
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion effects on molecular interaction between graphene oxide and organic molecules
Zilong
Liu
 *ab,
Tatiana
Rios-Carvajal
a,
Martin
P. Andersson
c,
Marcel
Ceccato
a,
Susan L. S.
Stipp
a and
Tue
Hassenkam
*a
*ab,
Tatiana
Rios-Carvajal
a,
Martin
P. Andersson
c,
Marcel
Ceccato
a,
Susan L. S.
Stipp
a and
Tue
Hassenkam
*a
aNano-Science Center, Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: zlliu89@gmail.com; tue@nano.ku.dk
bChemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
cChemical Engineering, Department of Chemical and Biochemical Engineering, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
First published on 20th June 2019
Abstract
Interactions between graphene oxide (GO) and organic molecules play a role in processes such as environmental remediation and water treatment. However, little is known about underlying molecular level processes with the presence of ions. In this study, we utilized atomic force microscopy (AFM) in chemical force mapping (CFM) mode to directly probe their adhesion interactions. AFM tips were functionalised to serve as models for nonpolar and polar organic molecules, i.e. with alkyl, –CH3, and carboxyl, –COO(H). For experiments with –COO(H) tips, adhesion between GO and tips decreased in the order: Ba2+ > Ca2+ > Mg2+ > Na+, whereas for the –CH3 tips, ion dependent adhesion was relatively low but followed the same: Ba2+ > Ca2+ > Mg2+ ≈ Na+. Calculations with Derjaguin–Landau–Verwey–Overbeek (DLVO) theory and the Schulze–Hardy rule could not account for the observations. We propose that ion bridging plays a definitive role in adhesion between –COO(H) tips and the GO surface. This is consistent with proposed models with density functional theory (DFT) calculations. Adhesion of –CH3 tips is a response to the hydrophilic interactions and the ion dependent part is suggested to arise from ion bridging between slightly negative charged –CH3 tips and the GO surface. High pH had a notable influence on the adhesion of the –COO(H) tip but a negligible effect on the –CH3 tip. These results offer important insights into interactions between solutions and mineral surfaces with adsorbed organic molecules.
Environmental significanceIon effect is important for the adhesion of organic materials on graphene oxide (GO) surface, such as in the processes of environmental remediation and water treatment. Herein, using chemical force mapping mode of atomic force microscopy, the underlying molecular interactions were directly measured. Adhesion behaviors of organics terminated with polar and nonpolar groups of –COO(H) and –CH3 followed similar order: Ba2+ > Ca2+ > Mg2+ ≈ Na+. Calculations with Derjaguin–Landau–Verwey–Overbeek (DLVO) theory and Schulze–Hardy rule could not explain the observed cation effect and we proposed ion bridging played a definitive role in the adhesions. This was consistent with density functional theory (DFT) calculations. It was also found that larger adhesion response was observed for the –COOH tip at high pH, while response was relatively low but significant for the –CH3 tip. These results provide important insights into the interaction processes between solutions and mineral surfaces with adsorbed organic molecules and offer clues for improving remediation strategies, such as the application of GO as adsorbents, membranes, catalysts and coating materials. |
1. Introduction
Graphene oxide (GO) is a two dimensional nanomaterial featuring a variety of chemically reactive functionalities, such as carboxyl, hydroxyl and epoxy at the edges and on the plane of GO sheets,1–3 which can be differentially functionalized.4 In spite of oxygen configurations, a significant proportion of the sp2 hybridized carbon network remains intact,5 allowing GO to remain a flexible planar sheet,6,7 with a high surface to volume ratio. Together with these structural characteristics, GO has wide potential applications in environmental remediation,8 water treatment,9,10 drug delivery,11 catalysis,4,12 energy storage.13,14 Among them, the large surface area (theoretical limit to 2630 m2 g−1)15 endows GO with excellent performance in adsorption of many kinds of organic compounds from aqueous solution, such as pesticides,16 aromatic compounds,17 antibiotics,18 and organic compounds.19 Therefore, understanding molecular interaction processes between organic molecule and GO is essential for determining the mobility and transport of organic contaminants and to provide clues for improving remediation strategies.The oxygen containing functional groups in GO tend to bind hydrophilic species through ionic interaction, hydrogen bonding, van der Waals, electrostatic interaction or Lewis acid–base interaction, while the graphitic parts of GO are affinitive to hydrophobic organic compounds by π–π stacking and hydrophobic effects.17,19–23 As one kind of the most common organic contaminants, polycyclic aromatic hydrocarbon and its derivatives can be adsorbed by GO and reduced graphene oxide (rGO) because of its aromatic character.17,20,23 Tetracycline antibiotics also have aromatic rings, except π–π interaction, and the cation–π bonding may happen between protonated amino groups and graphene π-electrons for the adsorption on GO.18 Cationic dyes such as methylene blue and rhodamine B could electrostatically bind to the negatively charged GO and hence removal is expected to be effective.24 Hydrophobic and oleophilic rGO-wrapped sponge are promising candidates for oil-spill remediation, enabling fast clean-up of viscous crude-oil spill.25
Although significant research has been conducted on organic material interacting with GO in water,17–25 few studies have considered their interactions in the presence of various ions. A complex mix of organic contaminants and ions often coexist in the aqueous solution.26 The adsorption of ions would affect the mobility and transport of organic compounds.27 To date, there are extensive study about the colloidal behaviour, adsorption, aggregation, dispersion and morphological transformation of GO in the presence of salt ions and heavy metal ions.28–31 It needs to understand the details how the ions affect the interaction processes between organic molecules and GO because the coexistence of metal ions and organic contaminants would cause different coadsorption mechanisms. We therefore constructed a model system to better understand the interaction and thus allow fine tuning for improved performance of organics adsorption. The atomic force microscopy (AFM) in chemical force mapping (CFM) mode was used to study the effect of ions on the interaction between GO and organic molecules. The AFM tips were coated with a self-assembled monolayer (SAM) with specific functionalities to interact with GO surface. As aromatic compounds have been extensively studied,17,20,23 this work mainly focus on common organics terminated with polar groups, –COO(H) and nonpolar groups, –CH3. Our group has widely applied CFM to investigate interactions between organics and various solid surfaces, such as sandstones,32,33 chalk34 and sapphire.35 GO itself could also be used as a model for other surfaces.
