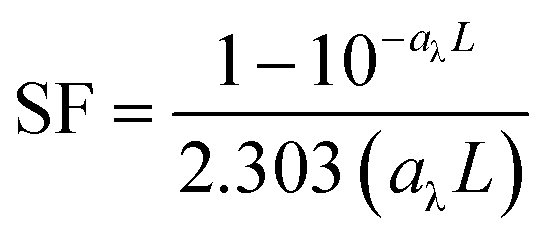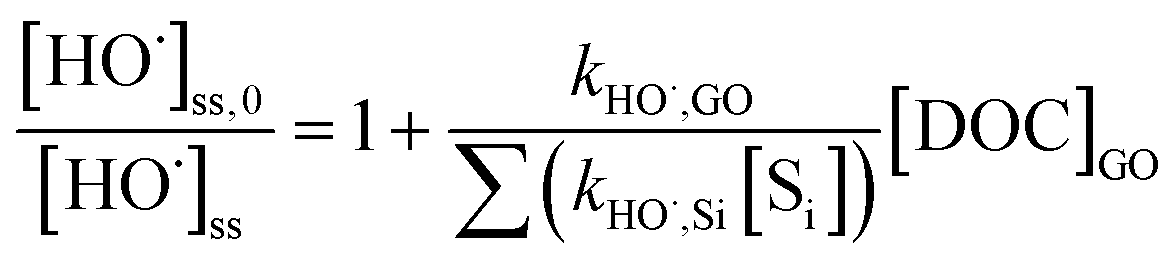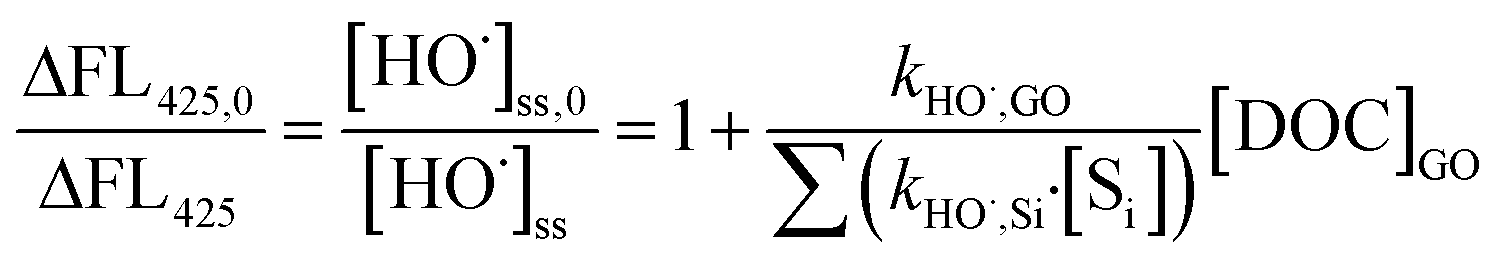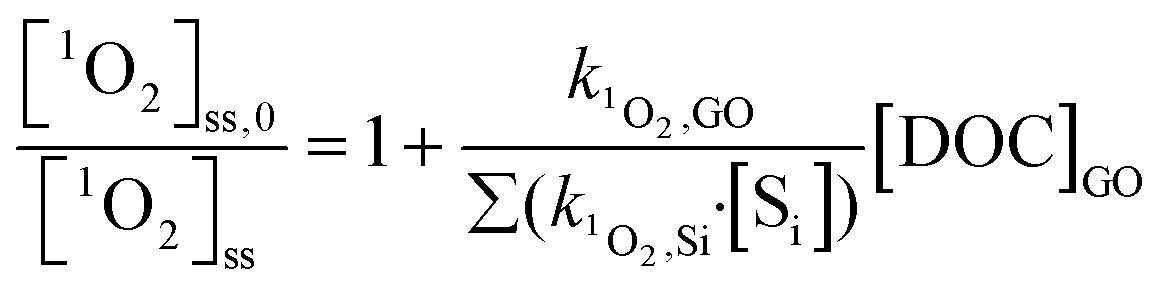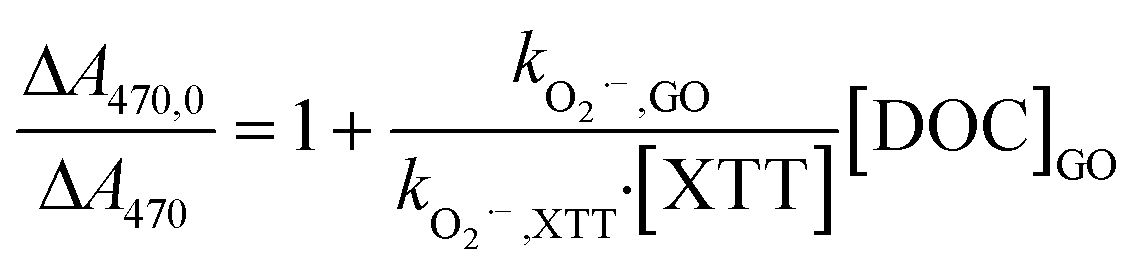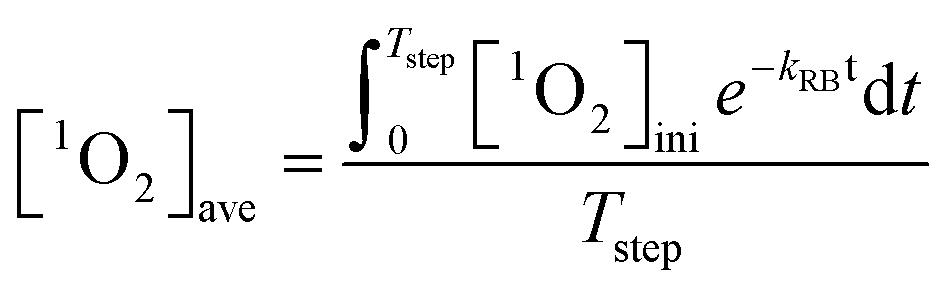Reactivity of graphene oxide with reactive oxygen species (hydroxyl radical, singlet oxygen, and superoxide anion)†
Received
22nd June 2019
, Accepted 9th October 2019
First published on 15th November 2019
Abstract
Increases in the production and applications of graphene oxide (GO), coupled with reports of its toxic effects, are raising concerns about its health and ecological risks. To better understand GO's fate and transport in aquatic environments, we investigated its reactivity with three major reactive oxygen species (ROS): HO˙, 1O2, and O2˙−. Second-order degradation rate constants were calculated on the loss of dissolved organic carbon (DOC) and steady-state concentration of individual ROS species. Absolute second-order rate constants were determined by competition kinetics to be 6.24 × 104, 8.65 × 102, and 0.108 mg-C−1 L s−1 for HO˙, 1O2, and O2˙−, respectively. Photoreduced GO products had a similar reactivity to HO˙ as GO, with rate constants comparable to polycyclic aromatic compounds, but about two times higher than dissolved organic matter on a per carbon basis. Reaction with HO˙ resulted in decomposition of GO, with loss of color and formation of photoluminescent products. In contrast, reaction with 1O2 showed no effect on DOC, UV-vis spectra or particle size, while reaction with O2˙− slightly reduced GO. These results demonstrate that interactions with ROS will affect GO's persistence in water and should be considered in exposure assessment or environmental application of GO.
Environmental significance
Major challenges in exposure assessment of graphene-based nanomaterials are evaluations of their stability and susceptibility to different reactive reagents in the environment. Our study demonstrated that reaction with singlet oxygen in the aquatic environment had a negligible effect on transformation of GO although it is normally two- to three-orders of magnitude more concentrated than HO˙. In contrast, hydroxyl radical attack quickly degraded not only GO, but also the less photoreactive rGO, the main photoproduct of GO. Together, these results suggest that an integrated understanding of ROS distribution and GO reactivity with ROS is needed to better assess long-term exposure to GO and rGO in the environment and in designing GO or rGO for environmental applications.
|
Introduction
Graphene-based nanomaterials have attracted attention from many disciplines including electronics, composite materials and energy, and from environmental and biomedical applications.1–3 Graphene oxide (GO) is of particular interest due to its economical production and its versatility for transformation into other graphene-based materials such as reduced graphene oxide or graphene quantum dots.4–6 Given the rapid advances in mass production of GO and potential increases in exposure, coupled with various toxic effects,7,8 health and ecological risk concerns have emerged. To assess exposure risk, there must be a better understanding of fate and transport of GO in aquatic environments, especially persistence under different environmental conditions.9 Research suggests that reactivity of GO in indirect photolysis or reactions with environmental reactive reagents (e.g., enzymatic oxidation) may be crucial to regulating long-term exposure.10,11
In natural waters, singlet oxygen (1O2), superoxide anion (O2˙−), and hydroxyl radical (HO˙) are three major reactive oxygen species (ROS). In sunlit waters, steady-state concentrations for 1O2, O2˙−, and HO˙ ranged from 10−12–10−13 M, 10−9–10−12 M, and 10−15–10−18 M, respectively.12 Additionally, HO˙ can be generated in reactions between reduced-form metals (e.g., Fe(II)) or minerals (e.g., iron oxide) with H2O2.13,14 Due to the extensive presence of H2O2 – some of which is produced by biological sources – metal-catalyzed HO˙ sources may dominate transformation of contaminants in waters where UV light irradiation is limited.15 ROS reactivity of various carbon nanomaterials has been reported. Fullerene (C60) reacts with 1O2 at a rate constant of (0.69 ± 0.28) mg-C−1 L s−1, presumably via physical quenching.16 Exposure of 1O2 to photo-excited C60 leads to formation of fullerene oxides.17 Scavenging abilities of fullerenols and carbon nanotubes (CNT) toward HO˙, 1O2, and O2˙− have been confirmed by spin trapping.18,19 Other product studies of CNT reactions show that reactions with 1O2 resulted in both surface oxide formation and decarboxylation, while reaction with HO˙ caused continuous loss of carboxyl groups and evolution of CO2.20
Reactivities of GO with ROS are interesting for their relevance to applications, toxicity, and transformation in biological and natural systems. For example, HO˙ was proposed as oxidant for degrading GO into graphene quantum dots, supported by evidences of microscopy (TEM and AFM), spectroscopy (XPS and Raman) and mass spectrometry.21,22 Additionally, HO˙ was suggested as the major species responsible for decomposition of GO in solar irradiation and ozonation.23–26 To estimate the reaction rate constant, Hou et al. irradiated 100 mM H2O2 to produce HO˙ and measured the resulting DOC decay of GO.10 The reported value is uncertain, however, due to coexistence of direct photolysis and HO˙ reaction. GO itself may generate 1O2via photolysis23,27,28 or by catalytic decomposition of H2O2,29 a ubiquitous ROS species in natural waters. While 1O2 was reported to mediate the photooxidation of graphene,30 reactivity of GO with 1O2 is not yet confirmed. Extracellular O2˙− was reported to reduce GO in saline solution in the dark,31 but there is also a knowledge gap on the rate of the reduction reaction. Only limited studies are available for quantifying the rate of reactions between GO and individual ROS, especially in the absence of direct photolysis.
