphere, 2013, 92, 941–948 CrossRef CAS PubMed.
- M. T. Garcia-Cubero, G. Gonzalez-Benito, I. Indacoechea, M. Coca and S. Bolado, Effect of ozonolysis pretreatment on enzymatic digestibility of wheat and rye straw, Bioresour. Technol., 2009, 100, 1608–1613 CrossRef CAS PubMed.
- P. Bose and D. A. Reckhow, The effect of ozonation on natural organic matter removal by alum coagulation, Water Res., 2007, 41, 1516–1524 CrossRef CAS PubMed.
-
J. Joos Lindberg, M. Sc. thesis, KTH Royal Institute of Technology, 2016 Search PubMed.
- T. D. Appleman, C. P. Higgins, O. Quiñones, B. J. Vanderford, C. Kolstad, J. C. Zeigler-Holady and E. R. Dickenson, Treatment of poly- and perfluoroalkyl substances in U.S. full-scale water treatment systems, Water Res., 2014, 51, 246–255 CrossRef CAS PubMed.
- C. Eschauzier, E. Beerendonk, P. Scholte-Veenendaal and P. D. Voogt, Impact of Treatment Processes on the Removal of Perfluoroalkyl Acids from the Drinking Water Production Chain, Environ. Sci. Technol., 2012, 46, 1708–1715 CrossRef CAS PubMed.
- C. Flores, F. Ventura, J. Martin-Alonso and J. Caixach, Occurrence of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in N.E. Spanish surface waters and their removal in a drinking water treatment plant that combines conventional and advanced treatments in parallel lines, Sci. Total Environ., 2013, 461–462, 618–626 CrossRef CAS PubMed.
- H. Lin, J. Niu, S. Ding and L. Zhang, Electrochemical degradation of perfluorooctanoic acid (PFOA) by Ti/SnO2–Sb, Ti/SnO2–Sb/PbO2 and Ti/SnO2–Sb/MnO2 anodes, Water Res., 2012, 46, 2281–2289 CrossRef CAS PubMed.
-
P. W. Atkins and J. de Paula, Physical chemistry, Oxford University Press, 2006 Search PubMed.
- H. Lin, J. Niu, J. Xu, H. Huang, D. Li, Z. Yue and C. Feng, Highly Efficient and Mild Electrochemical Mineralization of Long-Chain Perfluorocarboxylic Acids (C9-C10) by Ti/SnO 2-Sb-Ce, Ti/SnO 2-Sb/Ce-PbO 2, and Ti/BDD Electrodes, Environ. Sci. Technol., 2013, 47, 13039–13046 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ew00339h |
| ‡ Now at Rheinische-Friedrich-Wilhelms-University, Bonn, Germany. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Miriam Dorothea
Schäfers‡
*a,
Miriam Dorothea
Schäfers‡