In this study, we set out to improve understanding of the underlying molecular level processes of common environmental ions (Na+, Mg2+, Ca2+) and heavy metal ion (Ba2+) on interactions between GO and two types of tips that represent organic compounds, with physicochemical properties of the tested cations in Table 1.36,37 The divalent cations from the same group with different ion radii and hydrations could influence the adhesion between GO and organic molecules. Few studies also investigated the removal of heavy metal ion of Ba2+ from contaminated soil and produced water.38,39 The main objective of this study was: i) to gain insight into the processes that affect adsorption behavior on GO surfaces with presences of various ions, ii) to compare the results from –COO(H) and –CH3 tips that represented polar and nonpolar organic compounds and analyze the influence of pH, iii) to offer theoretical explanations (using DLVO theory and DFT calculations) and possible modifications to the adhesions.
| Cations | Ionic radii (Å) | Hydrated radii (Å) | Electronegativity | Polarizability (10−24 cm3) |
|---|---|---|---|---|
| Na+ | 0.95 | 3.58 | 0.93 | 24.08 |
| Mg2+ | 0.65 | 4.28 | 1.31 | 10.06 |
| Ca2+ | 0.99 | 4.12 | 1.01 | 25.00 |
| Ba2+ | 1.35 | 4.04 | 0.89 | 39.70 |
2. Experimental
2.1. Graphene oxide
Commercial graphite powder (Grade 3061) was obtained from Asbury Carbon Mills. H2SO4 (95–98%), KMnO4 and H2O2 (33%) were obtained from Sigma Aldrich and NaNO3 was obtained from Merck. All the chemicals were reagent grade or better and were used as received. GO was prepared by harsh oxidation of the graphite powder by the modified Hummers method described previously.3,40 For each sample, we dropped a 30 μL aliquot of GO solution on the silicon wafer with a speed of 3000 rpm and waited for one minute, then repeated the procedure six times. The full cover of GO on the silicon wafer ensured the force measurements conducted on the GO rather the silicon substrate.2.2. Solutions
All solutions were made with ultrapure deionized water (Milli-Q, resistivity >18.2 MΩ cm). To keep equivalent positive charge, the concentration of divalent cation solutions (Mg2+, Ca2+, Ba2+: 0.3 M) is half of NaCl solution of 0.6 M. Salinity for the NaCl solution is roughly equivalent to seawater. In our previous study,41 we investigated the effect of mixed cation solutions at high salinity (similar to seawater) and low salinity concentrations on adhesion. Here we focus on the effect of certain sets of cation on the adhesion. Maintaining moles of charge was important to investigate the effect of an ion exchange process where there was an equivalent amount of positive charge available for interactions with GO surface. The results based on the high concentration of divalent cation would not affect the conclusions about the proposed cation bridging mechanism. In an environmental context, the aqueous solution always contains mixed cations and our results provide important insights onto the role of certain types of divalent cation in the adhesion between GO and organic molecules. With low cationic strength, the degree of the contribution of cation bridging to the total adhesion could decrease due to the decreased number of bridges. For most of the experiments, the pH of solutions was adjusted to 5.5, while we increased the pH to 8.8 when we investigated the adhesion at higher pH, which the –COO(H) and functional groups of GO can be more deprotonated. All measurements were conducted at room temperature.2.3. AFM and CFM probes
The GO surface was scanned in tapping mode with standard silicon probes from Olympus (OMCL-AC240), that had a nominal spring constant of 2 nN nm−1 and resonance frequency of ∼80 kHz. To measure the adhesion force and generate force maps, we used Olympus biolever AFM probes. The chips are equipped with two cantilevers with nominal spring constants of 30 and 6 pN nm−1. Before each experiment, the deflection sensitivity of each tip was determined. Actual spring constants varied from 20 to 30 pN nm−1 for the stiff cantilevers and from 4 to 8 pN nm−1 for the soft cantilevers. In our experiments, we chose the stiff cantilevers. Before use, the biolevers were rinsed with ethanol, then dried with a jet of nitrogen, and UV ozone treated for 20 min. They were immediately submerged in an ethanol solution of ∼5 mM 1-undecanethiol (HS(CH2)10CH3) or 11-mercaptoundecanoic acid (HS(CH2)10COOH) for at least 24 h.32,33 The functionalised tips were rinsed with ethanol for half hour just prior to use to minimise the amount of molecules that were not firmly bonded.2.4. AFM imaging and force mapping
We used an MFP-3D atomic force microscope from Asylum Research, Santa Barbara, USA. AFM images, to show the topographic features of the surface, were acquired in tapping mode (512 × 512 pixels) in air. Chemical force maps were collected with 30 × 30 data points, over an area of 2 × 2 μm2, such as shown in the inset of Fig. 1. To collect the data for a force map, the tip and sample are brought into contact and then separated again, generating a force–distance curve such as that shown in Fig. 1. This is repeated for each point in a grid over the surface to produce the pixels required for a map. At each point, the tip starts ∼1 μm away from the surface. It moves toward the surface from a point where the cantilever and the surface are not in contact (the part A of Fig. 1a), comes into contact (the part B), and eventually stops when the surface resists with a force (the part C), that is predetermined in the software of the instrument, in this case, 600 pN.41 The tip is then retracted from the surface and as it moves away (the part D), adhesion between tip and sample causes the cantilever to deflect. At some displacement, the tip snaps free until the applied force is sufficient to separate the tip from the surface (the part E). The force at that point is the adhesion, which provides one pixel for the force map. The full set of adhesion force measurements is arrayed in x, y space and given false colour to reflect the relative adhesion force, which produces the map. | ||
| Fig. 1 (a) Schematic illustration of the tip movement during the approach and retraction processes in a force curve experiment. A: The cantilever starts from a point where it is not in contact with the surface, B: point of contact, C: the cantilever would be retracted after maximum given force is reached, D: point of zero deflection, E: maximum adhesion to separate the tip from the surface, F: retract. (b) A typical force–distance curve from oxidised graphene, measured with a –CH3 functionalised AFM tip. The red curve marks the approach and the blue curve shows the tip withdrawal from the surface. The adhesion force is measured where the tip springs free of the surface during retraction. The inset is a representative chemical force map, with each pixel showing one time of force curve. | ||
We performed different sets of experiments, with Milli-Q water, NaCl, MgCl2, CaCl2 and BaCl2 solutions. Each set has different orders of exchanging solutions. In the fluid cell, the sample and tip were initially in contact with ∼3 mL of original solutions that was one of above solutions and the force maps were repeated at least three times until the adhesion was relatively stable. For each experiment, the initial solution was then replaced by the alternate solution. This was done by extracting ∼2 mL from the liquid cell and then injecting ∼2 mL of the new solution. After five solution exchange cycles, 99.6% of the solution had been replaced. This kept the sample from drying and the tip from losing contact with the imaging location. After each solution exchange, which took around 5 minutes, the new force maps were acquired and the solution was changed again and so on. In this way, maps could be generated at precisely the same site on the sample, with the same tip during exposure to a series of solutions.