Here, we present a detailed study on reactivity of GO with three different types of ROS, using both competition kinetics of ROS and degradation kinetics of DOC measurement. The objective was to provide rate constants and equations for GO reactions with ROS to provide meaningful data for modeling fate and transport of this emerging carbon-based nanomaterial in aquatic environments.32
Materials and methods
Materials
Single-layer graphene oxide (GO) with a 2 mg mL−1 concentration in pure water was purchased from Cheap Tubes Inc. (Brattleboro, VT). According to the supplier, the GO was manufactured by the modified Hummer's method33 and contains 35–42% carbon content. Stock dispersions of GO were prepared by dispersing the purchased GO in water, with a low-energy ultrasonic bath (M5800, Branson Ultrasonics, Danbury, CT) for 5 min at room temperature. Photoreduced GO (rGO) was prepared by irradiating a 40 mg L−1 GO dispersion in an Atlas SunTest CPS+ solar simulator equipped with a 1 kW xenon arc lamp (Mount Prospect, IL) for different time intervals (1–7 d). The carbon contents of GO and rGO dispersions were determined by measuring the dissolved organic carbon (DOC) and using it as the concentration index. The DOC-normalized UV-vis absorbance of GO and rGO are provided in ESI† Fig. S1. Other characterizations of GO and rGO have been previously reported.34 All chemicals were reagent grade and used as received from Sigma-Aldrich (St. Louis, MO) or Fisher Sci. (Pittsburgh, PA); detailed information of chemicals is provided in ESI.† Aqueous solutions or dispersions were prepared with water purified by a Millipore Milli-Q Synthesis system (18.2 MΩ cm at 25 °C).
Reaction with hydroxyl radical
HO˙ was produced by photo-Fenton reaction in a rotating-turntable merry-go-round reactor (MGRR) equipped with a Hanovia medium pressure mercury lamp and a temperature-controlled circulating water bath (25 ± 2 °C).15 The 436 nm monochromatic light was isolated with a combination of Corning 3–73 and 5–58 filters. The light intensity at 436 nm was measured as 0.0063 mol photons per L h−1, using ferrioxalate actinometry within test tubes (Pyrex 9826, 5 mL sample).35 In the degradation experiments, reaction samples contained Fe(ClO4)3 (15 μM), H2O2 (10 mM), and GO (or rGO) (∼4 mg-C L−1) at pH 4. Fe concentration was confirmed by the 1,10-phenanthroline method after being reduced by hydroxylamine hydrochloride.36 The observed first-order degradation rate constant of GO, kobs,DOC (s−1), was measured by DOC decay in the samples. Steady-state HO˙ concentration, [HO˙]ss (M), was determined by dividing the observed pseudo-first-order decay rate constant of para-chlorobenzoic acid (pCBA, 1.5 μM), added in separate samples, by the rate constant of pCBA with HO˙ (5.2 × 109 M−1 s−1).37 The apparent degradation rate constant of GO with HO˙, kobsHO˙,GO (M−1 s−1), was calculated using eqn (1) and (2):| | | d[DOC]GO/dt = −kobsHO˙,GO × [HO˙]ss × [DOC]GO = −kobs,DOC × [DOC]GO | (1) |
| | | kobsHO˙,GO = kobs,DOC/[HO˙]ss | (2) |
where [DOC]GO (mg-C L−1) is the dissolved carbon content of GO or rGO.
In competition kinetics, with methanol (MeOH) as the major scavenger, the absolute second-order rate constant of GO with HO˙ was determined by adding varied amounts of GO to reaction solutions containing sodium oxalate (60 μM), Fe(ClO4)3 (1.5 μM), pCBA (0.5 μM), MeOH (50 μM) and H2O2 (50 μM) at pH 5.4 ± 0.1. The steady-state concentration of HO˙ was corrected with SFs to account for differences in inner-filtering effects, according to eqn (3) and (4):38
| | 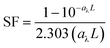 | (4) |
where
aλ (cm
−1) is the absorbance coefficient of samples at 436 nm and
L (cm) is the average optical pathlength for the test tubes (∼1 cm).
The second-order rate constant between GO and HO˙, kHO˙,GO (mg-C−1L s−1), was determined by the standard competition kinetics equation:
| | 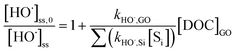 | (5) |
where [HO˙]
ss,0 is the steady-state HO˙ concentration in the absence of GO;
kHO˙,Si (M
−1 s
−1) is the rate constant of each HO˙ scavenger in the system; and [S
i] (M) is the concentration of each individual scavenger, including
pCBA, oxalate, H
2O
2, and MeOH (
kHO˙,Si = 5.2 × 10
9, 1.0 × 10
7, 2.7 × 10
7, and 9.7 × 10
8 M
−1 s
−1, respectively).
39
For comparison, kHO˙,GO was also examined by using terephthalic acid (TPA, 20 μM) to replace MeOH as the major HO˙ scavenger, and other reagents remained at the same concentrations without added pCBA.40 The relationship between HO˙ concentration and formation of 2-hydroxyterephthalic acid (HTA), a hydroxylated product of TPA by HO˙, was used to calculate the kHO˙,GO by:
| | 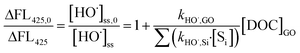 | (6) |
where ΔFL
425 and ΔFL
425,0 are the fluorescence intensities (counts) of HTA in samples with and without GO, respectively, and [S
i] is the concentration of oxalate, H
2O
2, and TPA (
kHO˙,TPA = 3.3 × 10
9 M
−1 s
−1).
40
Reaction with singlet oxygen
1O2 was generated by irradiating rose bengal (RB, 0.5 μM) as a photosensitizer. Solutions were adjusted to pH 5.3 ± 0.2 by 0.1 N NaOH solution and irradiated with a monochromatic light at 546 nm in the same MGRR, with a combination of Corning 3–68 and 4–72 filters. Light intensity was measured at 0.018 mol photons per L h−1 by the same ferrioxalate actinometry method.34 The half-life of RB upon exposure to light with this irradiance was 3.75 h, determined by the decay of its characteristic absorbance at 549 nm. Steady-state 1O2 concentrations, [1O2]ss (M), were obtained by dividing the pseudo-first-order decay rate of furfuryl alcohol (FFA, 0.1 mM) by its rate constant for reaction with 1O2 (k1O2,FFA = 1.2 × 108 M−1 s−1).40 [1O2]ss was also corrected with light-screening factors described in eqn (3), using the combined absorbance of RB and GO (or rGO) at 546 nm. The second-order rate constant between GO and 1O2, k1O2,GO, attributed to both physical and chemical quenching, was determined by eqn (7):| | 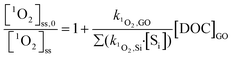 | (7) |
where [1O2]ss,0 is the steady-state 1O2 concentration in the absence of GO; k1O2, Si (M−1 s−1) is the rate constant of 1O2 scavenger in the system; and [Si] is the concentration of individual scavenger (k1O2,H2O·[H2O] = 2.5 × 105 s−1).41
To evaluate potential GO transformation due to reaction with 1O2, GO characterization was examined during a reaction course of 56 h. Considering that RB was gradually photobleached, extended 1O2 production was achieved by sequentially adding RB in a stepwise dose increment of 0.5 μM every 8 h.
Reaction with superoxide anion
O2˙− was chemically produced using phenazine methosulfate (PMS, 2 μM) to catalyze the oxidation of reduced-form β-nicotinamide adenine dinucleotide (NADH, 20–100 μM) in air-saturated solutions at pH 8.42 O2˙− production was confirmed by formation of XTT (35–75 μM) formazan, a product of the reaction between O2˙− and XTT.43 The second-order rate constant of GO with O2˙−, kO2˙−, GO (mg-C−1 L s−1), was calculated by the inhibitory effect of GO on formation of XTT formazan (eqn (8)):| | 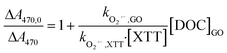 | (8) |
where kO2˙−, XTT (M−1 s−1) is the reaction rate constant between O2˙− and XTT; ΔA470 and ΔA470,0 are absorbances of XTT formazan via reaction with O2˙− in dispersions with and without GO, respectively. Superoxide dismutase (SOD), an enzyme that catalyzes the dismutation of O2˙− to H2O2, was used to determine whether the increased absorbance was due to O2˙−. In solutions of XTT, NADH, and PMS at pH 8, the self-disproportionation of O2˙− to H2O2 is negligible (k = 6 × 104 M−1 s−1)44 because of high reactivity of XTT (kO2˙−,XTT = 8.59 × 104 M−1 s−1)43 and slow production rate of O2˙− (NADH oxidation rate constant = 0.255 min−1). In the absence of scavengers, O2˙− would eventually disproportionate to H2O2. Therefore, the H2O2 yield in the NADH/PMS (without XTT) in the presence of varied amounts of GO was used as supportive evidence for the reaction between GO and O2˙−.
