
Adsorption of organic micropollutants onto biochar: a review of relevant kinetics, mechanisms and equilibrium

Abstract
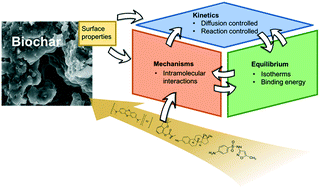
As an alternative to activated carbon, biochar has been considered for removal of organic micropollutants from water and wastewater via adsorption. This review elaborates on the fundamental basis of adsorption kinetics, mechanisms, and equilibrium with respect to biochar-based adsorption of micropollutants. The objectives include: 1) linking biochar surface properties with adsorption abilities, 2) categorizing the kinetics of adsorption of aqueous-phase organic compounds onto biochar, 3) categorizing the molecular-scale interactions between organic micropollutants and biochar, and 4) reviewing existing quantitative methods for characterizing adsorption equilibrium of organic micropollutants from water onto an adsorbent surface. To fulfill these goals, the relationships among biochar surface properties, adsorption kinetics, mechanisms, and equilibrium were clarified as current literature often lacks such discussion or may include conflicting descriptions. Due to its heterogeneous nature, research on biochar's adsorption potential for micropollutants is ambiguous. By adapting adsorption theories to biochar application specifically, this review helps to inform future research in terms of addressing knowledge gaps in characterizing and improving biochar adsorption.
- This article is part of the themed collection: Best Papers 2019 – Environmental Science: Water Research & Technology
 Please wait while we load your content...
Please wait while we load your content...