2.5. X-ray photoelectron spectroscopy
The chemical state and the element composition of GO were determined with XPS (Kratos Axis Ultra DLD), using monochromatized Al Kα (hν = 1486.6 eV) as the excitation source. The data were analyzed using commercial software, CasaXPS, and a Shirley background fit. The absolute energy scale was calibrated to the carbon C 1s peak of 284.5 eV. Uncertainty in XPS binding energy is about 0.1 eV. Uncertainty in the atomic percentages determined from XPS data is on the order of 5–10%.2.6. Derjaguin–Landau–Verwey–Overbeek (DLVO) theory
The DLVO theory is a continuum theory that describes the force between two surfaces or particles that interact through a liquid medium, such as an aqueous solution. There are attractive components to the adhesion force between the AFM tip and the substrate surface that come from van der Waals (vdW). To determine the vdW forces, we used the standard expression that describes these forces between a sphere and a planar surface42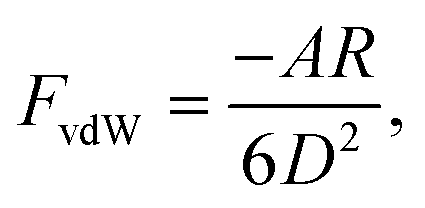 | (1) |
 | (2) |
 | (3) |
In this case, k represents Boltzmann's constant, T, temperature, e, the elemental charge, ρ∞i, the number density in the bulk (m−3) of the ion, i, and z represents the valence of ion i. The total adhesion (Fad) is the sum:
| Fad = FvdW + FEDL. | (4) |
2.7. Density functional theory (DFT) calculations
All DFT calculations were performed with the TURBOMOLE program, v6.5, using the Becke–Perdew (BP) functional, the triple-ζ valence plus polarization (TZVP) basis set, and the COSMO implicit solvent model.41,44 The COSMOtherm program with parameterization BP_TZVP_C30_1301 was used for all COSMO-RS calculations at 298 K. We modelled the –COO(H) tip using a dimer of octanoate, where we fixed the six carbon atoms furthest from the –COO(H) during the optimization. The length of nonterminal part of alkane chains will not affect the calculations. The adsorption energy for ions to the –COO(H) tip was determined in the reaction panel of COSMOtherm, which calculated the free energy of a reaction in solution (excluding the effect of the partition functions). The COSMOtherm flatsurf module was used to calculate free-energy differences that are required for transferring a molecule from a bulk solvent to an interface between two solvents.The interactions between the –COO(H) tip and the GO surface, in solutions with cations (i.e. –COO-M, where M = cations) and without (i.e. the –COOH dimer alone), were modelled. We used several types of molecule surfaces to model GO, which were the main functional groups of GO, as well as graphene. The models for the –COO(H) tip were the fully deprotonated carboxyl dimer with bound cations as well as the protonated acid. We compared the interaction energy of a dimer carrying a divalent ion in Fig. 2a to that of a fully protonated dimer in Fig. 2b, by calculating the differences in chemical potential, μ, between the dimers interacting with the surface phase, S, and the dimers solvated in the water phase, W. The difference in chemical potentials of the dimers carrying cations, ΔμCOO-M, and the protonated dimers, ΔμCOOH, in two phases (S and W) can be described as,
| ΔΔμ = ΔμCOO-M − ΔμCOOH = (μCOO-M(S) − μCOO-M(W)) − (μCOOH(S) − μCOOH(W)), | (5) |
 | ||
| Fig. 2 Octanoate dimers interacting with a surface in their (a) deprotonated form, –COO-M carrying a divalent ion, and (b) in their protonated form, –COOH. The difference in chemical potential between the dimers in the water phase and on GO surface is used to estimate the likelihood of ion bridging between the –COO(H) tip and GO surface. | ||
| Surface model (S) | Mg2+ | Ca2+ | Ba2+ |
|---|---|---|---|
| a We considered charged and neutral surfaces of GO according to the presence of possible functional groups. | |||
| Benzene | 37.3 | 37.6 | 45.7 |
| Benzoate | −3.8 | −13.6 | −22.1 |
| Benzoic acid | 9.8 | 9.6 | 8.7 |
| Phenolate | −4.6 | −17.7 | −29.1 |
| Phenol | 8.3 | 7.0 | 4.6 |
| Epoxide | 22.6 | 20.9 | 25.4 |
3. Results and discussion
3.1. Structural characterization of GO
Fig. 3a shows an AFM height image of a typical surface of GO, such as we used in the subsequent chemical force mapping. The height varies mostly with single layers of graphene, with the average deviation in height of 1.7 nm. Folds in the otherwise small patches of GO can be observed, such as a narrow bright curve indicated by the white arrow in the middle of the 2 μm × 2 μm image. The XPS spectrum of C 1s of GO is presented in Fig. 3b, with four most prominent deconvoluted components. One peak (blue) represents the main peak at 284.5 eV because of graphitic carbon and other peaks associated with oxygen functional groups such as C–O (red) in hydroxyl and epoxy at 286.5 eV, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (cyan) in carbonyl at 287.9 eV and O–CO (pink) in carboxylic and/or ester at 290.3 eV. Our results agree well with previously reported data for GO.3,9,21 The relative concentrations were 48.3% for C–C, 43.1% for C–O, 7.3% for CO and 1.3% for O–CO, indicating high oxidation of GO.
O (cyan) in carbonyl at 287.9 eV and O–CO (pink) in carboxylic and/or ester at 290.3 eV. Our results agree well with previously reported data for GO.3,9,21 The relative concentrations were 48.3% for C–C, 43.1% for C–O, 7.3% for CO and 1.3% for O–CO, indicating high oxidation of GO.