Analyses
pCBA and FFA were analyzed in an Agilent 1200 HPLC coupled with a diode array detector (DAD) at 230 nm and 219 nm, respectively. HTA was analyzed in the same HPLC with a fluorescence detector (FLD) (excitation 315 nm, emission 425 nm).40 XTT formazan was monitored at 470 nm in a Perkin Elmer Lambda 35 spectrophotometer equipped with 1 cm quartz cuvette. H2O2 concentration in the NADH/PMS systems was measured by the DPD/HRP assay at 551 nm, as described previously.45 DOC was analyzed in a TOC-VCPH analyzer (Shimadzu, Japan), using non-purgeable organic carbon (NPOC) mode with potassium hydrogen phthalate as the standard. Spectroscopic analyses of GO were carried out in Lambda 35 spectrophotometer for UV-vis absorbance and Horiba Fluorolog 3 fluorometer for photoluminescence (PL), both with 1 cm light path quartz cuvettes. Hydrodynamic size was measured on a Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK), using a dynamic light-scattering technique with a 633 nm He–Ne laser. Except in DOC measurement, samples used for GO characterization were filtered through Millipore Amicon Ultra-4 centrifugal filters (10k MWCO for RB and 3k MWCO for Fe(ClO4)3, H2O2, NADH, and PMS), via centrifugation at 4500 × g, for 40 minutes. Filtered GO concentrates were washed with water via the same centrifugal filtration procedure for 5 cycles, then re-dispersed in water to initial volume. The recovery of GO in this ultrafiltration process was 92.6% for 3k filter and 90.1% for 10k filter, found by comparing the 230 nm absorbance of filtered to non-filtered suspensions.
Results and discussion
Reactivity with hydroxyl radical (HO˙)
To minimize interference by direct photolysis of GO on determining its reactivity with HO˙, we chose a visible light of 436 nm for initiating the photo-Fenton reaction to generate HO˙ by reaction of Fe(II) (from photoreaction of Fe(III) complexes) with H2O2. Preliminary experiments showed there was no DOC decay for GO under 48 h irradiation at wavelengths longer than 405 nm in the MGRR (Fig. S2†).
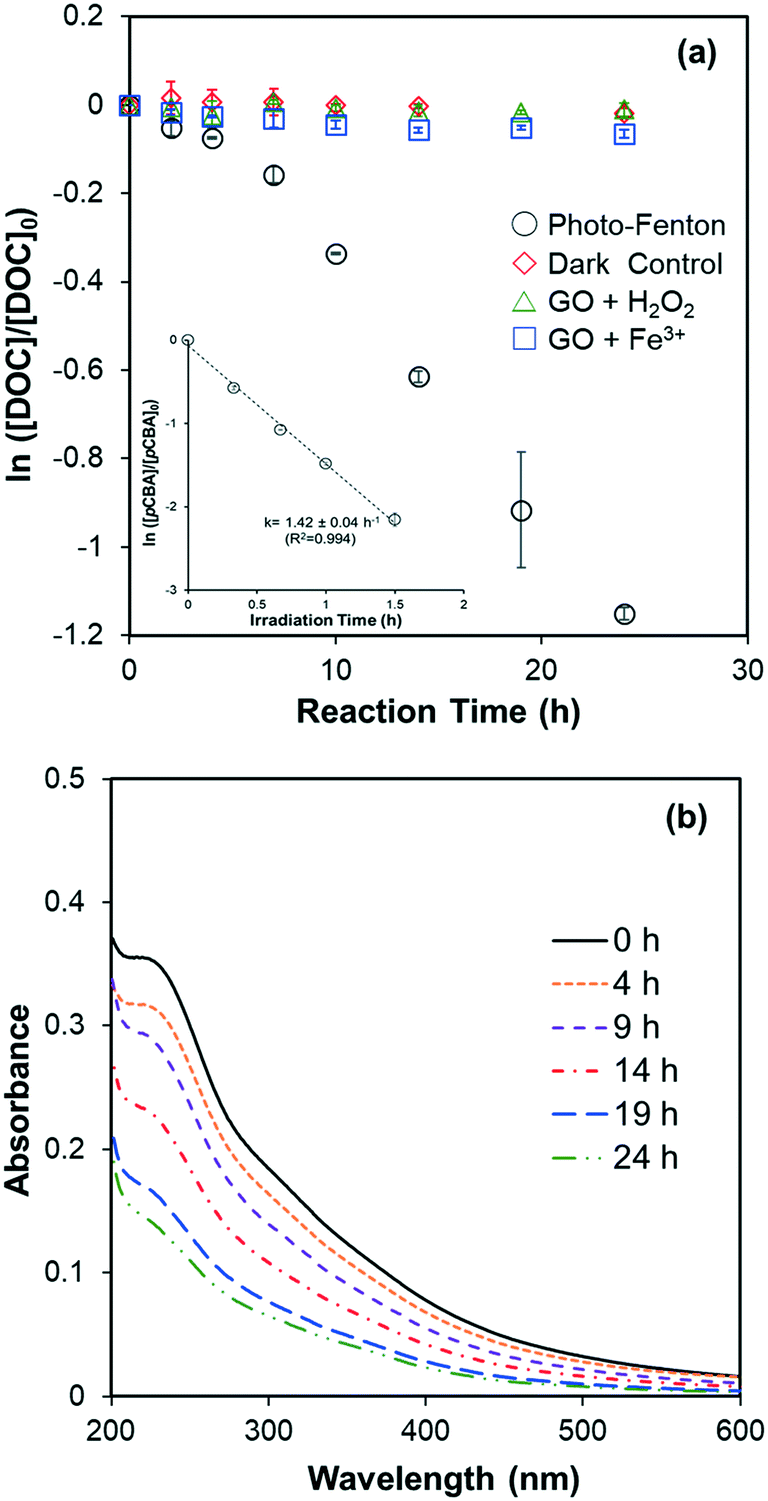
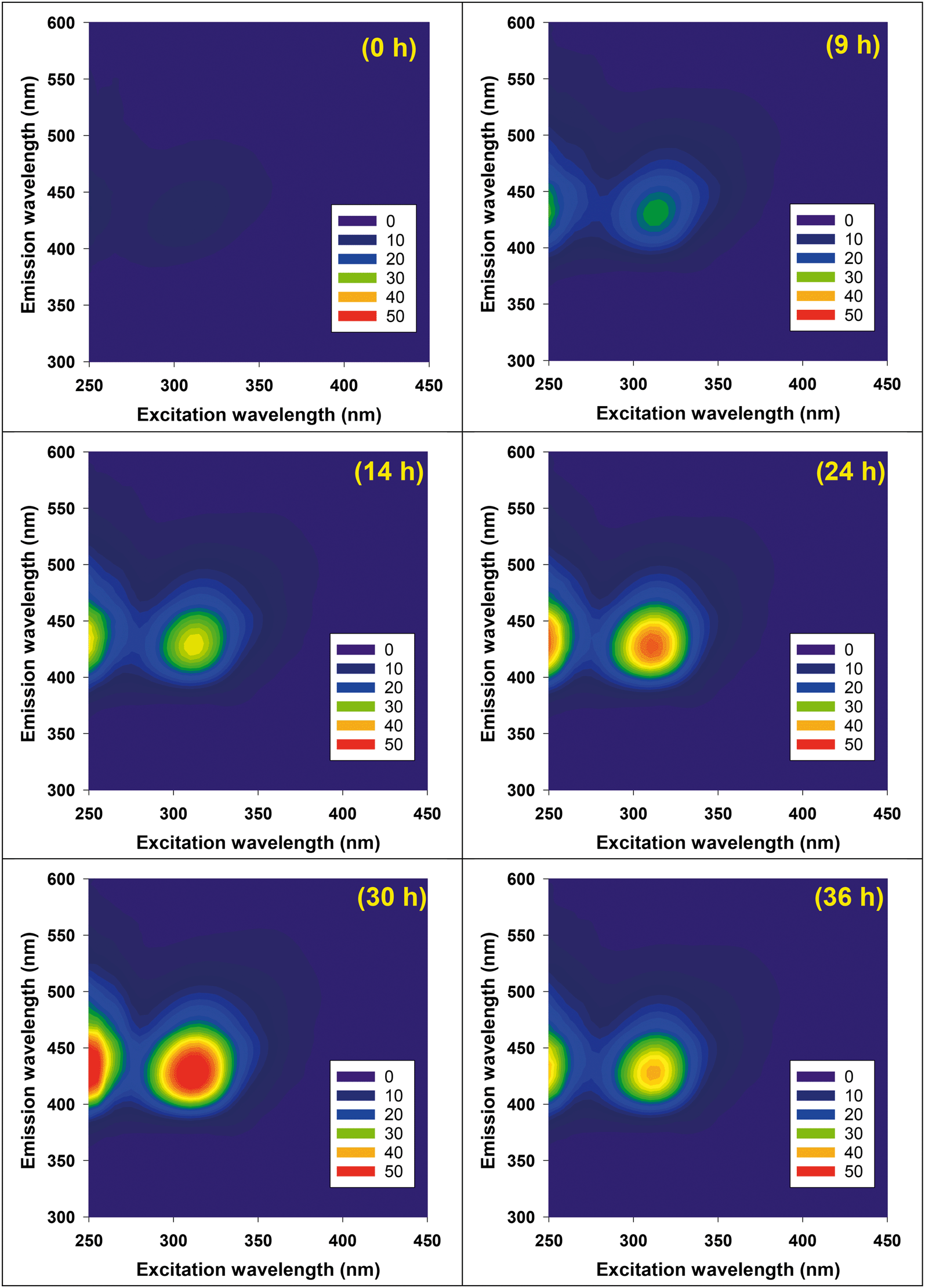
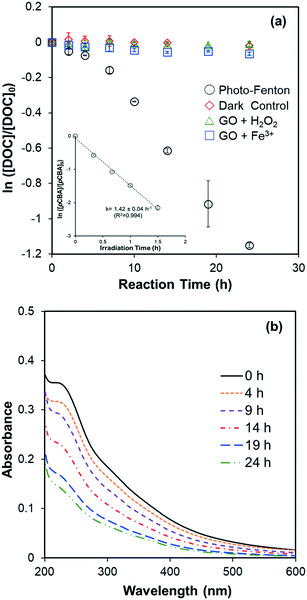
DOC decay of GO (10 mg L−1, ∼4 mg-C L−1) in pH 4 dispersions containing Fe(ClO4)3 and H2O2, under irradiation at 436 nm are shown in Fig. 1a. No DOC decrease was observed in the dark control or in systems without added Fe (i.e., H2O2 alone). In systems with added Fe(ClO4)3 alone, a slight reduction of DOC was observed, presumably caused by Fenton reaction related to the small yield of H2O2, a self-disproportionated product of O2˙−, produced from photolysis of GO.23 The pseudo-first-order decay rate constant of DOC decay in this control, however, is 25 times smaller than in the experiment with both Fe(ClO4)3 and H2O2. These results suggested that direct photolysis is negligible and DOC reduction was mainly due to the reaction with HO˙ generated from photo-Fenton reaction. Indeed, the steady-state concentration of HO˙ was calculated as 7.6 × 10−14 M using the observed pseudo-first-order degradation rate of pCBA (see inset in Fig. 1a). The reaction of GO with HO˙ resulted in a continuous decrease in the UV-vis absorbance, as shown in Fig. 1b; this differs from the previous indirect photolysis study in which absorbance increased at the beginning due to direct photolysis.10 It is consistent with the argument that observed DOC decay was due to GO oxidation by HO˙, and is further supported by the inhibitory effect of MeOH, a HO˙ scavenger, on the decrease of GO absorbance at 230 nm (Fig. S3†). Note that addition of MeOH did not totally prevent degradation, which could be due to the locally-high concentration of Fe on the GO surface (see later discussion). Even though both DOC and UV-vis spectra decreased, Fig. 2 shows that reacted GO products exhibited strong photoluminescence (in the range of 400–480 nm) upon excitation by light with wavelengths between 250–450 nm. A similar fluorescence pattern has been reported previously10,21 from the fragmentation of GO flakes by HO˙, confirmed with qualitative characterization (e.g., TEM, AFM, and XPS etc.) and mass spectrometry.