 | ||
| Fig. 3 (a) AFM height image of GO surface, with white arrow indicating a fold structure. (b) XPS spectrum of the C 1s region from a sample of GO. The black solid line shows the original data. The individual peaks represent the intensity of photoelectrons originating from bonds of C–C (blue), C–O (red), CO (cyan) and O–OC (pink). | ||
3.2. The –COO(H) interaction with GO
Fig. 4 shows the average adhesion measured between the –COO(H) terminated tip and GO surface in different solutions at pH 5.5. Each bar in the plot represents the adhesion averaged from five force maps as we repeated the measurements five times for each kind of solution, with each map comprising 900 force curves generated over a 2 × 2 μm2 area, such as the inset force map in Fig. 1b. The error bar therefore represents the standard deviations of 4500 force curves in adhesion measured over the same surface with the same AFM tip collected over five force mapping measurements. They do not represent the error in the true sense of uncertainty but rather reflects the range of variability in the adhesion over the surface. For example, the adhesion in CaCl2 solution of –COO(H) set 3 is 275 ± 6 pN. The experiments show that the adhesion with –COO(H) tip is highest in CaCl2 solution, almost twice the adhesion in NaCl solution and minimum in pure water. Even though there were negligible ions in control (H2O) scenarios, adhesion forces were ∼100 pN. In our recent study,45 we derived hydration forces of ∼62 pN for the –COO(H)–COO(H) hydrophilic system, which was consistent with the reported value from Butt et al.43 The source of adhesions in the current setup could therefore originate from hydration forces between the –COO(H)–GO hydrophilic surfaces.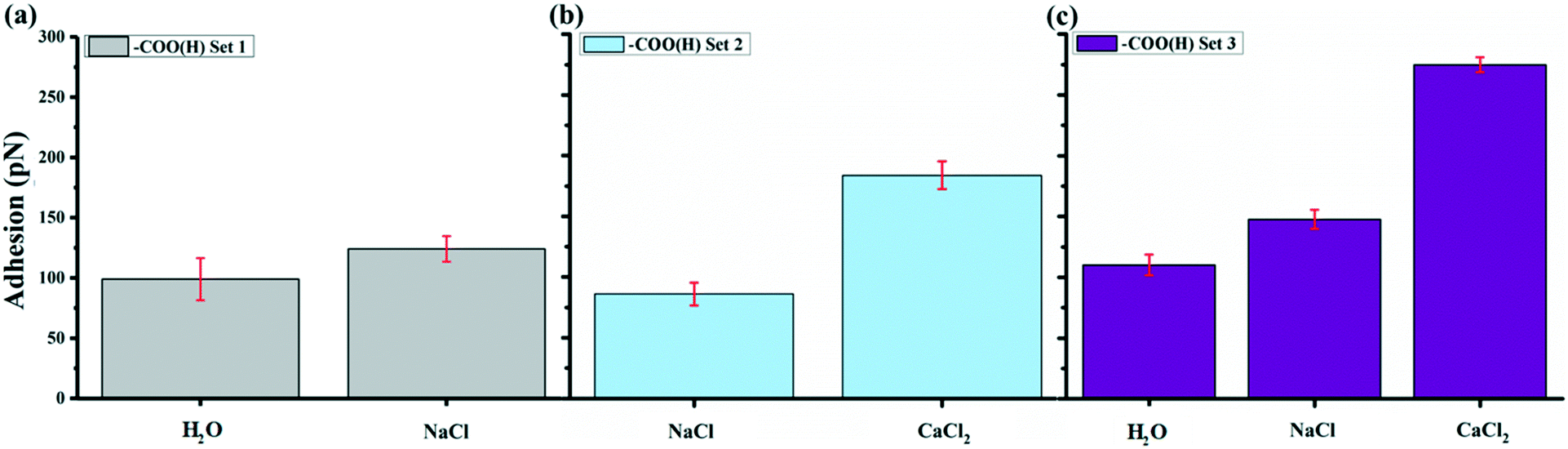 | ||
| Fig. 4 Adhesion of –COO(H) functionalized tips in different salt solutions on GO surface with pH 5.5. (a) –COO(H) set 1 started with H2O, then NaCl. (b) –COO(H) set 2 started with NaCl, then CaCl2. (c) Set 3 in the order of H2O, NaCl and CaCl2. Each set conducted the measurements with –COO(H) tips on different GO areas. We provided a step-by-step investigation on ion effects at different experiment sets by separated comparisons between two or more kinds of cations and/or changing measurement order. The error bars represent the standard deviation of the mean adhesion collected at each solution, which repeated five times. | ||
To determine van der Waals force FvdW with eqn (1), R, the tip radius of curvature, is reported by the manufacturer to be 30 nm. The Hamaker constant was 6.26 × 10−21 J for GO–GO nanomaterials30 and 5.02 × 10−21 J for –COO(H) SAMs46 so that using the geometric mean we got a Hamaker constant in our system to be 5.6 × 10−21 J. D, the distance between the tip and surface, could not be determined from our measurements so we estimated it from experiments with the –CH3 tip and core plug sandstone grains, i.e. ∼1 nm.33 Our GO is highly hydrophilic; thus, it could result in a larger distance, i.e. as much as 2.5 nm. The space was likely filled with a water film and hydrated or partially hydrated cations and anions. Using eqn (1), we calculated FvdW to be in the range of 4 to 13 pN, which was much lower than the experimental results (mostly above 100 pN) in –COO(H) set 1–3. It is therefore unlikely that the adhesion was caused by van der Waals force alone.
Using eqn (3), the Debye length in the NaCl solution was 0.39 nm and it was 0.32 nm in the CaCl2 solution, meaning that the EDL thickness was suppressed to length scales comparable to single water molecules in these high salinity solutions. To be able to use eqn (2) to calculate FEDL, we had to estimate the surface charge densities of the tip and GO surface. For our surfaces, there were two possible charging processes,
(I) Deprotonation of carboxylic groups
| –COOH + H2O ⇄ –COO− + H3O+, | (6) |
(II) Deprotonation of enolic and phenolic groups
| C–OH + H2O ⇄ C–O− + H3O+. | (7) |
The reaction I affords negative surface charges for the –COO(H) tip, while the surface charge of GO could be affected by both reactions. In previous study we have estimated the surface charge of the tip σT in similar solutions to −0.26 C m−2.41 Bei et al.29 found a zeta potential of −10 mV for GO surface in high salinity solutions. If we combine this result with Grahame equation, σS of GO surface in Na+ and Ca2+ solution should be around −0.018 and −0.023 C m−2. We derived the repulsive FEDL from eqn (2), producing a range of 0–24 pN, which might have reduced the adhesion a bit but not to a level that could have affected the overall tendency with the different levels of increased adhesion with different ions. Since FvdW and FEDL cannot explain the adhesion we observed, the DLVO model does not account for cation effect on adhesion, suggesting that there must be additional contribution for adhesion. The obvious alternative is bridging formed by the divalent cations between the negative surfaces.
The monovalent cation of Na+ should not have specific interactions with functional groups on GO surface or tip, because the free energy of interaction with carboxylic groups is most likely weak (−2.98 kJ mol−1).47 The higher adhesion with the presence of Na+ in solution than in pure water can be explained by the thicker double layer in the pure water which causes an increased EDL repulsion. The increased charge screening that the added Na+ provided to the charge surface decrease the double layer repulsion between GO and tip. Based on Schulze–Hardy rule,48 Ca2+ could produce more charge screening than Na+. It is also important to consider the effect of water molecules, which align themselves around cations form a hydration shell. The cations with relatively smaller ionic radii have higher hydration numbers and larger hydrated radii, whereas cations with larger ionic radii have weaker hydration shells and tend to more easily detach their hydration layers.49,50 Thus, Na+ with small ionic radii can only form outer-sphere complexes and cannot serve as bridging agents. However, the calculated values from the DLVO model and the hydration forces, assuming that this is all the forces involved, fits well with the observed value for the adhesion in NaCl solution of ∼100 pN.
In the presence of Ca2+, the adhesion picture gets more complicated because divalent cations can not only screen the surface charge but also bind to surface functional groups of GO, forming inner-sphere complexes. It is well known that Ca2+ can form complexes with carboxylic acids, such as m-hydroxybenzoic acid and 3,5-dihydroxybenzoic acid,51 and with carboxyl groups on humic and fulvic acids.52 Accordingly, Ca2+ could result in strong cation bridge by forming complexes with surface functional groups of GO and –COO(H) tip. There are three types of cation bridging that may cause the higher adhesion: (1) bridging the –COO(H) located in edge and/or plane of GO with –COO(H) tip, (2) bridging the enolic and phenolic groups located in plane of GO with –COO(H) tip, (3) cation–π interaction through residual π-conjugated domains in GO and –COO(H) tip. The first two types come from the deprotonated or partially deprotonated oxygen functional groups by reaction I and II bridging the –COO(H) tip that was identified with model surfaces of benzolate and phenolate in our DFT calculations in Table 2. From our benzene model surface with rich π electron donors, this kind of cation–π interaction seems negligible and ion bridging from (deprotonated) negatively charged surface sites appear to be the major contribution to the adhesion.
To further test this theory and its implications, we performed a new set of experiments where we tested solutions with other sets of divalent ions. Fig. 5a show a plot of the average adhesion from sequential force maps obtained with the addition of MgCl2 compared to Fig. 4. The adhesions in the different solutions were found to have the order CaCl2 > MgCl2 > NaCl. With the presence of BaCl2 in Fig. 5b, the adhesion became even stronger than in a CaCl2 solution in the three experimental sets where we included a BaCl2 solution. The adhesion followed the order BaCl2 > CaCl2 > MgCl2 > NaCl. Table 3 summarizes the adhesion difference (ΔFad) between NaCl and divalent salt solutions and the relative increase in adhesion (% Inc) defined as,
| ΔFad = Fad(MeCl2) − Fad(NaCl), | (8) |
| % Inc = 100 × (Fad(MeCl2) − Fad(NaCl))/Fad(NaCl). | (9) |
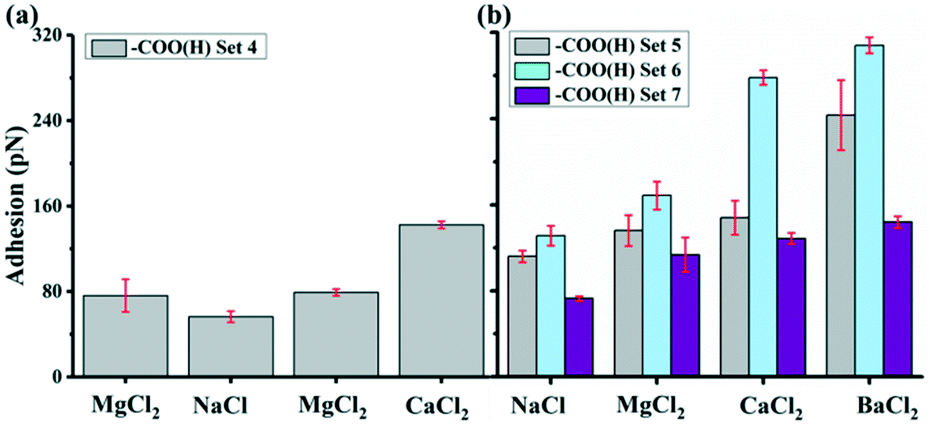 | ||
| Fig. 5 Ion effects on adhesion of –COO(H) tips on GO surface with pH 5.5. (a) –COO(H) set 4 was in the order of MgCl2, NaCl, MgCl2 and CaCl2. The twice tests in MgCl2 before and after NaCl were to verify reproducible data and that different measurement orders would not affect the results we obtained. (b) –COO(H) set 5–7 were in the order of NaCl, MgCl2 CaCl2 and BaCl2. These sets included Ba2+ and measurements were performed on different samples with the same kind of tip. | ||
| With –COO(H) of ΔFad (% Inc) | With –CH3 of ΔFad (% Inc) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Set 5 | Set 6 | Set 7 | Set 8 | Set 9 | Set 1 | Set 2 | Set 3 | Set 4 | Set 5 | |
| Mg2+ | 24 (21) | 37 (28) | 41 (56) | 108 (172) | 105 (108) | 18 (13) | 5 (4) | −20 (−13) | −4 (−3) | −46 (−27) |
| Ca2+ | 36 (32) | 147 (112) | 56 (77) | 234 (372) | 186 (192) | 50 (38) | 10 (8) | 19 (11) | 38 (36) | 35 (21) |
| Ba2+ | 131 (117) | 177 (135) | 71 (97) | 288 (460) | 250 (257) | 119 (89) | 62 (54) | 92 (57) | 84 (78) | 181 (107) |
The adhesion increased in all divalent cation solutions. The increase percentage for Mg2+ was smaller ranging from 21 to 56% in experiment set 5–7 and for Ca2+, it was 32–112%. While Ba2+ had strongest increase ratio as high as 135%, indicating a specific cation dependent response.