 |
| | Fig. 1 (a) Degradation of GO (10 mg L−1) in the pH 4 dispersions with: Fenton reagent (15 μM Fe(ClO4)3 and 10 mM H2O2) (○), 10 mM H2O2 (△), 15 μM Fe(ClO4)3 (□) under irradiation at 436 nm. The dark control samples containing the same Fenton reagents were placed in the dark (◇). The inset shows the pseudo-first-order decay of added pCBA (1.5 μM) in the photo-Fenton sample. (b) UV-vis spectra of HO˙ reacted GO after removing Fenton reagents by ultrafiltration. | |
 |
| | Fig. 2 Excitation-emission matrices of HO˙ reacted GO (10 mg L−1 initially) in the condition described in Fig. 1a. Intensity units are quinine sulfate equivalents after removing water Raman and Rayleigh scattering peaks. | |
Fig. S4a† shows the DOC decay of rGO and Fig. S4b† shows the decay of pCBA, obtained from separated experiments in which HO˙ was produced by irradiating dispersions containing rGO, Fe(ClO4)3 and H2O2 at 436 nm. Both DOC and pCBA decays are well-fitted with pseudo-first-order kinetics in dispersions containing rGO, indicating that HO˙ reached steady state status in the systems and contributed to first-order decay of the DOCs of GO and rGO (eqn (1) and (2)). The observed first-order DOC decay rate (kobs,DOC), steady-state HO˙ concentration ([HO˙]ss), and the computed apparent second-order degradation rate constants of GO and rGO in reaction with HO˙, are summarized in Table 1. We analyzed DOC data in Fig. 1a by calculating rate constants for the initial loss (0–7 h) and subsequent more rapid loss (during the 7–24 h interval). Degradation rate constants for GO are 0.8 × 108 M−1 s−1 for the first 7 h, and 2.2 × 108 M−1 s−1 thereafter. The time-dependence indicates that different oxidation mechanisms may be involved, presumably due to susceptibility of different functionalities to HO˙ attack. For rGO, the second-order degradation rate constant appeared to be dependent on previous irradiation time in the solar simulator. The value decreased from 1.78 × 108 M−1 s−1 for 1D-rGO to 0.74 × 108 M−1 s−1 for 7D-rGO, based on DOC data in the 0–13 h interval. We suspect this is because GO was photoreduced to rGO quickly (<1 d), and further irradiation caused photooxidation of rGO to form more oxidized functionalities (e.g., carboxyl and phenyl) which are normally less reactive with HO˙. Further investigation is warranted, however. Compared to the previous study which used solar irradiated H2O2 as HO˙ source, second-order degradation rate constants reported here are only 0.14–0.43 times the previous one (5.1 × 108 M−1 s−1).10 This difference may be a result of error in determining the rate of direct photolysis in a previous study where the DOC decay did not follow a first-order pattern very well. In other words, the rate of direct photolysis might be underestimated when a pseudo-first-order decay model was used to fit the DOC decay for a long period. In contrast, the experiment presented here avoided direct photolysis of GO by using photo-Fenton reaction at 436 nm, which certainly improved the accuracy of the determined indirect rate constant.
Table 1 Apparent degradation rate constants for the reactions of GO and photo-reduced GO (rGO) with HO˙ produced in the pH 4 photo-Fenton system described in Fig. 1a. Standard errors are calculated from the linear regression of ln(C/C0) where C is [DOC] and [pCBA] separately
|
|
k
obs,DOC
(h−1) |
k
obs,pCBA
(h−1) |
[HO˙]ss (×10−14 M) |
k
obsHO˙,GO
(×108 M−1 s−1) |
|
Pseudo-first-order decay rates for DOC and pCBA, respectively.
The second-order degradation rate constant was calculated by eqn (2).
|
| GO (0–7 h) |
0.022 ± 0.002 |
1.42 ± 0.04 |
7.6 ± 0.2 |
0.81 ± 0.08 |
| GO (7–24 h) |
0.059 ± 0.003 |
1.42 ± 0.04 |
7.6 ± 0.2 |
2.16 ± 0.13 |
| 1D-rGO |
0.036 ± 0.002 |
1.04 ± 0.03 |
5.6 ± 0.2 |
1.80 ± 0.11 |
| 2D-rGO |
0.033 ± 0.002 |
1.17 ± 0.05 |
6.3 ± 0.3 |
1.47 ± 0.11 |
| 4D-rGO |
0.026 ± 0.001 |
1.51 ± 0.02 |
8.0 ± 0.1 |
0.90 ± 0.04 |
| 7D-rGO |
0.027 ± 0.002 |
1.89 ± 0.09 |
10.1 ± 0.5 |
0.74 ± 0.07 |
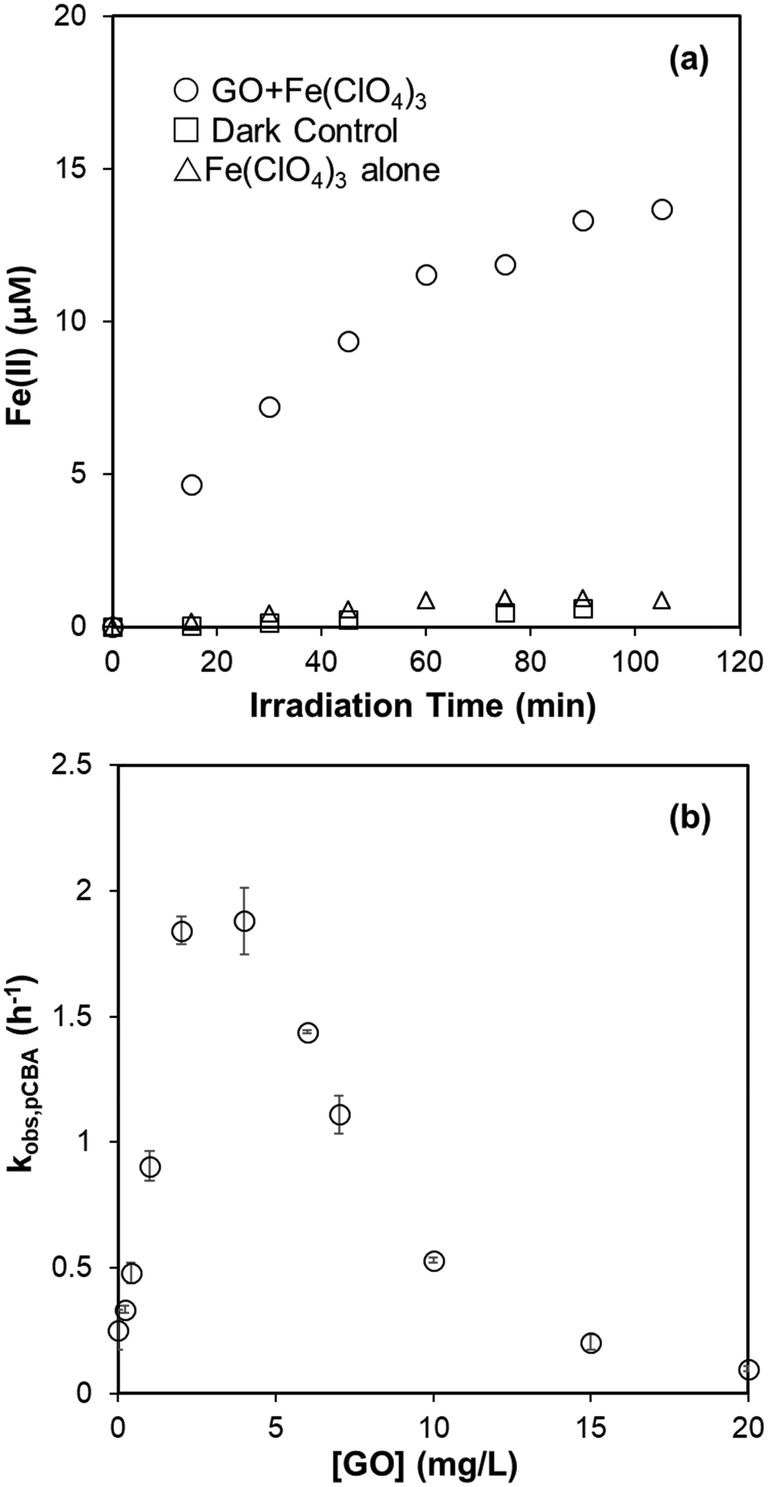
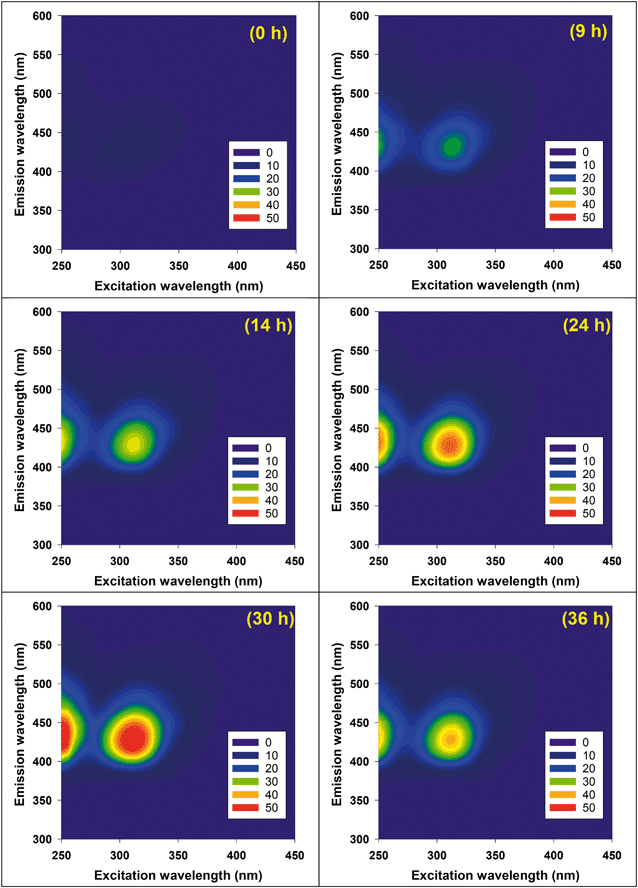