According to the Schulze–Hardy rule,48 the same valent cations should produce similar charge screening effects. In our experiments, divalent cations displayed different adhesion behaviors, indicating that the interaction between tip and GO was a more complex process rather than only a simple EDL suppression process. This specificity could be linked to the hydration shell thickness of cations. As discussed above, cations with small ionic radii have large hydrated radii and can only form outer-sphere complexes, whereas cations with large ionic radii tend to form inner-sphere complexes.50 The Mg2+ is a smaller ion with a valence of 2 and can strongly hold its first hydration shell with 6 water molecules and it also has 9 to 12 water molecules coordinated in its second hydration shell, whereas Ca2+ has only 3 to 6.53 The larger divalent cation of Ba2+ holds its hydration relatively less strongly. Mg2+ is much more hydrated than Ca2+ and Ba2+ is less hydrated than Ca2+. So the order of adhesion coincides with the order of hydration. It therefore makes sense that the ion bridging is more or less strong depending on the amount of water surrounding the ions. Our DFT calculations of the chemical potentials shown in Table 2 confirmed this order. To estimate the contribution of ion bridging to the total adhesion, the calculated surface charge of GO in Ca2+ solution was −0.023 C m−2, which was assumed the same for Ba2+ and Mg2+ solutions. If the coverage of the tip with divalent ion is 100% and the estimated contact area between the tip and the sample was 365 nm2,34 we approximated the number of ion bridges to be 52. Taking the benzoate model surface for GO, the ΔΔμ for Mg2+, Ca2+ and Ba2+ is −3.8, −13.6, −22.1 kJ mol−1, respectively. Thus, it would give a total energy −197.6, −707.2, −1149.2 kJ mol−1, which amount to a surface energy of 0.9, 3.2, 5.2 mJ m−2. The Johnson–Kendall–Roberts theory gives the relation between the adhesion and surface energy (W),43

| (10) |
3.3. The –CH3 interaction with GO
To quantitatively compare the results obtained with –CH3 tips, we plotted average adhesion as a function of experiment sets, as shown in Fig. 6. Throughout each set, we kept the tip and substrate the same and varied only the salt solution. For all measurements, the adhesion varies from solution to solution and for most it follows the same order as observed for the –COO(H) terminated tip with: Ba2+ > Ca2+ > Mg2+ ≈ Na+. There is also similar average adhesion in Na+ solution around 130 pN. Mg2+ causes a slight increase in adhesion up to 140–150 pN in one experiment but with a decrease in adhesion in another experiment. Ca2+ causes a bigger increase up to 180 pN, while Ba2+ induces highest adhesion up to 250 pN. The adhesion response for the solution containing Mg2+ shows an interesting behavior: it can be both positive and negative but without much difference to Na+. Compared to –COO(H) set 5–7, the adhesion responses in –CH3 set 1–3 are generally less for each divalent salt solutions, as shown in Table 3. | ||
| Fig. 6 Adhesion of –CH3 functionalized tips in different salt solutions on GO surface with pH 5.5. (a) –CH3 set 1 in the order of MgCl2, NaCl, MgCl2, CaCl2 and NaCl. The reoccurrence of the NaCl test showed that the adhesion was reversible. (b) –CH3 set 2 and 3 in the order of NaCl, MgCl2 CaCl2 and BaCl2. | ||
We again use DLVO theory as starting point and the van der Waals force FvdW is calculated with eqn (1). R is given by the manufacturer to be 30 nm. The Hamaker constant was 6.26 × 10−21 J for GO–GO nanomaterials30 and for the tip we assume the Hamaker constant can substituted for hydrophobic dodecane surface to 5 × 10−21 J.42 The average Hamaker constant in the system is therefore 5.6 × 10−21 J. D, the distance between the tip and surface, is assumed to be around ∼1 nm.33 The attractive van der Waals FvdW was therefore ∼23 pN, which could explain small contribution to the adhesion in –CH3 set 1–3. The –CH3 terminated tip is often assumed to be neutral and carries no surface charge. As we used the same setup of solution as in –COO(H) case, the repulsive EDL force FEDL should still be negligible compared to the measured adhesions. Therefore, the forces predicted by the traditional DLVO model do not fit our observations. The –CH3 tip is hydrophobic and ions should not specifically bind to the tip. Because GO contains abundant oxidized groups as shown by our XPS analysis, the surface should be highly hydrophilic. Consequently, the hydrophilic interactions could play the primary role for the interaction between –CH3 tip and hydrophilic GO surface, which cannot be determined easily theoretically.54 However, we could not rule out the possible contributions from the hydrophobic interactions between aromatic regions of GO and –CH3 for the adhesion. The hydrophilic forces are influenced by the adsorbed cations as well as the intrinsic surface properties of GO. The cations most likely form a thin hydrated layer on the surface of GO, with the thickness depending on hydrated radius of the ions: Mg2+ > Ca2+ > Ba2+ in Table 1. On the surface the part of the hydration layer that is towards the GO surface is most likely stripped off. Thus, Ba2+ has the shortest interaction distance with –CH3 tip, leading to highest adhesion.
Moreover, Schwierz et al. calculated a negative surface charge of 0.0035 C m−2 for the –CH3 terminated SAM similar to our –CH3 tip,55 which was much smaller than the surface charge of –COO(H) tip. As a result, the –CH3 tip can be considered as poorly deprotonated –COO(H) tip carrying low negative charge that is important for the attractive adhesion for GO. To use the same model that we explained ion bridging for –COO(H) tip, we approximated the number of ion bridges to be 8 with the –CH3 tips. The contributions from this type of ion bridge can therefore be calculated to be 20, 70, 113 pN for Mg2+, Ca2+ and Ba2+ using the benzoate model surface. The values and their differences from this model fall well within the same order of magnitude as the measured differences in adhesion. This serves to show that the crude model with ion bridges to –CH3 terminated tip is consistent with observed ion specific differences.
3.4. Adhesions in high pH
Fig. 7 shows the adhesions with –COO(H) and –CH3 tips at pH 8.8. As expected for –COO(H) tip, there are significant differences of –COO(H) set 8 and 9 compared to –COO(H) set 5–7 in Table 1. Each divalent cation almost doubles their adhesion responses compared to Na+ and the adhesion still follows the pattern Ba2+ > Ca2+ > Mg2+ > Na+. The situation for –CH3 tip does not change much compared to –CH3 set 1–3. We can also find negative adhesion response for Mg2+, a low positive response of Ca2+ and a little higher response for Ba2+. The pKa for a surface-bound carboxyl44 has been estimated to be in the range 4.7–5.5 that is close to the previous low pH solutions. We expect the –COO(H) functionalized tip to be more deprotonated at high pH. The surface charges of GO could be affected by pH through the ionization of the oxygen functional groups on GO of eqn (4) and (5), which is also favored at high pH. It is further supported by the fact of larger zeta potential of GO at high pH.30,56 In comparison with –COOH groups, the deprotonation of –OH is much weaker and may not contribute much to the surface charge development on GO when pH increases.57,58 Combined above-mentioned discussions with –COO(H) tips and ion bridging model, it could have stronger ion bridge effects in high pH, leading to a higher adhesion response. However, there should be negligible influence on adhesion with –CH3 tip that is consistent with the observed low adhesion response. | ||
| Fig. 7 Ion effects on average adhesion of –COO(H) tips (a) and –CH3 tips (b) on GO surface at pH 8.8. | ||
4. Conclusions
The natural aquatic environments contain abundant electrolytes, various cations and many different organic contaminants. The findings of this study demonstrate that these ions, which vary in charge, size and complexing capability, can greatly affect the adhesion of organic materials on GO surface, especially for organics terminated with polar groups of –COO(H). For experiments with –COO(H) tips, adhesion decreased in the order: Ba2+ > Ca2+ > Mg2+ > Na+, whereas for –CH3 tips, ion dependent adhesion was relatively low but followed the same: Ba2+ > Ca2+ > Mg2+ ≈ Na+. An important observation of the study was that classic DLVO theory and Schulze–Hardy rule could not account for cation effect on adhesion. We propose that ion bridging plays a definitive role in adhesion between –COO(H) tips and the GO surface. This is consistent with density functional theory (DFT) calculations. Adhesion of –CH3 tips is a response to the hydrophilic interactions and the ion dependent part is suggested to arise from ion bridging between slightly negative charged –CH3 tips and the negative surface. It is also found that larger adhesion response can be observed for the –COOH tip at high pH, while response is relatively low but significant for the –CH3 tip. These results provide important insight into interaction processes between solutions and mineral surfaces with adsorbed organic molecules and offer clues for improving remediation strategies, such as environmental remediation and water treatment with GO, which can be used as adsorbents, membranes, catalysts and coating materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank AJ and XF for valuable discussions. The project was supported by the running budget of the NanoGeoScience Research Group through a bonus grant to SLSS from the University of Copenhagen. ZL acknowledges the PhD support from China Scholarship Council.Notes and references
- A. Lerf, H. He, M. Forster and J. Klinowski, Structure of graphite oxide revisited, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, New insights into the structure and reduction of graphite oxide, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- Z. Liu, K. Nørgaard, M. H. Overgaard, M. Ceccato, D. M. Mackenzie, N. Stenger, S. L. Stipp and T. Hassenkam, Direct observation of oxygen configuration on individual graphene oxide sheets, Carbon, 2018, 127, 141–148 CrossRef CAS.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Harnessing the chemistry of graphene oxide, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- K. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Determination of the local chemical structure of graphene oxide and reduced graphene oxide, Adv. Mater., 2010, 22, 4467–4472 CrossRef CAS PubMed.
- L. J. Cote, F. Kim and J. Huang, Langmuir− Blodgett assembly of graphite oxide single layers, J. Am. Chem. Soc., 2008, 131, 1043–1049 CrossRef PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Precise and ultrafast molecular sieving through graphene oxide membranes, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- S. Wang, H. Sun, H.-M. Ang and M. O. Tadé, Adsorptive remediation of environmental pollutants using novel graphene-based nanomaterials, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Adsorption of divalent metal ions from aqueous solutions using graphene oxide, Dalton Trans., 2013, 42, 5682–5689 RSC.
- R. K. Upadhyay, N. Soin and S. S. Roy, Role of graphene/metal oxide composites as photocatalysts, adsorbents and disinfectants in water treatment: a review, RSC Adv., 2014, 4, 3823–3851 RSC.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano-graphene oxide for cellular imaging and drug delivery, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- D. R. Dreyer, H.-P. Jia and C. W. Bielawski, Graphene oxide: a convenient carbocatalyst for facilitating oxidation and hydration reactions, Angew. Chem., 2010, 122, 6965–6968 CrossRef.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, The role of graphene for electrochemical energy storage, Nat. Mater., 2015, 14, 271 CrossRef CAS PubMed.
- Z. L. Liu, F. J. Xia, Q. Z. Xue, Y. G. Du and Q. K. Guo, in Applied Mechanics and Materials, Trans Tech Publ, 2015, vol. 719, pp. 119–122 Search PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: synthesis, properties, and applications, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- S. M. Maliyekkal, T. S. Sreeprasad, D. Krishnan, S. Kouser, A. K. Mishra, U. V. Waghmare and T. Pradeep, Graphene: a reusable substrate for unprecedented adsorption of pesticides, Small, 2013, 9, 273–283 CrossRef CAS PubMed.
- L. Ji, W. Chen, Z. Xu, S. Zheng and D. Zhu, Graphene nanosheets and graphite oxide as promising adsorbents for removal of organic contaminants from aqueous solution, J. Environ. Qual., 2013, 42, 191–198 CrossRef CAS PubMed.
- Y. Gao, Y. Li, L. Zhang, H. Huang, J. Hu, S. M. Shah and X. Su, Adsorption and removal of tetracycline antibiotics from aqueous solution by graphene oxide, J. Colloid Interface Sci., 2012, 368, 540–546 CrossRef CAS PubMed.
- N. Cai, D. Peak and P. Larese-Casanova, Factors influencing natural organic matter sorption onto commercial graphene oxides, Chem. Eng. J., 2015, 273, 568–579 CrossRef CAS.
- J. Wang, Z. Chen and B. Chen, Adsorption of polycyclic aromatic hydrocarbons by graphene and graphene oxide nanosheets, Environ. Sci. Technol., 2014, 48, 4817–4825 CrossRef CAS PubMed.
- T. Xia, Y. Qi, J. Liu, Z. Qi, W. Chen and M. R. Wiesner, Cation-inhibited transport of graphene oxide nanomaterials in saturated porous media: the Hofmeister effects, Environ. Sci. Technol., 2017, 51, 828–837 CrossRef CAS PubMed.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biomedical applications, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- X. Yang, J. Li, T. Wen, X. Ren, Y. Huang and X. Wang, Adsorption of naphthalene and its derivatives on magnetic graphene composites and the mechanism investigation, Colloids Surf., A, 2013, 422, 118–125 CrossRef CAS.
- G. K. Ramesha, A. V. Kumara, H. B. Muralidhara and S. Sampath, Graphene and graphene oxide as effective adsorbents toward anionic and cationic dyes, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS PubMed.
- J. Ge, L.-A. Shi, Y.-C. Wang, H.-Y. Zhao, H.-B. Yao, Y.-B. Zhu, Y. Zhang, H.-W. Zhu, H.-A. Wu and S.-H. Yu, Joule-heated graphene-wrapped sponge enables fast clean-up of viscous crude-oil spill, Nat. Nanotechnol., 2017, 12, 434 CrossRef CAS PubMed.
- J. Chen, D. Zhu and C. Sun, Effect of heavy metals on the sorption of hydrophobic organic compounds to wood charcoal, Environ. Sci. Technol., 2007, 41, 2536–2541 CrossRef CAS PubMed.
- J. Wang and B. Chen, Adsorption and coadsorption of organic pollutants and a heavy metal by graphene oxide and reduced graphene materials, Chem. Eng. J., 2015, 281, 379–388 CrossRef CAS.
- K. Yang, B. Chen, X. Zhu and B. Xing, Aggregation, adsorption, and morphological transformation of graphene oxide in aqueous solutions containing different metal cations, Environ. Sci. Technol., 2016, 50, 11066–11075 CrossRef CAS PubMed.
- F. Bei, X. Hou, S. L. Chang, G. P. Simon and D. Li, Interfacing colloidal graphene oxide sheets with gold nanoparticles, Chem. – Eur. J., 2011, 17, 5958–5964 CrossRef CAS PubMed.
- I. Chowdhury, M. C. Duch, N. D. Mansukhani, M. C. Hersam and D. Bouchard, Colloidal properties and stability of graphene oxide nanomaterials in the aquatic environment, Environ. Sci. Technol., 2013, 47, 6288–6296 CrossRef CAS PubMed.