Useful information for evaluating the persistence of GO and rGO subjected to attack by HO˙ is provided in Table 1. The use of DOC, however, cannot explicitly represent reactivity of GO with HO˙ because HO˙ attack does not necessarily involve mineralization of carbon grids. Competition kinetics was therefore used to determine the absolute second-order rate constant for reaction with HO˙. For competition kinetics to be valid, it is assumed that production of HO˙ was not affected by the target material. In acid solutions of Fe(ClO4)3, however, GO was found to catalyze the photoreduction of Fe(III) to Fe(II) under irradiation at 436 nm (Fig. 3a), presumably due to electron transfer from irradiated GO to the complexed Fe(III). An increased steady-state concentration of HO˙ was subsequently observed when H2O2 was present (Fig. 3b). This may explain why adding MeOH (10 mM) did not totally prevent GO degradation in the photo-Fenton system (Fig. S3†), since the HO˙ generated by GO-catalyzed Fenton reaction is spatially close to GO and readily reacts with it before diffusing to be scavenged by MeOH. This also suggests that complexation of Fe with GO may be an important pathway for degradation of GO in aquatic environments containing high concentrations of Fe.
 |
| | Fig. 3 (a) Production of Fe(II) in the pH 4 suspension of 20 mg L−1 GO and 100 μM Fe(ClO4)3 under irradiation at 436 nm. Fe(II) concentration was determined by 1,10-phenanthroline complexing method, using the absorbance at 510 nm. (b) Pseudo-first-order decay rate of pCBA (1 μM) in the photo-Fenton system (5 μM Fe(ClO4)3 and 20 μM H2O2, pH = 4) under irradiation at 436 nm. | |
Based on eqn (5), the catalytic effect of GO on Fe reduction resulted in an unrealistic, negative second-order rate constant for the reaction between GO and HO˙ when GO concentration was below 5 mg L−1 (∼2 mg-C L−1). The catalytic effect of GO was then eliminated by adding sodium oxalate (60 μM) to chelate Fe, and the Fe concentration was decreased to 1.5 μM to avoid complexation between GO and Fe. The Fenton reaction was mainly driven by ligand-to-metal charge transfer from oxalate to Fe, which efficiently reduced Fe(III) to Fe(II) and produced HO˙ at a constant rate under irradiation at 436 nm.15 Reactivity of HO˙ scavengers may limit application of competition kinetics when HO˙ production is not uniform. As a benchmark, we used tert-butanol (t-BuOH) as a control; a linear relationship between added tert-butanol and [HO˙]ss,0/[HO˙]ss was observed in the presence of 50 μM MeOH (Fig. S5a†). The calculated kHO˙,t-BuOH of 6.15 ± 0.32 × 108 M−1 s−1 agreed well with the previously reported value of 6 × 108 M−1 s−1,38 indicating that, in this oxalate/Fe/H2O2 system, the error from using pCBA-measured [HO˙]ss for competition kinetics is minimized.
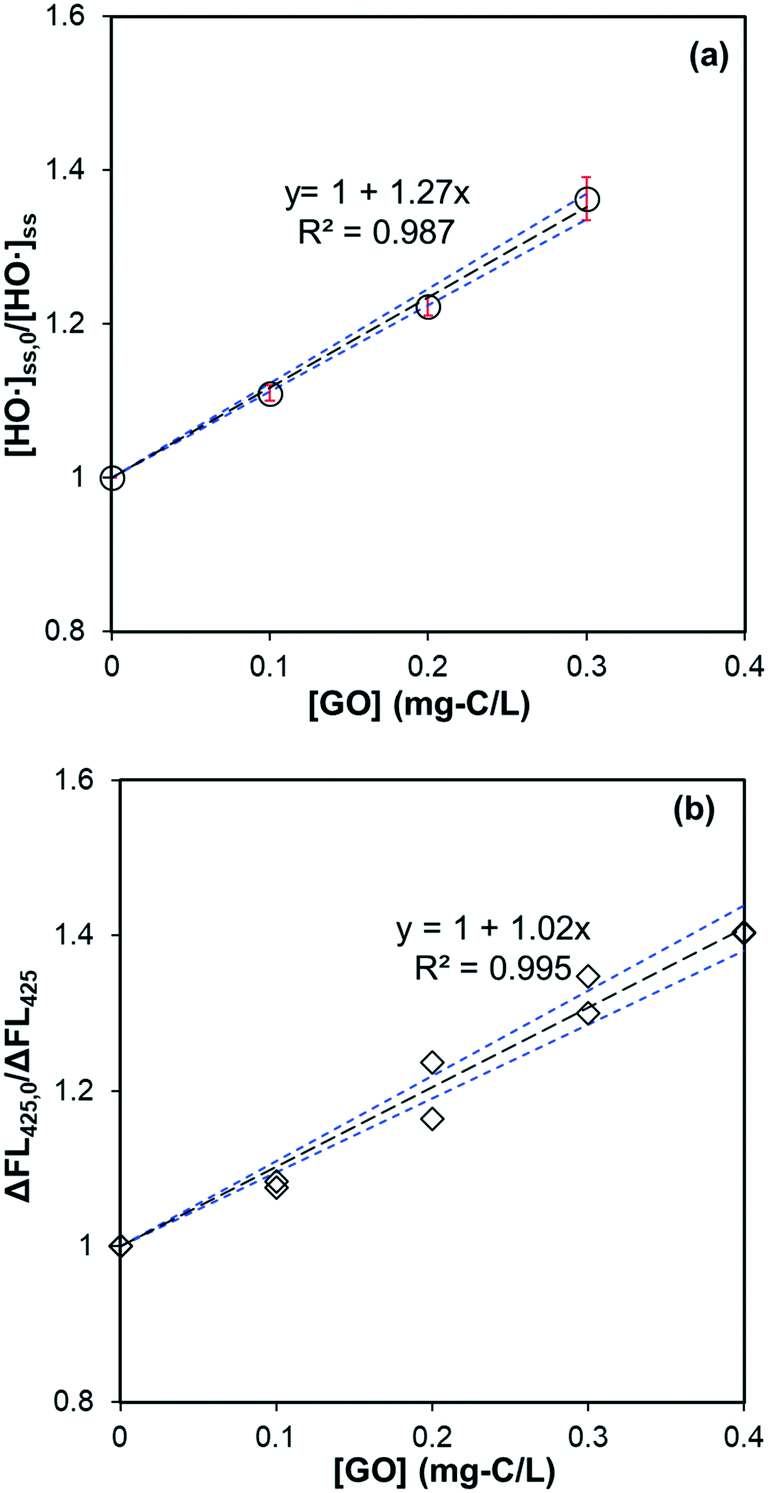
A linear relationship between dosed GO amount and [HO˙]ss,0/[HO˙]ss in the same oxalate/Fe/H2O2 system – when pCBA (0.5 μM) was used to measure steady-state HO˙ concentration – is shown in Fig. 4a, supporting that GO's catalytic effect was negligible. A similar linear relationship was observed for HTA fluorescence when TPA was used as the major HO˙ scavenger (Fig. 4b). Results of competitive kinetics for rGO are provided in Fig. S5b–e.† Second-order rate constants (calculated by eqn (5)) for the reaction between GO (or rGO) with HO˙ are summarized in Table 2. The rate constant for GO is (6.24 ± 0.13) × 104 mg-C−1 L s−1. When HTA fluorescence (Fig. 4b) and eqn (6) are used, the rate constant is calculated to be (6.93 ± 0.22) × 104 mg-C−1 L s−1. The two values are within ∼10% error, but much smaller than that reported for graphene (3.67 × 105 mg-C−1 L s−1).46 Rate constants for rGO were in the range of (8.59–5.89) × 104 mg-C−1 L s−1, decreasing with extended duration in the previous solar irradiation period and agreeing with the trend of DOC degradation rate constants listed in Table 1. Overall, these values are comparable to most polycyclic aromatic hydrocarbons (e.g., 7.8 × 104 mg-C−1 L s−1 for pyrene and 7.5 × 104 mg-C−1 L s−1 for biphenyl),47 but two to three times higher than dissolved organic matter (DOM) in water systems ((2.7 ± 0.77) × 104 mg-C−1 L s−1).48
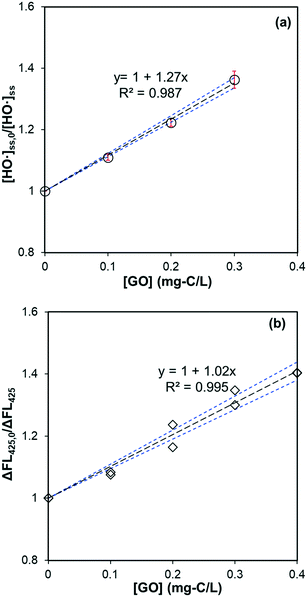 |
| | Fig. 4 (a) Effect of GO on the steady-state HO˙ concentration in the system of sodium oxalate (60 μM), Fe(ClO4)3 (1.5 μM), H2O2 (50 μM), MeOH (50 μM), and pCBA (0.5 μM) under irradiation at 436 nm. The slope is significantly different from zero (p = 0.002) and the standard deviation of the slope is 0.04. (b) Effect of GO on the fluorescence of HTA produced in the system of sodium oxalate (60 μM), Fe(ClO4)3 (1.5 μM), H2O2 (50 μM), TPA (20 μM) under irradiation at 436 nm. The slope is significantly different from zero (p < 0.001) and the standard deviation of the slope is 0.03. Black dotted lines represent the linear regression fit, and the blue dotted lines are 95% confidence bands. | |
Table 2 Absolute second-order rate constants and calculated R squared value for GO and rGO with HO˙ in the pH 5.4 photo-Fenton system described in Fig. 4a
|
|
k
HO˙,GO (×104 mg-C−1 L s−1) |
R squared |
| GO |
6.24 ± 0.13 |
0.987 |
| 1D-rGO |
8.59 ± 0.32 |
0.966 |
| 2D-rGO |
7.43 ± 0.48 |
0.894 |
| 4D-rGO |
6.98 ± 0.39 |
0.927 |
| 7D-rGO |
5.89 ± 0.31 |
0.917 |
Reactivity with singlet oxygen (1O2)
Irradiation of 0.5 μM RB at 546 nm light generated a steady-state 1O2 concentration ([1O2]ss) of 1.33 × 10−12 M within 80 minutes. As irradiation extended, RB photo-bleached (first-order rate constant of 0.185 h−1) and the 1O2 production rate decreased accordingly. Therefore, competition kinetics experiments were carried out in the span of 80 minutes. The effect of GO concentration on the [1O2]ss in the solutions of pH 5.3 ± 0.2 is shown in Fig. 5. Multiplying the slope and its standard deviation by the combined rate constant of 1O2 with H2O and FFA, the absolute second-order rate constant for the reaction of GO with 1O2 was calculated to be 865 ± 183 mg-C−1 L s−1, which is about two orders of magnitude lower than the reaction with HO˙. Because concentrations of 1O2 in natural water could be two or three orders of magnitude higher than HO˙, we further examined whether reaction with 1O2 would result in any transformation of GO. In the continuous production of 1O2via stepwise dose of RB into the irradiated sample, the average 1O2 concentration in the system containing 10 mg L−1 GO was calculated as:| | 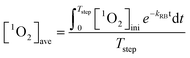 | (9) |
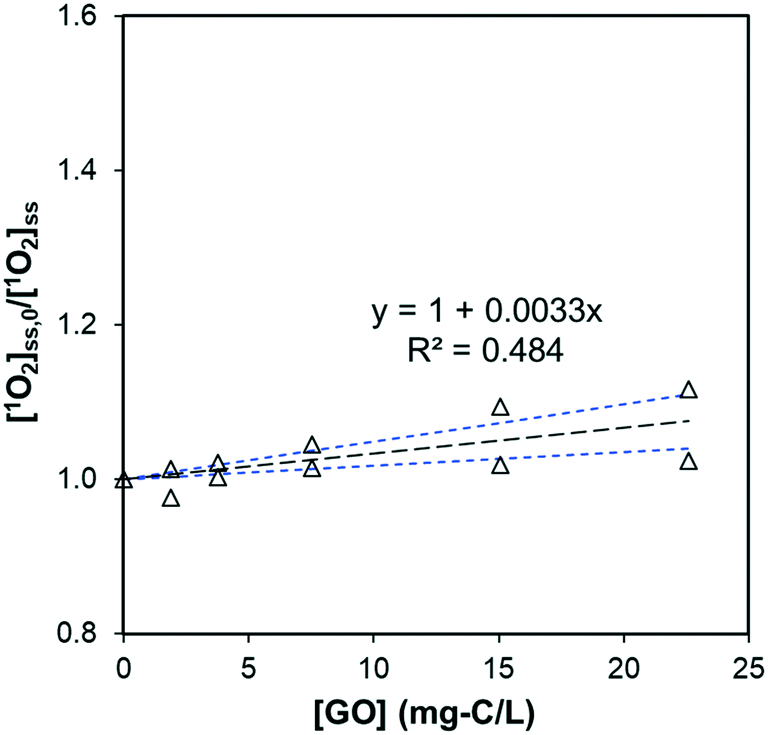
where [1O2]ini is the steady-state 1O2 concentration at the first 80 minutes (1.33 × 10−12 M), kRB is the observed photo-bleaching rate of RB (0.185 h−1), and Tstep (h) is the time interval between each dose. With a stepwise dose every 8 h, the [1O2]ave is calculated to be 6.9 × 10−13 M. In 56 h experiments, there was no DOC decrease for GO after subtracting the DOC in the control of RB alone from the DOC in GO + RB solutions to (kobs,DOC < 10−4 h−1), as shown in Fig. S6a;† this resulted in an apparent second-order degradation constant less than 4 × 104 M−1 s−1. Besides DOC, measurements of UV-vis spectra, hydrodynamic size, and photoluminescence also show that GO remained intact (Fig. S6b and S7†), suggesting the chemical reaction between GO and 1O2 is negligible. In contrast, the effect of GO on decreasing the steady-state 1O2 concentration produced by RB was mainly due to physical quenching. This is similar to the reported reaction between C60 and 1O2, despite the three-order discrepancy in second-order rate constant (865 mg-C−1 L s−1vs. 0.69 mg-C−1 L s−1),16 yet different from results of CNTs in which 1O2 was reported to cause surface oxidation.20 We believe this is the first report of GO being chemically inert to photo-generated 1O2.
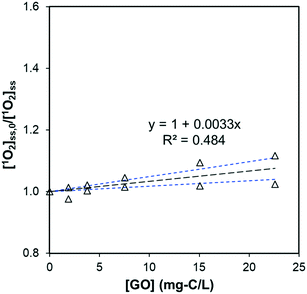 |
| | Fig. 5 Effect of GO on the steady-state concentration of 1O2, generated by irradiating 0.5 μM RB at 546 nm (pH = 5.3 ± 0.2). The slope is significantly different from zero (p = 0.017) and the standard deviation of the slope is 0.0007. Black dotted lines represent the linear regression fit, and the blue dotted lines are 95% confidence bands. | |
Reactivity with superoxide anion (O2˙−)
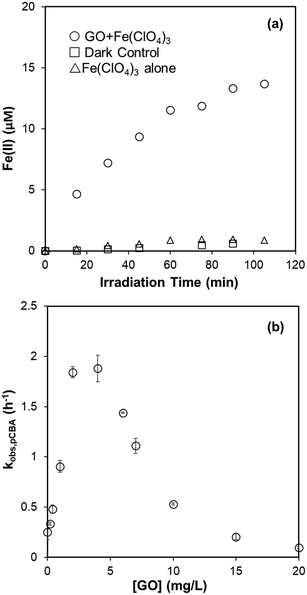
Reactivity of GO with O2˙− was investigated by its competitive effect on the formation of soluble XTT formazan. We chose XTT as the O2˙− probe rather than nitro blue tetrazolium (NBT2+) because the negatively-charged formazan product prevents coagulation with GO. That GO inhibited the formation of O2˙−-reduced XTT formazan in the NADH/PMS is shown in Fig. 6a. The second-order rate constant for the reaction of GO with O2˙−, kO2˙−,GO, was calculated to be 0.108 ± 0.004 mg-C−1 L s−1 by multiplying the slope in Fig. 6a by the rate constant of XTT with O2˙−, based on eqn (8). To limit direct reaction between XTT and reduced PMS and obtain measurable change in the absorbance, the XTT concentration used in these experiments was not high enough for eqn (8) to be exactly valid (i.e., [XTT] was not constant during reaction). If the decrease of XTT over time was considered, the kO2˙−,GO was calculated as 0.078 mg-C−1 L s−1 by using least square-fitting to minimize the sum of squared error (SSE) between predicted and measured ΔA470,0/ΔA470. Interested readers are referred to ESI† for further details.
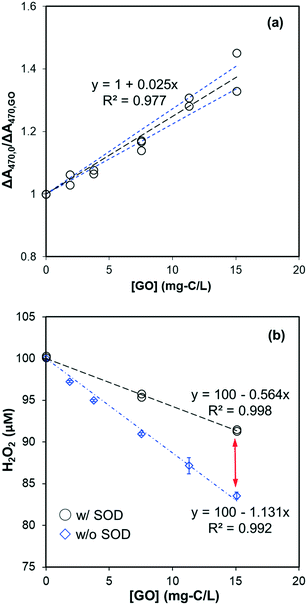 |
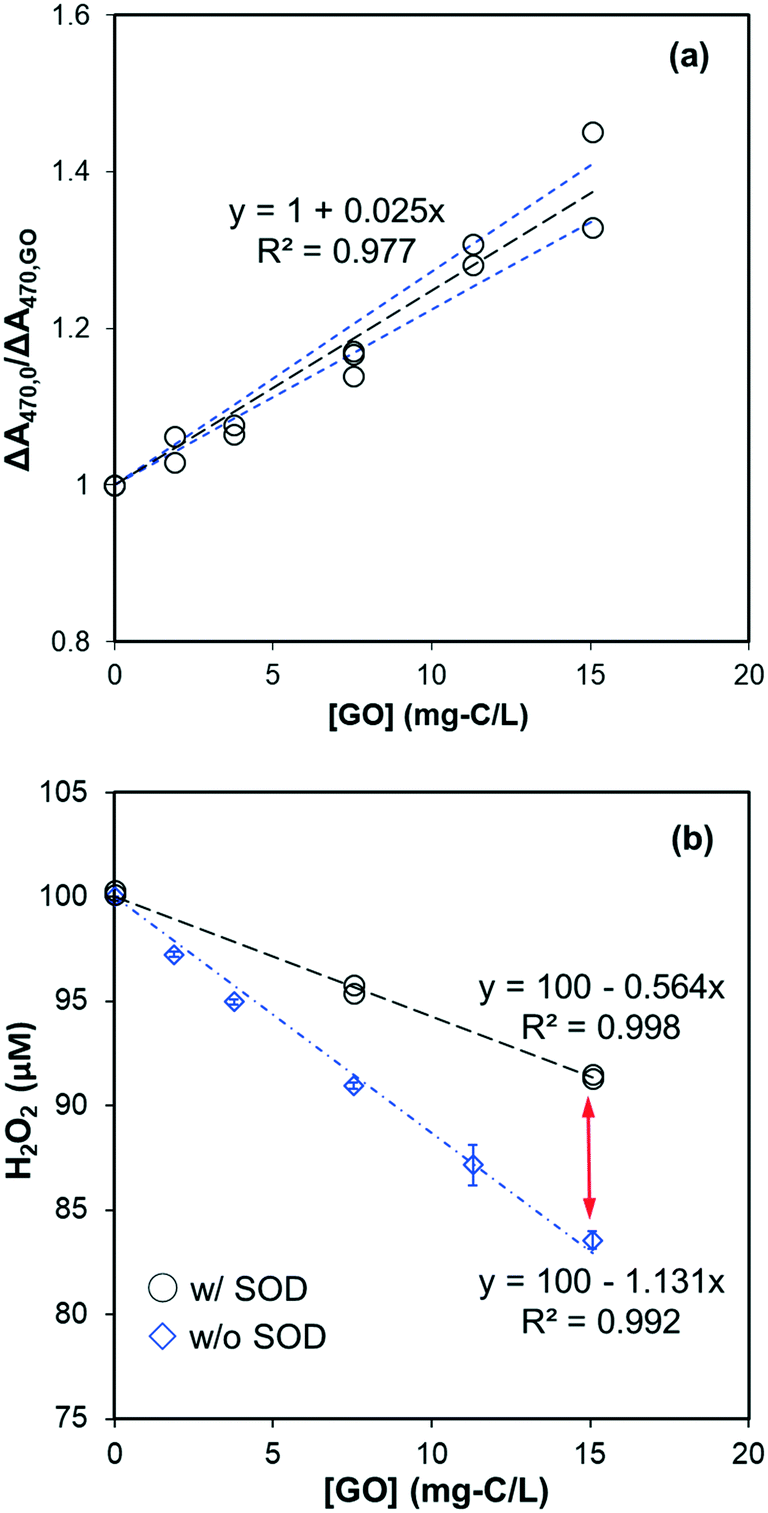
| | Fig. 6 (a) GO effect on XTT formazan formation by O2˙− in the system of NADH (30 μM), PMS (2 μM), and XTT (50 μM) at pH 8. The slope is significantly different from zero (p < 0.001) and the standard deviation of the slope is 0.002. Black dotted lines represent the linear regression fit, and the blue dotted lines are 95% confidence bands. (b) Effect of SOD (40 U mL−L) on the H2O2 formation in the pH 8 phosphate buffered dispersions containing NADH (100 μM), PMS (2 μM), and GO. The difference indicated by the red arrow is attributed to scavenging of O2˙− by GO. | |
To observe the transformation of GO caused by O2˙− reaction, GO was found to be slowly reduced with increasing absorbance in the visible wavelength region (Fig. S8†). Spectral change was only partially attributed to O2˙−, however, as the addition of SOD (40 U mL−1) did not totally inhibit absorbance increase which indicates a direct reaction between GO and reduced PMS or NADH. This raised the question of whether inhibition of XTT formazan (Fig. 6a) was due to decreased O2˙− yield in the presence of GO. To test it, we compared the effect of XTT concentration on formation of XTT formazan. If inhibition on XTT formazan was due to decreased production of O2˙−, the slope of ΔA470,0/ΔA470 should remain constant at different XTT concentrations. Increasing XTT from 35 μM to 75 μM resulted, however, in a proportional decrease in slope from 0.0345 to 0.018 mg-C−1 L (as shown in Fig. S9†), suggesting that inhibition on XTT formazan was mainly due to competition between GO and XTT for O2˙−. Using slopes in Fig. S9,† the calculated rate constant was in the range of 0.095–0.116 mg-C−1 L s−1, consistent with the value obtained from Fig. 6a, despite different NADH concentrations (30 μM vs. 20 μM).
The effect of GO on the final H2O2 concentration in the NADH/PMS system (without XTT) is shown in Fig. 6b. H2O2 concentration linearly decreased with increased GO, indicating GO acted as an electron sink in the system. This excludes the possibility that GO inhibited the formation of XTT formazan by simply shutting down electrons from O2˙− to form H2O2. Meanwhile, decreased H2O2 concentration was significantly recovered by adding SOD to the system, suggesting that O2˙− was competitively consumed by GO – presumably by forming chemical bonding or one-electron reduction of GO, consistent with increased GO absorbance in the range of visible wavelength (Fig. S8†). A similar finding based on XPS, FTIR and Raman analysis31 demonstrated the O2˙−-reduction of GO. Since reduction of GO to rGO could greatly change its hydrophilicity and bioaccumulation properties, further study is needed to clarify evolution of GO from exposure to superoxide anion.
Environmental implications
Major challenges in exposure assessment of graphene-based nanomaterials are their stability and susceptibility to different reactive reagents in environment. Our study demonstrated that reaction with 1O2 had a negligible effect on transformation of GO, although its steady-state concentration in aquatic environments is normally two- to three-orders of magnitude higher than HO˙. In contrast, HO˙ attack quickly degraded not only GO, but also the less photoreactive rGO. Together, these results suggest that an integrated understanding of ROS distribution and GO reactivity with ROS is needed to better assess long-term exposure of GO and rGO. It may also prove valuable in designing GO or rGO in environmental applications because high HO˙ reactivity indicates stability of these types of nanomaterials cannot be overlooked when they are used as catalysts or carriers. Nevertheless, further study is still needed to clarify the detailed mechanisms of GO degradation by individual ROS, including the identification of reacted products.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This article has been reviewed in accordance with the U.S. Environmental Protection Agency's (U.S. EPA) peer and administrative review policies and approved for publication. Mention of trade names or commercial products does not constitute an endorsement or recommendation for use by the U.S. EPA. We thank F. Rauschenberg, Senior Service America, Inc., for editorial assistance.
References
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, A roadmap for graphene, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Graphene-like two-dimensional materials, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- F. Perreault, A. Fonseca de Faria and M. Elimelech, Environmental applications of graphene-based nanomaterials, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- D. Chen, H. Feng and J. Li, Graphene oxide: preparation, functionalization, and electrochemical applications, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and Graphene Oxide: Synthesis, Properties, and Applications, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Chemical methods for the production of graphenes, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- Q. Zhang, Z. Wu, N. Li, Y. Pu, B. Wang, T. Zhang and J. Tao, Advanced review of graphene-based nanomaterials in drug delivery systems: Synthesis, modification, toxicity and application, Mater. Sci. Eng., C, 2017, 77, 1363–1375 CrossRef CAS PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Duran, Nanotoxicity of graphene and graphene oxide, Chem. Res. Toxicol., 2014, 27, 159–168 Search PubMed.
- J. Zhao, Z. Wang, J. C. White and B. Xing, Graphene in the aquatic environment: adsorption, dispersion, toxicity and transformation, Environ. Sci. Technol., 2014, 48, 9995–10009 CrossRef CAS PubMed.
- W.-C. Hou, W. M. Henderson, I. Chowdhury, D. G. Goodwin, X. Chang, S. Martin, D. H. Fairbrother, D. Bouchard and R. G. Zepp, The contribution of indirect photolysis to the degradation of graphene oxide in sunlight, Carbon, 2016, 110, 426–437 CrossRef CAS.
- G. P. Kotchey, B. L. Allen, H. Vedala, N. Yanamala, A. A. Kapralov, Y. Y. Tyurina, J. Klein-Seetharaman, V. E. Kagan and A. Star, The Enzymatic Oxidation of Graphene Oxide, ACS Nano, 2011, 5, 2098–2108 CrossRef CAS PubMed.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake, S. A. Rusak, A. L. Rose and T. D. Waite, Methods for reactive oxygen species (ROS) detection in aqueous environments, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS.
- W. P. Kwan and B. M. Voelker, Rates of hydroxyl radical generation and organic compound oxidation in mineral-catalyzed Fenton-like systems, Environ. Sci. Technol., 2003, 37, 1150–1158 CrossRef CAS PubMed.
- B. A. Southworth and B. M. Voelker, Hydroxyl Radical Production via the Photo-Fenton Reaction in the Presence of Fulvic Acid, Environ. Sci. Technol., 2003, 37, 1130–1136 CrossRef CAS PubMed.
- R. G. Zepp, B. C. Faust and J. Hoigne, Hydroxyl Radical Formation in Aqueous Reactions (Ph 3-8) of Iron(II) with Hydrogen-Peroxide - the Photo-Fenton Reaction, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef CAS.
- J. W. Arbogast, A. P. Darmanyan, C. S. Foote, F. N. Diederich, R. L. Whetten, Y. Rubin, M. M. Alvarez and S. J. Anz, Photophysical properties of sixty atom carbon molecule (C60), J. Phys. Chem., 1991, 95, 11–12 CrossRef CAS.
- D. I. Schuster, P. S. Baran, R. K. Hatch, A. U. Khan and S. R. Wilson, The role of singlet oxygen in the photochemical formation of C60O, Chem. Commun., 1998, 2493–2494, 10.1039/A806603E.
- J.-J. Yin, F. Lao, P. P. Fu, W. G. Wamer, Y. Zhao, P. C. Wang, Y. Qiu, B. Sun, G. Xing, J. Dong, X.-J. Liang and C. Chen, The scavenging of reactive oxygen species and the potential for cell protection by functionalized fullerene materials, Biomaterials, 2009, 30, 611–621 CrossRef CAS PubMed.
- I. Fenoglio, M. Tomatis, D. Lison, J. Muller, A. Fonseca, J. B. Nagy and B. Fubini, Reactivity of carbon nanotubes: Free radical generation or scavenging activity?, Free Radical Biol. Med., 2006, 40, 1227–1233 CrossRef CAS PubMed.
- X. Qu, P. J. Alvarez and Q. Li, Photochemical transformation of carboxylated multiwalled carbon nanotubes: role of reactive oxygen species, Environ. Sci. Technol., 2013, 47, 14080–14088 CrossRef CAS PubMed.
- H. Bai, W. T. Jiang, G. P. Kotchey, W. A. Saidi, B. J. Bythell, J. M. Jarvis, A. G. Marshall, R. A. S. Robinson and A. Star, Insight into the Mechanism of Graphene Oxide Degradation via the Photo-Fenton Reaction, J. Phys. Chem. C, 2014, 118, 10519–10529 CrossRef CAS PubMed.
- X. J. Zhou, Y. Zhang, C. Wang, X. C. Wu, Y. Q. Yang, B. Zheng, H. X. Wu, S. W. Guo and J. Y. Zhang, Photo-Fenton Reaction of Graphene Oxide: A New Strategy to Prepare Graphene Quantum Dots for DNA Cleavage, ACS Nano, 2012, 6, 6592–6599 CrossRef CAS PubMed.
- T. Du, A. S. Adeleye, T. Zhang, C. Jiang, M. Zhang, H. Wang, Y. Li, A. A. Keller and W. Chen, Influence of light wavelength on the photoactivity, physicochemical transformation, and fate of graphene oxide in aqueous media, Environ. Sci.: Nano, 2018, 5, 2590–2603 RSC.
- T. Du, A. S. Adeleye, T. Zhang, N. Yang, R. Hao, Y. Li, W. Song and W. Chen, Effects of ozone and produced hydroxyl radicals on the transformation of graphene oxide in aqueous media, Environ. Sci.: Nano, 2019, 6, 2484–2494 RSC.
- L. Duan, T. Zhang, W. Song, C. Jiang, Y. Hou, W. Zhao, W. Chen and P. J. J. Alvarez, Photolysis of graphene oxide in the presence of nitrate: implications for graphene oxide integrity in water and wastewater treatment, Environ. Sci.: Nano, 2019, 6, 136–145 RSC.
- Y. Gao, X. Ren, X. Zhang and C. Chen, Environmental fate and risk of ultraviolet- and visible-light-transformed graphene oxide: A comparative study, Environ. Pollut., 2019, 251, 821–829 CrossRef CAS PubMed.
- A. S. Adeleye, X. Wang, F. Wang, R. Hao, W. Song and Y. Li, Photoreactivity of graphene oxide in aqueous system: Reactive oxygen species formation and bisphenol A degradation, Chemosphere, 2018, 195, 344–350 CrossRef CAS PubMed.
- F. F. Zhao, S. C. Wang, Z. L. Zhu, S. G. Wang, F. F. Liu and G. Z. Liu, Effects of oxidation degree on photo-transformation and the resulting toxicity of graphene oxide in aqueous environment, Environ. Pollut., 2019, 249, 1106–1114 CrossRef CAS.
- D. M. Wang, Y. Zhang, L. L. Zheng, X. X. Yang, Y. Wang and C. Z. Huang, Singlet Oxygen Involved Luminol Chemiluminescence Catalyzed by Graphene Oxide, J. Phys. Chem. C, 2012, 116, 21622–21628 CrossRef CAS.
- X. G. Hu, M. Zhou and Q. X. Zhou, Ambient Water and Visible-Light Irradiation Drive Changes in Graphene Morphology, Structure, Surface Chemistry, Aggregation, and Toxicity, Environ. Sci. Technol., 2015, 49, 3410–3418 CrossRef CAS PubMed.
- H. Zhao, C. Zhang, Y. Wang, W. Chen and P. J. J. Alvarez, Self-Damaging Aerobic Reduction of Graphene Oxide by Escherichia coli: Role of GO-Mediated Extracellular Superoxide Formation, Environ. Sci. Technol., 2018, 52, 12783–12791 CrossRef CAS PubMed.
- Y. Han, C. D. Knightes, D. Bouchard, R. Zepp, B. Avant, H. S. Hsieh, X. Chang, B. Acrey, W. M. Henderson and J. Spear, Simulating graphene oxide nanomaterial phototransformation and transport in surface water, Environ. Sci.: Nano, 2019, 6, 180–194 RSC.
- W. S. Hummers and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- W. C. Hou, I. Chowdhury, D. G. Goodwin, Jr., W. M. Henderson, D. H. Fairbrother, D. Bouchard and R. G. Zepp, Photochemical transformation of graphene oxide in sunlight, Environ. Sci. Technol., 2015, 49, 3435–3443 CrossRef CAS.
- C. G. Hatchard and C. A. Parker, A New Sensitive Chemical Actinometer. II. Potassium Ferrioxalate as a Standard Chemical Actinometer, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS.
- J. W. Stucki and W. L. Anderson, The Quantitative Assay of Minerals for Fe2+ and Fe3+ Using 1,10-Phenanthroline: I. Sources of Variability1, Soil Sci. Soc. Am. J., 1981, 45, 633–637 CrossRef CAS.
- M. S. Elovitz and U. von Gunten, Hydroxyl Radical/Ozone Ratios During Ozonation Processes. I. The Rct Concept, Ozone: Sci. Eng., 1999, 21, 239–260 CrossRef CAS.
- C. Hu, F. E. Muller-Karger and R. G. Zepp, Absorbance, absorption coefficient, and apparent quantum yield: A comment on common ambiguity in the use of these optical concepts, Limnol. Oceanogr., 2002, 47, 1261–1267 CrossRef.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in Aqueous Solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- M. Saran and K. H. Summer, Assaying for hydroxyl radicals: Hydroxylated terephthalate is a superior fluorescence marker than hydroxylated benzoate, Free Radical Res., 1999, 31, 429–436 CrossRef CAS.
- W. R. Haag and J. Hoigne, Singlet Oxygen in Surface Waters. 3. Photochemical Formation and Steady-State Concentrations in Various Types of Waters, Environ. Sci. Technol., 1986, 20, 341–348 CrossRef CAS.
- V. Ponti, M. U. Dianzani, K. Cheeseman and T. F. Slater, Studies on the reduction of nitroblue tetrazolium chloride mediated through the action of NADH and phenazine methosulphate, Chem.-Biol. Interact., 1978, 23, 281–291 CrossRef CAS.
- M. W. Sutherland and B. A. Learmonth, The Tetrazolium Dyes MTS and XTT Provide New Quantitative Assays for Superoxide and Superoxide Dismutase, Free Radical Res., 1997, 27, 283–289 CrossRef CAS.
- B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, Reactivity of HO2/O−2 Radicals in Aqueous Solution, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS.
- H. S. Hsieh, R. Wu and C. T. Jafvert, Light-independent reactive oxygen species (ROS) formation through electron transfer from carboxylated single-walled carbon nanotubes in water, Environ. Sci. Technol., 2014, 48, 11330–11336 CrossRef CAS.
- J. G. Radich and P. V. Kamat, Making Graphene Holey. Gold-Nanoparticle-Mediated Hydroxyl Radical Attack on Reduced Graphene Oxide, ACS Nano, 2013, 7, 5546–5557 CrossRef CAS.
- M. E. Lindsey and M. A. Tarr, Inhibition of hydroxyl radical reaction with aromatics by dissolved natural organic matter, Environ. Sci. Technol., 2000, 34, 444–449 CrossRef CAS.
- P. L. Brezonik and J. Fulkerson-Brekken, Nitrate-induced photolysis in natural waters: Controls on concentrations of hydroxyl radical photo-intermediates by natural scavenging agents, Environ. Sci. Technol., 1998, 32, 3004–3010 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9en00693a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Richard G.
Zepp
a and
Richard G.
Zepp