- Y. Gao, X. Ren, X. Tan, T. Hayat, A. Alsaedi and C. Chen, Insights into key factors controlling GO stability in natural surface waters, J. Hazard. Mater., 2017, 335, 56–65 CrossRef CAS PubMed.
- T. Hassenkam, C. S. Pedersen, K. Dalby, T. Austad and S. L. S. Stipp, Pore scale observation of low salinity effects on outcrop and oil reservoir sandstone, Colloids Surf., A, 2011, 390, 179–188 CrossRef CAS.
- E. Hilner, M. P. Andersson, T. Hassenkam, J. Matthiesen, P. A. Salino and S. L. S. Stipp, The effect of ionic strength on oil adhesion in sandstone–the search for the low salinity mechanism, Sci. Rep., 2015, 5, 9933 CrossRef CAS PubMed.
- T. Hassenkam, L. L. Skovbjerg and S. L. S. Stipp, Probing the intrinsically oil-wet surfaces of pores in North Sea chalk at subpore resolution, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6071–6076 CrossRef CAS PubMed.
- K. M. S. Juhl, N. Bovet, T. Hassenkam, K. Dideriksen, C. S. Pedersen, C. M. Jensen, D. V. Okhrimenko and S. L. S. Stipp, Change in organic molecule adhesion on α-alumina (sapphire) with change in NaCl and CaCl2 solution salinity, Langmuir, 2014, 30, 8741–8750 CrossRef CAS PubMed.
- E. R. Nightingale Jr, Phenomenological theory of ion solvation. Effective radii of hydrated ions, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, itd: CRC Press, Boca Raton, FL, DR 2003–2004 Search PubMed.
- I. de O. da Mota, J. A. de Castro, R. de Góes Casqueira and A. G. de Oliveira Junior, Study of electroflotation method for treatment of wastewater from washing soil contaminated by heavy metals, J. Mater. Res. Technol., 2015, 4, 109–113 CrossRef.
- A. Zhang, V. Cortes, B. Phelps, H. van Ryswyk and T. Srebotnjak, Experimental analysis of soil and mandarin orange plants treated with heavy metals found in Oilfield-Produced wastewater, Sustainability, 2018, 10, 1493 CrossRef.
- W. S. Hummers Jr and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- Z. L. Liu, T. Rios-Carvajal, M. P. Andersson, M. Ceccato, S. L. S. Stipp and T. Hassenkam, Insights into the Pore-Scale Mechanism for the Low-Salinity Effect: Implications for Enhanced Oil Recovery, Energy Fuels, 2018, 32, 12081–12090 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Academic press, 2011 Search PubMed.
- H.-J. Butt, B. Cappella and M. Kappl, Force measurements with the atomic force microscope: Technique, interpretation and applications, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- M. P. Andersson, M. H. M. Olsson and S. L. S. Stipp, Predicting the p K a and Stability of Organic Acids and Bases at an Oil–Water Interface, Langmuir, 2014, 30, 6437–6445 CrossRef CAS PubMed.
- T. Rios-Carvajal, N. R. Pedersen, N. Bovet, S. L. S. Stipp and T. Hassenkam, Specific ion effects on the interaction of hydrophobic and hydrophilic self-assembled monolayers, Langmuir, 2018, 34, 10254–10261 CrossRef CAS PubMed.
- T. C. Preston, M. Nuruzzaman, N. D. Jones and S. Mittler, Role of hydrogen bonding in the pH-dependent aggregation of colloidal gold particles bearing solution-facing carboxylic acid groups, J. Phys. Chem. C, 2009, 113, 14236–14244 CrossRef CAS.
- A. Y. Mehandzhiyski, E. Riccardi, T. S. van Erp, T. T. Trinh and B. A. Grimes, Ab initio molecular dynamics study on the interactions between carboxylate ions and metal ions in water, J. Phys. Chem. B, 2015, 119, 10710–10719 CrossRef CAS PubMed.
- M. Sano, J. Okamura and S. Shinkai, Colloidal nature of single-walled carbon nanotubes in electrolyte solution: the Schulze− Hardy rule, Langmuir, 2001, 17, 7172–7173 CrossRef CAS.
- B. Tansel, J. Sager, T. Rector, J. Garland, R. F. Strayer, L. Levine, M. Roberts, M. Hummerick and J. Bauer, Significance of hydrated radius and hydration shells on ionic permeability during nanofiltration in dead end and cross flow modes, Sep. Purif. Technol., 2006, 51, 40–47 CrossRef CAS.
- M. Pham, E. A. Mintz and T. H. Nguyen, Deposition kinetics of bacteriophage MS2 to natural organic matter: Role of divalent cations, J. Colloid Interface Sci., 2009, 338, 1–9 CrossRef CAS PubMed.
- M. M. Emara, N. A. Farid, A. M. Wasfi, M. M. Bahr and H. M. Abd-Elbary, Thermodynamics of ion association in aqueous solutions of calcium-and magnesium-substituted hydroxybenzoates using an ion-selective electrode technique, J. Phys. Chem., 1984, 88, 3345–3348 CrossRef CAS.
- A. Ouatmane, M. Hafidi, M. E. Gharous and J. C. Revel, Complexation of calcium ions by humic and fulvic acids, Analusis, 1999, 27, 428–431 CrossRef CAS.
- H. Ohtaki and T. Radnai, Structure and dynamics of hydrated ions, Chem. Rev., 1993, 93, 1157–1204 CrossRef CAS.
- S. H. Donaldson Jr, A. Røyne, K. Kristiansen, M. V. Rapp, S. Das, M. A. Gebbie, D. W. Lee, P. Stock, M. Valtiner and J. Israelachvili, Developing a general interaction potential for hydrophobic and hydrophilic interactions, Langmuir, 2014, 31, 2051–2064 CrossRef PubMed.
- N. Schwierz, D. Horinek and R. R. Netz, Anionic and cationic Hofmeister effects on hydrophobic and hydrophilic surfaces, Langmuir, 2013, 29, 2602–2614 CrossRef CAS PubMed.
- A. Terracciano, J. Zhang, C. Christodoulatos, F. Wu and X. Meng, Adsorption of Ca2+ on single layer graphene oxide, J. Environ. Sci., 2017, 57, 8–14 CrossRef PubMed.
- R. L. Whitby, A. Korobeinyk, V. M. Gun'Ko, R. Busquets, A. B. Cundy, K. Laszlo, J. Skubiszewska-Zięba, R. Leboda, E. Tombacz and I. Y. Toth, pH-driven physicochemical conformational changes of single-layer graphene oxide, Chem. Commun., 2011, 47, 9645–9647 RSC.
- T. Szabó, E. Tombácz, E. Illés and I. Dékány, Enhanced acidity and pH-dependent surface charge characterization of successively oxidized graphite oxides, Carbon, 2006, 44, 537–545 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |