
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe atmospheric chemistry of indoor environments
Jonathan P. D.
Abbatt
 * and
Chen
Wang
* and
Chen
Wang
Department of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: jonathan.abbatt@utoronto.ca; irenechen.wang@mail.utoronto.ca
First published on 31st October 2019
Abstract
Through air inhalation, dust ingestion and dermal exposure, the indoor environment plays an important role in controlling human chemical exposure. Indoor emissions and chemistry can also have direct impacts on the quality of outdoor air. And so, it is important to have a strong fundamental knowledge of the chemical processes that occur in indoor environments. This review article summarizes our understanding of the indoor chemistry field. Using a molecular perspective, it addresses primarily the new advances that have occurred in the past decade or so and upon developments in our understanding of multiphase partitioning and reactions. A primary goal of the article is to contrast indoor chemistry to that which occurs outdoors, which we know to be a strongly gas-phase, oxidant-driven system in which substantial oxidative aging of gases and aerosol particles occurs. By contrast, indoor environments are dark, gas-phase oxidant concentrations are relatively low, and due to air exchange, only short times are available for reactive processing of gaseous and particle constituents. However, important gas–surface partitioning and reactive multiphase chemistry occur in the large surface reservoirs that prevail in all indoor environments. These interactions not only play a crucial role in controlling the composition of indoor surfaces but also the surrounding gases and aerosol particles, thus affecting human chemical exposure. There are rich research opportunities available if the advanced measurement and modeling tools of the outdoor atmospheric chemistry community continue to be brought indoors.
 Jonathan P. D. Abbatt | Jon Abbatt is a Professor in the Department of Chemistry at the University of Toronto. He obtained his BSc and PhD from University of Toronto and Harvard University, respectively, and taught at the University of Chicago for 8 years before returning to Toronto. His research interests are atmospheric chemistry, indoor chemistry, aerosol and cloud processes in the atmosphere, and the chemistry of remote regions such as the Arctic. |
 Chen Wang | Chen Wang is a postdoctoral fellow in the Department of Chemistry at the University of Toronto. She obtained a B.Sc and M.Sc in Environmental Science from Nankai University and Peking University, respectively, and a PhD in Environmental Chemistry from University of Toronto. Chen's research interests include indoor chemistry, atmospheric chemistry, and phase partitioning of chemicals. |
Environmental significanceHumans increasingly spend much of their time indoors and so it is important to understand at a fundamental level the factors that control our chemical exposure in indoor environments. This exposure may arise by breathing airborne particles or by direct uptake of chemicals through the skin. This review article takes an atmospheric chemistry perspective to summarize recent advances in our understanding of the different partitioning and reactive processes that occur indoors, contrasting the behavior with that which occurs outdoors. Particularly important in controlling the chemical state of the indoor environment, and our pollutant exposure, is the chemistry involved in the interplay of molecules moving between surfaces, the gas phase, and particles. |
1. Introduction
Humans spend most of their time indoors. This is increasingly true as the global population becomes more urban. The built environment is also changing, with rising wealth leading to a higher prevalence of air conditioning and heating. As well, as climate change progresses there is the need to minimize air exchange with the outdoors to make air-handling processes more energy efficient. This may lead to increases in human exposure to the indoor chemical environment.1 In particular, little is known about how the dynamic multiphase chemistry that occurs indoors affects exposure via inhalation of contaminated aerosol particles, or by sorption through the skin or contact with contaminated surfaces.2Needing to understand the factors that determine exposure, the field of indoor chemistry addresses the chemical processes that occur in the air, aerosol particles, and surface reservoirs of the indoor environments. General research questions include: do gas-phase chemicals infiltrating from outdoors or emitted indoors partition to surfaces or particles, or are they flushed outdoors instead? When transient events occur, what are the timescales for air–particle–surface exchange? And, in which phase do chemical contaminants predominantly reside? Are molecules chemically transformed indoors, via oxidative or other mechanisms? What are the reaction products, and to what phase do they partition? What are the roles of humans and building practices in indoor chemistry?
Indoor chemistry can be viewed as a subset of the larger atmospheric chemistry discipline, which has traditionally explored the chemical diversity of different atmospheric environments, starting with the stratosphere and urban regions but now extending to rural, polar, forested and marine settings. However, the mainstream atmospheric chemistry community has traditionally paid much less attention to the chemistry of indoor environments than to outdoor settings, largely leaving this domain to the building science and persistent organic pollutant communities.
There is much to be gained if the modeling and measurement techniques used in outdoor environments are brought indoors.3,4 In particular, although public agencies regulate outdoor air quality, indoor air quality is largely unregulated despite our need to understand all the mechanisms by which we receive our chemical exposure. For example, why do negative health outcomes correlate with the particulate mass loading of outdoor air whereas we do most of our breathing indoors? What is the connection to indoor air exposure?5,6 Do indoor aerosol loadings scale with outdoor values, and to what degree is the chemical composition of indoor aerosol particles modulated when they infiltrate from outdoors? Another issue is that there is clear evidence that emissions of chemical consumer products and indoor cooking have a direct impact on outdoor air.7,8 What chemistry occurs with these compounds before they are transported outdoors? There are many similar issues that the outdoor atmospheric chemistry community with expertise on short-lived and reactive species can help address.
The goal of this article is to assess our understanding of indoor chemistry from an atmospheric chemistry perspective. It is distinguished from past reviews and feature articles on the subject9–16 by focusing largely on new insights and findings from the past decade or so of research and by presenting a molecular-based perspective. The article starts with a brief introduction to indoor environments and chemical sources, followed by summaries of our current understanding of indoor oxidants and reactive processes in the gas phase. The largest sections of the paper address the many recent studies of multiphase chemistry, defined as the collection of non-reactive partitioning and reactive processes that occur between gases, aerosol particles and surfaces.17 The article then concludes by contrasting the chemistry of indoor and outdoor environments, and by presenting directions for future research. It is hoped that this article will be useful to atmospheric chemists who are interested in addressing chemistry that occurs indoors. However, the article is also written for scientists and engineers working on the built environment, environmental chemistry, and human exposure to chemical contaminants.
Finally, much of the work in the field of indoor chemistry has been performed in built environments characteristic of the industrially developed world. While many of the processes described in the article will be fundamental to any indoor environment, it is nevertheless important to emphasize that some indoor chemistry of particular importance to industrially developing countries – for example, the chemistry associated with the use of inefficient cookstoves – is not addressed in this article.
2. The physical nature of the indoor environment
Despite being highly heterogeneous in function and form, indoor spaces have common features of importance to indoor atmospheric chemistry. To start, the building structure acts as a transport barrier that inhibits flow to and from outdoors. Yet, when the doors and windows are closed, and with no mechanical ventilation system operating, there is nevertheless infiltration of air through leaky walls and windows. In a multi-unit dwelling, there is also air exchange between neighbouring units.18 The air exchange is driven by pressure gradients across the building, arising from wind or differential heating. A typical residence time for indoor air is on the order of an hour or two, with a lot of variability. For example, in a wide survey of US residences the median air exchange rate has been reported to be 0.5 h−1, with a standard deviation of 0.9 h−1.19 As well, there can be different mixing zones within indoor spaces. Rapid mixing may occur intrazonally within one indoor compartment, such as the floor of a multi-level house, but interzonal mixing with the rest of the house may be slower.20,21Fig. 1 is an illustration of well-defined mixing rates within three different compartments within a residence, as measured using simultaneous release and fast timescale measurement of multiple tracers (deuterated alkenes) within the house.20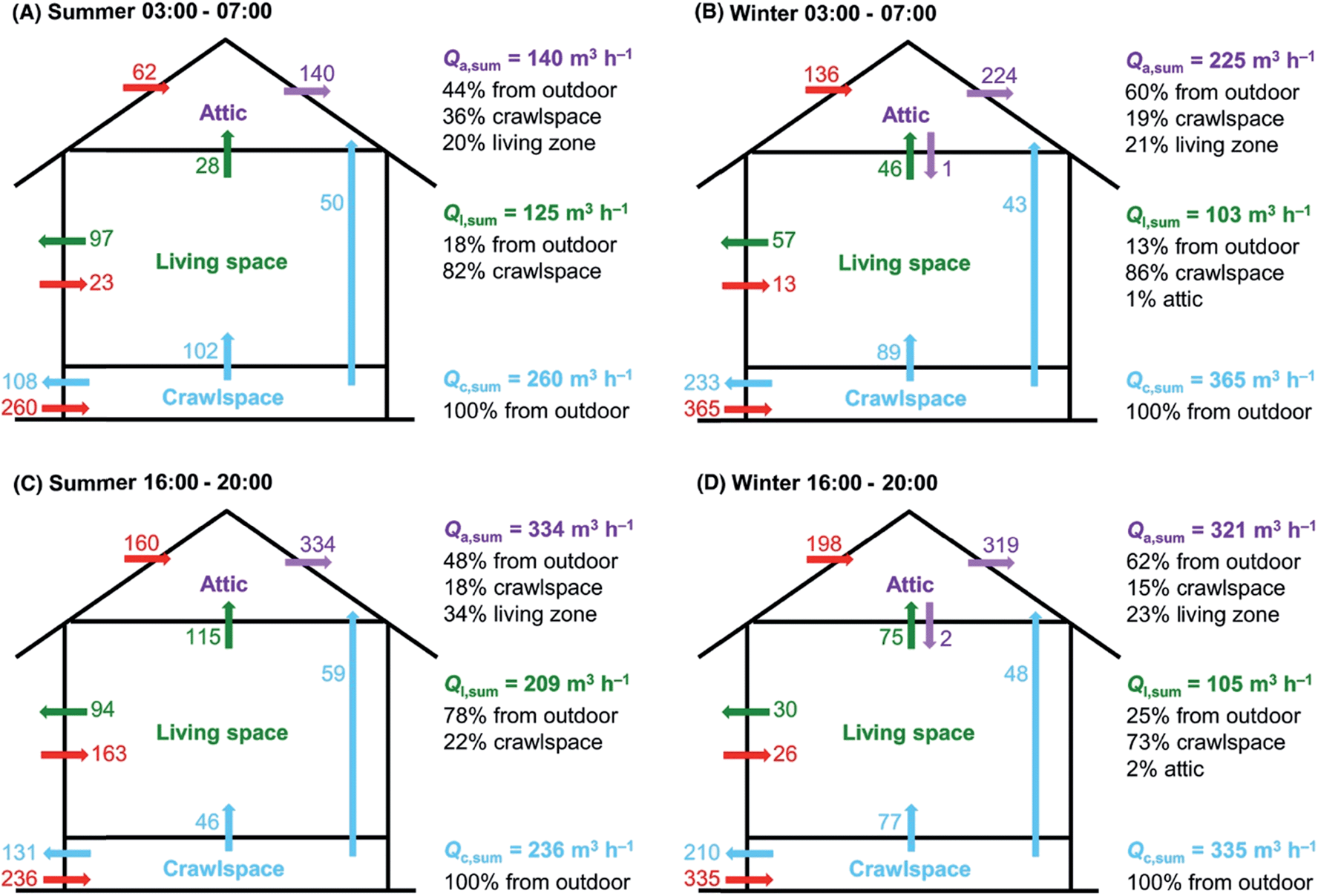 | ||
| Fig. 1 Volumetric flow rates (Q) within different spaces (crawlspace, living space, attic) during different times and seasons in a house as determined using continuous measurements of conservative tracers, i.e. molecules released indoors that were not naturally present and do not react rapidly or partition strongly to surfaces. The long blue arrow indicates a direct transport route from the crawlspace to the attic.20 Reproduced with permission of the publisher from Liu et al., Indoor Air, 2018, 28, 572–584. | ||
A second common feature is the very high surface-area-to-volume ratio (S/V) of indoor spaces, on the order of 3 m−1.22 Calculated by considering only macroscopic surface areas, this is a lower limit to the value at the microscopic scale given that surfaces may be porous or rough. As well, building materials, furnishings and paint may have sufficiently low viscosity or high porosity that molecules can diffuse into them.23,24 As described in Section 6b, these surface reservoirs drive important non-reactive partitioning processes and reactive chemistry. In particular, many volatile species in outdoor environments instead exhibit semivolatile behavior indoors by partitioning to the surfaces.
Third, indoor photon fluxes are significantly lower than outdoors, especially for ultraviolet light.25,26 Indeed, the human eye has a remarkable ability to adapt to low light levels. Fig. 2 presents average spectral irradiances from 290 to 750 nm for a number of settings in Toronto in July at roughly 1.5 m height from the floor: (i) after sunset in the middle of a semi-detached townhouse kitchen illuminated with ceiling halogen lights (‘kitchen’), (ii) 16:00 in the middle (i.e. 3 m away from a window) of a laboratory illuminated with both fluorescent strip lighting and with large (2.9 m2 each) north-facing windows (‘room B’), (iii) 12:00 in a meeting room with large (2.9 m2 each) south-facing windows at 1 m and 3 m distances from a window (‘room A’). As well, the irradiance for outside sunlight is included for comparison. The blackbody spectra from the halogen kitchen lights provide only 0.65 W m−2 illumination and there is no ultraviolet light, whereas the Sun provides 600 W m−2 and significant UV. The mid-day spectra in the laboratory and meeting room are superpositions of the light from the fluorescent bulbs and solar radiation that has passed through the windows. These locations are brighter than the kitchen but their photon fluxes are still much lower than outdoors. In particular, the total irradiance very close to a south-facing window is only 12 W m−2, and that value quickly drops to 2.5 W m−2 two meters further into the room. The light intensity and spectra are strongly dependent on the transmission efficiency of sunlight through the glass, glass cleanliness, time of day, number of windows present, distance from the window, outside cloudiness and types of indoor lights.25–27
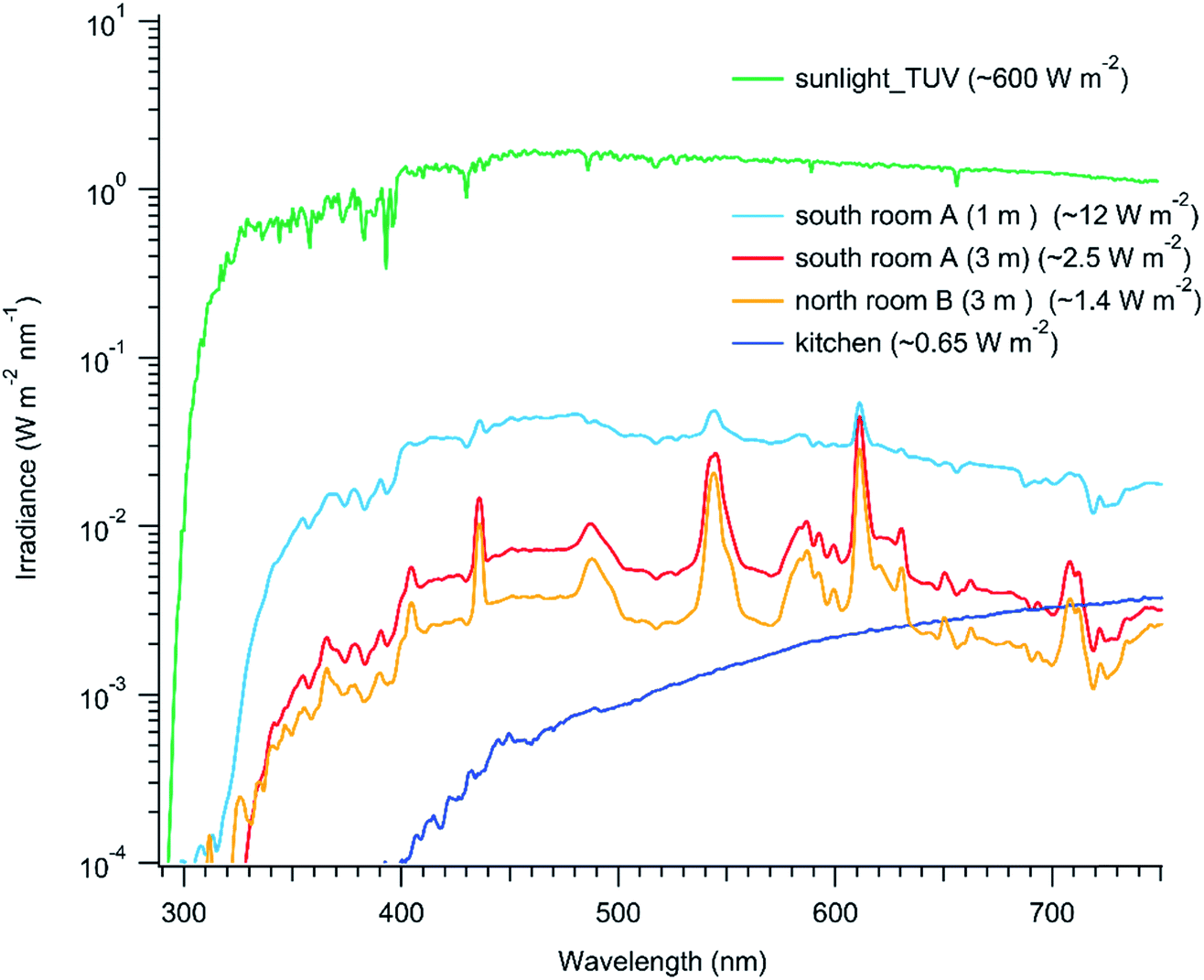 | ||
| Fig. 2 Irradiance measurements conducted in different indoor settings in Toronto in July, using a Black Comet, Stellar Net Inc. spectral radiometer. Measurements were made in room A 1 m or 3 m away from a south-facing window at noon (light blue and red), in room B 3 m away from a north-facing window at 4 pm (orange), and a kitchen with halogen bulbs after sunset. The spectra show the average irradiances measured with the spectral radiometer facing six different directions (up, down, front, back, left and right). The number in the bracket is the total irradiance between 290 and 750 nm. The sunlight values are the clear-sky outdoor solar irradiance for July in Toronto at noon local time, 300 DU ozone, 0.1 surface albedo (NCAR TUV model, http://cprm.acom.ucar.edu/Models/TUV/Interactive_TUV/). Note that average irradiance is plotted, not actinic flux. | ||
Fourth, unlike outdoors, indoor temperature and relative humidity are frequently controlled and wet deposition does not occur. Exceptions are in the kitchen during cooking and in bathrooms during showering. Even without heating or cooling systems, the temperature and relative humidity variations indoors are frequently smaller than those outdoors.
Finally, a common feature in all indoor environments is the presence of humans. Not only are our activities, such as cooking and cleaning, important but humans also have direct effects through their emissions and via multiphase chemistry that occurs on clothing and skin.28 These effects can be pronounced in heavily populated settings.29–32
3. Sources of indoor chemical constituents
Primary chemical sources are those emitted indoors or that infiltrate from outdoors whereas secondary sources arise from reactive chemistry occurring indoors. Secondary sources15 are discussed in Sections 5 and 6c. Examples of some primary sources include emissions from:– Building materials and furnishings: a wide range of volatile organic compounds (VOCs) including formaldehyde, terpenes, small carboxylic acids, and alcohols from insulation, resins, wood, paint, carpet, upholstery, other furnishings.33–38
– Food and cooking: triglycerides, fatty acids, proteinaceous material, terpenes, ethanol.39–41
– Cleaning products: terpenes, chlorinated molecules, acetic acid, ammonia.42–46
– Humans: ammonia, amino acids, small VOCs such as isoprene and lactic acid, unsaturated oils such as squalene, triglycerides, and fatty acids, personal care products such as siloxanes.29–31,47–51
– Microbes: VOCs.52
– Consumer products: phthalates and many others, including volatile chemical products (VCPs).53
– Combustion activities such as cigarette smoking, gas stoves, candle/incense burning: carbonaceous aerosol with black carbon and organic carbon components, VOCs, reactive nitrogen oxides (NOx), nitrous acid (HONO), isocyanic acid (HNCO).54–56
As well, species that infiltrate from outdoors include ozone, NOx, and numerous aerosol components, such as those in photochemical smog and mineral dust.
Many molecules have multiple sources. Reactive nitrogen oxides such as nitric oxide (NO) and nitrogen dioxide (NO2) can have elevated mixing ratios indoors compared to outdoors when a gas stove is in operation.57–59 However, when there are no indoor combustion sources, the net flow of NOx (i.e. NO + NO2) will be from the polluted outdoor environment.60 HONO can also be formed from gas stoves, giving mixing ratios of 10's of ppb in some cases.21,59,61 However, even in the absence of combustion, reactive mechanisms (see Section 6c) can give rise to HONO mixing ratios in residential settings that are typically a few ppb, at least an order of magnitude higher than outside. Similarly, many VOCs have much higher mixing ratios indoors than outdoors, such as the monoterpenes which have multiple sources such as plants, specific foods and fragrances, organic cleaning fluids, tobacco, and cannabis.62 Indeed, terpene mixing ratios can be very high, with values in the ppb to 10's of ppb range routinely reported.63,64
Sources can be either sustained or episodic, as demonstrated recently in a house where human activities demonstrated highly transient signals whereas release from building materials, such as from decaying wood, was more sustained.38
4. Indoor oxidants
Oxidants react with more chemically reduced molecules in a thermodynamically favorable manner. In the outdoor atmosphere, the major oxidants – O3, OH, NO3 and Cl – are only present because of the input of energy from the Sun; OH, NO3 and Cl are radicals whereas O3 has radical character. In particular, OH and NO3 are formed in the presence of ozone, but ozone is usually not generated in the dark. With low light levels indoors, the rate of oxidant production in indoor environments is generally much smaller than outdoors.65Ozone is viewed as the major gas-phase oxidant in indoor environments, transported inside after being photochemically generated in the outdoor troposphere.65–68 Indoor mixing ratios are typically 0.1 to 0.8 of outdoor mixing ratios, with lower values observed in less well-ventilated spaces.67,69 The fraction of outdoor ambient ozone that makes its way through walls and windows (i.e. the penetration factor) is on the order of 0.8.70 Low indoor mixing ratios (i.e. as low as a few ppb) are indicative of efficient reaction with indoor surfaces, and to a lesser degree via gas-phase reactions. There is little generation of ozone indoors, aside from localized sources near some photocopying machines and air purifiers.71–73 Ozone generators are also sold as air purifiers, whose use should be avoided.
The gas-phase OH radical is too short lived to be transported from outdoors. Its major indoor sources are ozonolysis reactions of alkenes, such as terpenes, which can lead to OH formation in the dark.74–76 OH is generated by the unimolecular decay of excited Criegee intermediates that form from decomposition of primary ozonides.77 As well, the photolysis of HONO can also occur to form OH, particularly when direct sunlight is present.78,79 OH concentrations are on the order of 105 molecules per cm3 in most indoor settings,32,74,80 whereas concentrations of 106–107 molecules per cm3 have been reported via photolysis of HONO in sunlit air or via the use of cleaning agents close to the inlet of the OH detector.78,81
NO3 is formed by the reaction of NO2 with ozone.82 It is an important outdoor oxidant during the night but not during the day because it is readily photolyzed. Indoors, the gas-phase photolytic lifetime of NO3 will be long (e.g. >104 s in an art gallery)32 but low ozone mixing ratios and short residence times can lead to unfavorable formation conditions. Given its high reactivity, its deposition velocity to indoor surfaces is likely very high, at least as large as that of ozone, but has not been measured in genuine indoor settings. The only report of indoor gas-phase NO3 is from a study where ozone was artificially added to a house, to increase the NO3 formation rate.83 As well, there has been a study of the sum of NO3 and N2O5 in an office.84
Hydrogen peroxide (H2O2) and chlorine cleaning agents are used episodically. High mixing ratios of H2O2 and ultraviolet light are employed in some hospitals to remove drug-resistant infectious agents.85 Similarly, chlorine dioxide (OClO) has been used to disinfect mold-ridden environments, for example houses that were flooded after Hurricane Katrina.86 These are specialized situations, hopefully without humans present. Moreover, chlorine bleach is a widely used anti-microbial cleaning agent. When used to wash surfaces, the solution releases a large number of chlorinated species to the air,45,87 including high mixing ratios of HOCl as well as Cl2, ClNO2, NCl3, NH2Cl, NHCl2 and chlorinated organics such as CHCl3. With indoor illumination, HOCl and Cl2 can photolyze into reactive OH and Cl radicals.45 There is also the potential for chlorinated gas release with the use of other chlorinated cleaning materials, as in dishwashers.42,44
5. Gas-phase chemistry
The short air residence time and low oxidant levels limit the degree to which the lifetimes of gas-phase molecules are controlled by gas-phase oxidants. This was recently demonstrated, for example, in an indoor museum setting.32 To illustrate, a molecule such as nicotine, which reacts with the OH radical at close to the gas-kinetic collision rate constant of ≈10−10 cm3 per molecule per s,88 has a lifetime of over a day if OH is at 105 molecules per cm3. This is much longer than typical residence times and so only a small fraction will be oxidized before it is mixed outdoors. Similarly, many monoterpenes react with ozone with rate constants of ≈10−16 cm3 per molecule per s.89,90 Indoor ozone mixing ratios can be as low as 5 ppb (1011 molecules per cm3), in which case the lifetimes of these terpenes are also much longer than the air residence time. There are some exceptions to these generalizations. For example, some terpenes react an order of magnitude faster than assumed above.90 Also, with high HONO or HOCl concentrations, the OH concentrations can rise for short periods to 106–107 molecules per cm3 provided there is enough sunlight present.45,78 However, the impact on the overall VOC lifetimes is usually constrained by the small volume of indoor air that is sufficiently bright to drive this level of radical production.Even if gas-phase chemistry does not normally dominate the fate of most VOCs, important gas-phase chemistry nevertheless occurs including substantial radical cycling and organic nitrate formation.74 As well, secondary organic aerosol (SOA) formation can occur by gas-phase oxidation of a variety of precursors, including monoterpenes,91–94 unsaturated compounds arising from skin95,96 and cooking oils,97–99 and cigarette smoke.100 For the most part indoor SOA is not the major component of indoor aerosol but its importance rises in special circumstances when ozone levels are high and the air exchange rate is low.101,102 Episodic events of very high precursor concentrations, such as washing with a terpene-based cleaner or cigarette smoking are also able to promote ultrafine particle formation. In the case of cigarette smoking, new particle formation would occur from the high loading of gas-phase precursors, despite there being a large condensation sink for condensable vapors.100
SOA formation is promoted by gas-phase autoxidation mechanisms.103–105 This mechanism is initiated by organic radical formation, which is then followed by multiple sequential steps of O2 addition and isomerization to form hydroperoxy functional groups. Autoxidation generally requires low radical oxidant concentrations, as present indoors, so that radical–radical reactions do not terminate the intramolecular isomerization reactions that lead to the formation of highly oxygenated products. As well, NOx levels should be low. Highly functionalized products tend to form rapidly, sometimes on the second timescale,104 after oxidation by OH or ozone.
Gas-phase radical chemistry with NO3 radicals may also lead to important products. NO3 addition reactions with terpenes are a major source of SOA in the outdoor environment,106 but similar chemistry has not yet been reported indoors. As well, H-atom-abstraction reactions involving NO3 may be an important indoor source of HNO3 in addition to the multiphase hydrolysis of N2O5.16 In general, the chemistry of NO3 and N2O5 are coupled through an equilibrium with NO2.
Gas-phase photochemistry primarily occurs in directly sunlit volumes, and more slowly with some indoor light sources. Photolysis of O3 to form O(1D) and then OH is unimportant given the lack of ultraviolet light close to 300 nm. Similarly, the low OH concentrations and long photolytic lifetime for NO2 make photochemical production rates of ozone generally negligible. As mentioned above, some oxidant precursors, such as HONO, H2O2 and formaldehyde (HCHO) may photolyze in specific situations, forming radicals78,79 at a rate slower than outside and dependent on the spectrum of the indoor light sources.26,27 The light intensity falls off linearly with distance for strip lighting and as the reciprocal of distance squared for point sources, so the radical production will be highly spatially localized. Finally, Cl2, released from bleach washing, is easily photolyzed with indoor light leading to the production of Cl atoms that will efficiently react with most VOCs.45 Using indoor lights, rapid photolysis of Cl2 (and perhaps HOCl) of the products of the dark reaction between Cl2/HOCl and limonene were shown to produce a high yield of ultrafine particles.107
6. Multiphase processes
(a) The nature of indoor surfaces and aerosol particles
Very high indoor surface area-to-volume ratios (typically 3 m−1)22 differentiate indoor from outdoor environments. By comparison, the outdoor planetary boundary layer has S/V values of 10−3 m−1 to 10−2 m−1 for boundary layer heights of 1000 m and 100 m, when considering the ground geometric surface area. Overall, there is much more condensed-phase area and volume available for multiphase chemistry in indoor surfaces whereas partitioning to aerosol particles is more important in an outdoor environment. Collectively, the accessible volumes of the building materials and furnishings, the gas–surface interface, and the organic- and water-rich surface films described below are referred to as surface reservoirs, or sometimes simply surfaces.The chemical and physical properties of indoor surfaces are highly heterogeneous. Some building materials are porous, such as wallboard or upholstery, and others are impervious, such as glass or stone. Many building materials, such as paint,23,24 have sufficiently low viscosity that molecules can diffuse into and out of them over environmentally relevant timescales. To add additional complexity, all building material and furnishings surfaces are coated with chemical constituents that have accumulated either by gas or particle deposition. The presence of such nm-thick films is well documented.108–110 It is predicted that the growth rate of these organic films is on the order of roughly a few nm per month,111,112 consistent with experiments conducted by exposing clean substrates to genuine indoor air over a period up to a year long.110 Chemical analyses of films developing on windows have identified suites of carboxylic acids, dicarboxylic acids, aromatic acids and alkanes.109 However, there is likely significantly more chemical complexity present given the numerous indoor sources and that multiphase processing occurs. The chemical complexity in the deposited organic film substrates is probably analogous to that of SOA.
Condensed-phase water molecules are present in surface reservoirs in a variety of forms,113 with measurements of roughly 10−6 g cm−2 present on surfaces in a house.114 It is well known that water sorbs to the interfaces of typical indoor materials, such as metal oxides (e.g. silica) with roughly a couple of monolayers of water present at a relative humidity of 50%. The average thickness grows as the relative humidity increases.115–117 Other hygroscopic indoor materials also have adsorbed water, such as gypsum (i.e. interior of wallboard)118 and cellulose (e.g. in cotton fabrics).119 Water may also diffuse into organic matrices, such as paint,120 which will affect their viscosity. The proton activity (i.e. pH) of surface reservoir water, either sorbed into weakly polar or hygroscopic surface reservoirs, is not well known. Given the high concentrations of ammonia in indoor environments (10's of ppb or higher)121 as well as the presence of some alkaline building materials such as concrete, the pH may be considerably higher than outdoor aerosol particles.
Multiphase reactions and partitioning also occur with skin and clothing of humans. Skin oil is reactive, being composed of a wide array of unsaturated compounds, including fatty acids, triglycerides, sterols, and squalene.122,123 Skin flakes and oil are shed sufficiently rapidly, at the rate of 10's to 100's of mg per hour per human,124,125 that squalene and cholesterol can be important components of indoor dust.126 Cholesterol can also arise from meat cooking.126
Indoor aerosol particles have their sources either indoors or outdoors. As described in Section 5, SOA can form indoors, mostly via ozone oxidation of terpenes and cooking/skin oils. Other indoor particle sources arise via combustion processes. A gas stove or burning candle greatly increases the number density of ultrafine particles,127,128 whereas cooking and smoking lead to the aerosol mass loading being substantially higher.129,130 Another common source of indoor particles is via mechanical processes, such as humans walking or air flow, which leads to the generation and/or resuspension of large particles such as dust, carpet fibres, etc.131,132 In the absence of in situ particle production, the major source of indoor particulate matter (PM) is infiltration of outdoor aerosol particles through walls and windows. This leads to PM mass loadings being typically lower than those outdoors.133 The infiltration factors of outside aerosol particles are size dependent, as are their indoor deposition rates.134,135
(b) Multiphase partitioning
Many studies have been conducted in test chambers filled with a wide range of representative indoor materials. Using substances such as carpet, wallboard, acrylic and glass, they have illustrated the propensity for gas-phase molecules to partition to indoor surface reservoirs.136–140 The decay to steady state while sorption occurs is monitored after the gas-phase molecules are injected into a static chamber, or desorption can be studied after exposure ceases. Alternatively, the breakthrough of molecules through a chamber containing indoor surface materials can be monitored. Specific chemical interactions have been highlighted, such as the ability of polar compounds to sorb to wallboard and non-polar compounds to carpet.137 Recent work has focused, for example, on partitioning of contaminants to clothing.141 In all of these cases and in the partitioning discussion below, the processes being described occur in the absence of chemical reactions. Multiphase reactions are discussed in Section 6c.The temporal responses of gas-phase species to enhanced-ventilation (usually by opening doors and windows) within genuine indoor spaces have also been monitored.21,144,145 Gas-phase mixing ratios of most gas phase species (i.e. VOCs, HONO, NH3) drop significantly upon enhanced ventilation because the outdoor air is typically cleaner than that indoors, and the mixing ratios then rebound to their previous steady state values upon closing the doors and windows. The rebound effect is indicative of a source of these molecules from a labile surface reservoir. An example is shown in Fig. 3 for HONO and HCOOH, whose mixing ratios rapidly rebound with the same time constant after a house no longer experiences enhanced-ventilation. In another example, repeated enhanced-ventilation experiments in a house demonstrated this rebound effect on each experiment.145 This indicates that the surface reservoirs are large, with much more material sorbed to these reservoirs than in the gas phase.
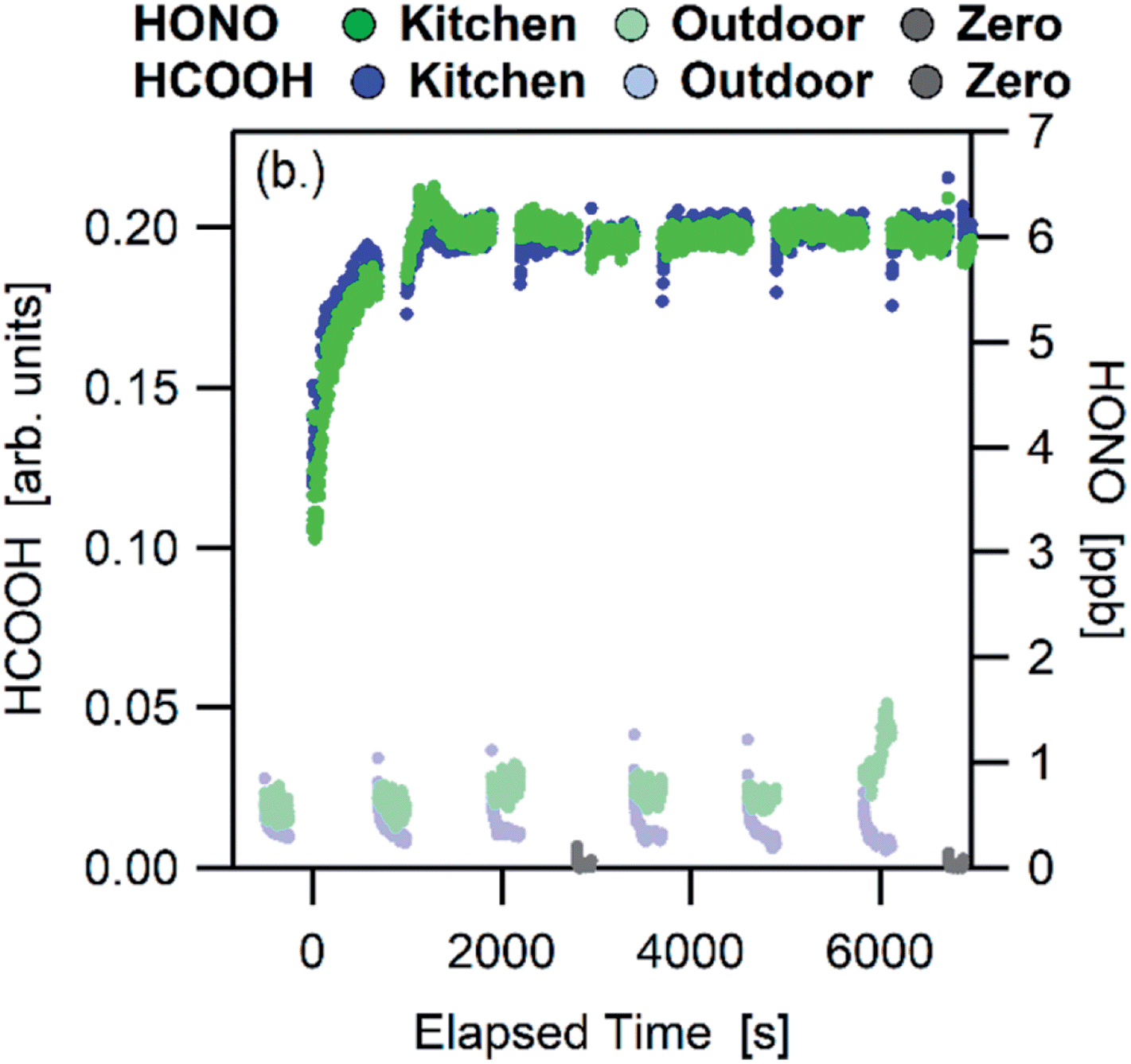 | ||
| Fig. 3 Rapid response of gas-phase HONO (green points) and HCOOH (blue points) in a house kitchen after enhanced-ventilation ceases at 0 seconds.21 Reprinted with permission from D. Collins et al., Environ. Sci. Technol., 2018, 52, 12419–12427. Copyright 2018 American Chemical Society. | ||
The environmental significance of this rebound phenomenon is that the positive effects of short-term ventilation to flush out a house or apartment are likely less than anticipated because many gas-phase species rapidly assume their pre-ventilation mixing ratios after the enhanced ventilation ceases. This is illustrated by the third-hand smoke phenomenon,146–148 where the smell of cigarette smoke lingers long after many full air exchanges. The lingering is due to the large pool of semi-volatile material sorbed to surface reservoirs. Similarly, it is likely that there are residual effects arising from semi-volatile material residing on surfaces after cooking and cleaning.
Other partitioning effects on the indoor environment arise with air conditioning, which typically cycles on and off. For air passing over the cool cooling coils, there can be significant phase change if liquid water is present. This effect has been demonstrated by measurements of the temporal behavior of water-soluble organic gases, such as formic and acetic acid, whose gas-phase mixing ratios decreased when air conditioning cycled on.149
The diffusion timescales of sorbed molecules deep within a building material may be longer. For example, paint can be formulated to be sufficiently porous to permit air exchange with the interior spaces in the walls and ceilings.23 The diffusion coefficients for a range of species with C* saturation concentrations (i.e. vapor pressures) of between 109 and 106 μg m−3 are between 10−7 and 10−8.5 cm2 s−1.24 Dried paint thickness is on the order of 50 μm.153 Thus, the diffusion times through a full paint layer are on the order of an hour. This is slower than for thin surface films but still sufficiently rapid that sorbed molecules may respond to changing ventilation conditions.
Mass transfer timescales may be even longer, as with diffusion out of viscous or porous media such as vinyl flooring or concrete.137,154 Very low volatility compounds, such as the large phthalate plasticizers or PBDE flame retardants,140,155 will also slowly volatilize. As well, there may be semivolatile species in enclosed spaces within a structure, such as associated with insulation in a wall cavity,156 that exchange with the larger volume of indoor air on a slower timescale.
As an example, some third-hand smoke constituents146–148,158 that sorb to indoor surfaces are sufficiently volatile that they affect the mass loading and composition of aerosol particles, via surface-to-gas-to-particle exchange. The dynamics of this process have been demonstrated in a university building impacted by nearby smokers.146 It was observed that the organonitrogen component of aerosol particles was much higher indoors than outdoors in the summer but not the winter (see Fig. 4). It was inferred that acid–base interactions between acidic particles and basic third-hand smoke components occur only in deliquesced aqueous particles which are present in the summer, but not in the winter when particles had effloresced. This partitioning behavior was confirmed in a Teflon chamber whose walls were coated with deposited smoke materials.158 Partitioning from the walls of organonitrogen molecules to seed particles occurred with liquid ammonium sulfate particles but not with solid particles. As well, it was demonstrated that there is a hydrocarbon-like component of third-hand smoke that participates in surface-to-gas-to-particle partitioning processes.
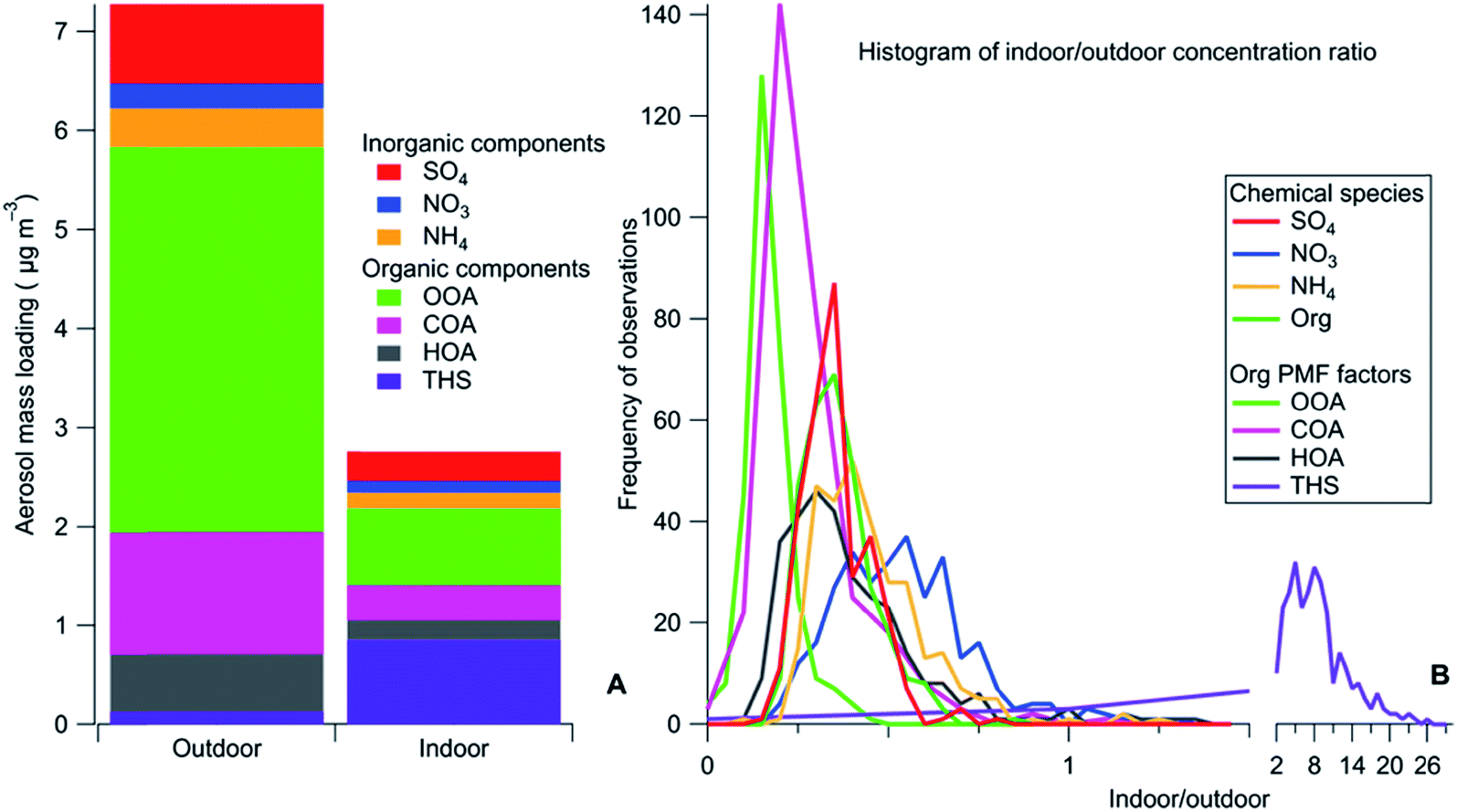 | ||
| Fig. 4 Mass loadings for different aerosol particle components in a university building impacted by third-hand smoke (THS), which is seen to have much a higher indoor/outdoor ratio than all other components.146 Reprinted from Decarlo et al., Sci. Adv., 2018, 4, eaap8368. © The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC) http://creativecommons.org/licenses/by-nc/4.0/. | ||
Aerosol mass spectrometry measurements have also demonstrated the chemical modification of infiltrating aerosol particles, such as the loss of semi-volatile organics and nitrate when aerosol warms upon coming indoors.159 As well, there is potential for high relative humidity in HVAC systems to change the oxidation state of the particles through aqueous phase chemistry.
To expand on this prior work,157,160Fig. 5 presents two dimensional thermodynamic partitioning model predictions. The model assumes simultaneous equilibria between the gas phase and specified volumes of two surface reservoirs, one polar and one weakly polar.145,161 Non-reactive interactions involving significant H-bonding occur in the polar reservoir. Although the model assumes the polar reservoir is liquid water, these interactions more likely occur in thin water-rich films present on building materials or furnishings, and within the building materials themselves, such as with hydrated wallboard. Acid–base effects are treated as they occur in liquid water. Likewise, the weakly polar reservoir is modeled by liquid octanol, to represent the interactions within the less polar, more organic-rich surface reservoir components. Molecules are placed within the 2D-partitioning space plot based on their octanol–air (Koa) and water–air (Kwa) partitioning constants, which indicate whether they are predicted to reside predominantly in the gas phase, or in one of the two surface reservoirs. Despite its simplicity, this model provides a valuable conceptual framework by which to view indoor gas–surface partitioning.
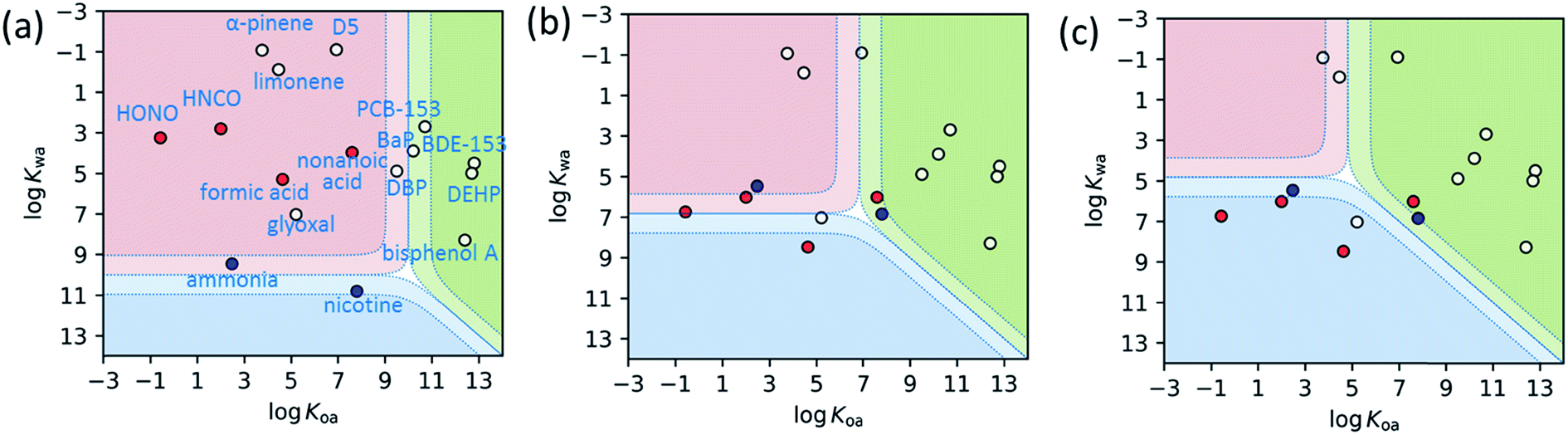 | ||
Fig. 5 Two-dimensional phase partitioning plots for common atmospheric molecules. Species in the red region reside largely in the gas phase, whereas those in the blue and green regions are predicted to reside in the polar and weakly-polar reservoirs, respectively. The solid boundaries and dotted boundaries represent 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 and 90:10 partitioning, respectively. Acid–base effects are considered in the polar reservoir as they would occur in water. Non-dissociating molecules are indicated by white circles, acids by red circles and bases by blue circles. (a) is for outdoor polluted conditions, with the equivalent of 100 μg m−3 of both polar (pH 3) and weakly-polar aerosol mass loading. (b) represents conditions for 50 nm-thick water (i.e. polar, assumed pH 7) and organic (i.e. weakly polar) films in an indoor space of S/V = 3 m−1. (c) represents conditions for 100× larger surface reservoir volumes than those in (b), to model partitioning into building materials and furnishings. Chemical names are labeled in (a), with HONO, HNCO, D5, BaP, DBP, DEHP representing nitrous acid, isocyanic acid, decamethylcyclopentasiloxane, benzo[a]pyrene, dibutyl phthalate, and bis(2-ethylhexyl)phthalate, respectively. The physical chemical properties are from literature.160,162–166 The locations of the acidic and basic chemicals change in (b) and (c) from (a) due to different assumed polar phase pH values. The same model has been applied to understand observations of gas–surface partitioning in a house.145 :50 and 90:10 partitioning, respectively. Acid–base effects are considered in the polar reservoir as they would occur in water. Non-dissociating molecules are indicated by white circles, acids by red circles and bases by blue circles. (a) is for outdoor polluted conditions, with the equivalent of 100 μg m−3 of both polar (pH 3) and weakly-polar aerosol mass loading. (b) represents conditions for 50 nm-thick water (i.e. polar, assumed pH 7) and organic (i.e. weakly polar) films in an indoor space of S/V = 3 m−1. (c) represents conditions for 100× larger surface reservoir volumes than those in (b), to model partitioning into building materials and furnishings. Chemical names are labeled in (a), with HONO, HNCO, D5, BaP, DBP, DEHP representing nitrous acid, isocyanic acid, decamethylcyclopentasiloxane, benzo[a]pyrene, dibutyl phthalate, and bis(2-ethylhexyl)phthalate, respectively. The physical chemical properties are from literature.160,162–166 The locations of the acidic and basic chemicals change in (b) and (c) from (a) due to different assumed polar phase pH values. The same model has been applied to understand observations of gas–surface partitioning in a house.145 | ||
Fig. 5a presents predictions for a wide range of compounds with different physical properties under polluted outdoor conditions with high aerosol mass loading of both polar (pH 3) and weakly-polar aerosol components. Except for the least volatile or most basic species, the compounds reside in the gas phase. By contrast, Fig. 5b assumes partitioning to surface films (50 nm-depth), one organic-rich (i.e. weakly polar) and one water-rich (i.e. polar, pH 7). Many species that were in the gas phase under outdoor conditions now exhibit semi-volatile behavior close to the boundaries between the gas phase and the surface film reservoirs. Fig. 5c presents results for polar and weakly-polar reservoir volumes 100 times larger, to conceptually illustrate the potential to partition to large effective volumes within the building materials and furnishings. For example, paint layers are about 1000 times thicker than the films assumed in Fig. 5b.153 This plot illustrates that most chemical constituents in an indoor environment largely partition to the condensed-phase surface reservoirs. Even the monoterpenes lie close to the boundary between the gas-phase and the weakly polar, organic reservoir. A more accurate model that better characterizes the chemical properties and sizes of the surface reservoirs is needed.
(c) Multiphase reactions
In addition to thermodynamic partitioning, chemical reactions also occur with constituents of surface reservoirs. These transformations may proceed via heterogeneous chemistry with gas-phase oxidants or photochemistry, or involve only condensed-phase species.For heterogeneous uptake of species X from the gas phase, the uptake is expressed in terms of a deposition velocity (vD) which is the proportionality constant between flux to a surface (FX) and the gas phase concentration of X ([X]):167
| FX = vD × [X] | (1) |
The magnitude of the deposition velocity is determined by the efficiency with which molecules can undergo gas-phase mass transfer from the room (i.e. beyond the fluid dynamical boundary layer alongside a surface) and by the efficiency of uptake to the surface.167 The surface uptake efficiency (or uptake coefficient, γ) is the fraction of collisions with the surface of a gas-phase molecule that leads to loss from the gas phase. In the sections below, the uptake is driven by reactive processes on the surface. However, uptake can also occur via non-reactive partitioning, as described above in Section 6b.
When the surface is reactive (γ roughly 10−4 or larger), the deposition velocity is independent of the uptake coefficient because mass transfer through the laminar boundary layer adjacent to the surface is rate limiting. This is illustrated in Fig. 6 which expresses calculated values for the ozone deposition velocity to carpet as a function of both the uptake coefficient and the velocity of air at the surface (u*).167 Conversely, when the uptake coefficient is low, the surface chemistry is rate-limiting and vD scales linearly with γ.
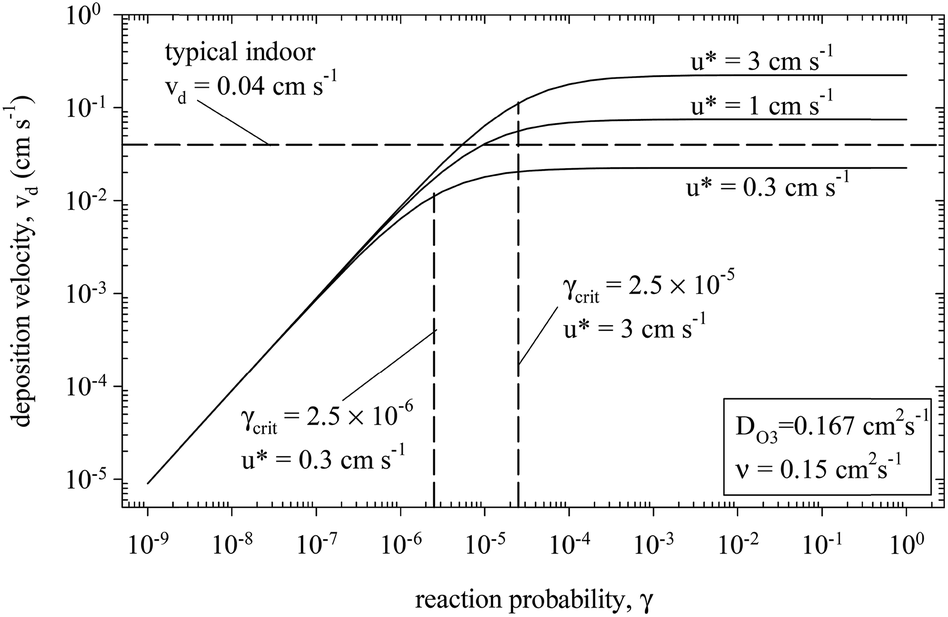 | ||
| Fig. 6 Modelled dependence of the deposition velocity on the reaction probability (or reactive uptake coefficient, γ). The calculations are performed for the uptake of ozone to carpet.167 Republished with permission of Elsevier Science and Technology Journals, from G. C. Morrison and W. W. Nazaroff, Atmos. Environ., 2002, 36, 1749–1756; permission conveyed through Copyright Clearance Center, Inc. | ||
Studies of ozone uptake have focussed on a wide range of indoor materials.168 Known to be reactive are carpet,169,170 clothing and fabric,170–172 some green building materials,173 human hair, cooking oil and skin oil substituents,174–177 insulation materials,156 and ventilation filters and ducts.178–180 A general observation is that carbonyls, especially aldehydes, are formed in high yield as a result of the reactive uptake. This effect is shown in Fig. 7 where it is seen that oxygenated VOCs rise and the ozone concentrations drop by a factor of two when two people enter a test chamber.175 The emissions of carbonyls occur not only under ambient ozone conditions but also when very high mixing ratios of ozone are used for indoor disinfection,181 with emissions occurring from some materials (e.g. fibreboard) long after the disinfection period is over. Studies of the time-dependent reactivity of select materials yield variable results, with some surfaces becoming gradually less reactive to ozone. For example, duct systems exposed to ambient ozone levels became less reactive over a 10 day exposure period,178 new carpets are known to be more reactive than old carpets,182 and some building materials such as ceiling tile and painted drywall also lose reactivity with time.183 By contrast, kitchen countertops were shown in a multiple home study extending over two years to remain consistently reactive.182 This is probably because of the constant addition of cooking oils to such surfaces. As well, addition of other reactive materials such as skin oils and flakes, essential oils, and cleaning agents may sustain the reactivity of some surfaces. Lastly, work has been done to assess the utility of different materials for engineered, passive removal of ozone.184
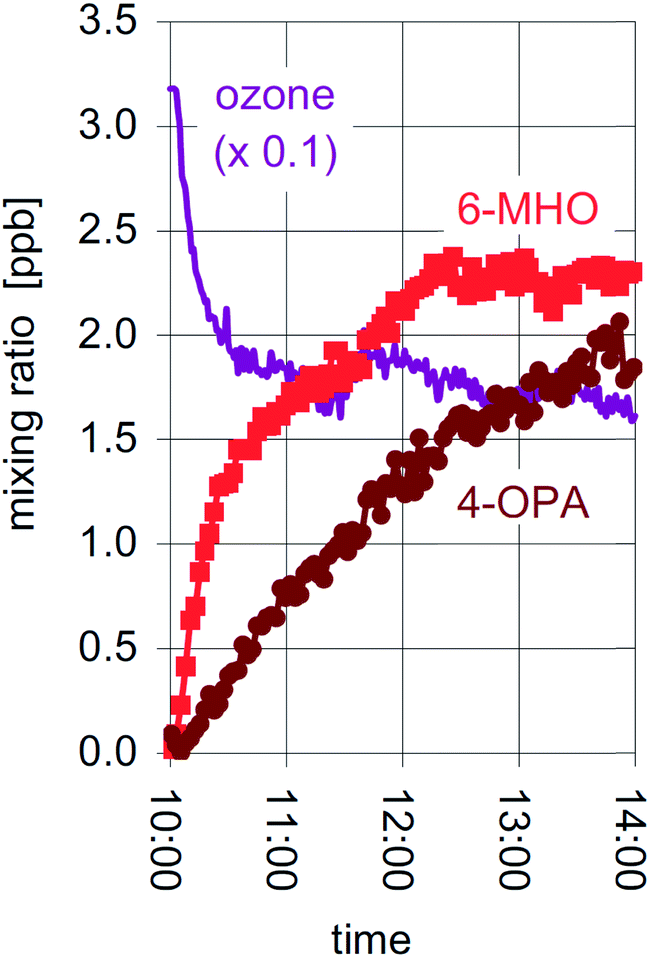 | ||
| Fig. 7 The mixing ratios of ozone and two carbonyls (6-MHO (6-methyl-5-hepten-2-one) and 4-OPA (4-oxopentanal)) when two individuals enter a test chamber at 10:00.175 Reproduced with permission of the publisher from Wisthaler and Weschler, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 6568–6575. | ||
The ozone deposition velocity is large for two reasons. First, on solid inorganic surfaces such as mineral dust, ozone decomposes to form molecular oxygen.185 The surfaces deactivate with high ozone exposure, but some degree of the reactivity regenerates with time. This mechanism is the chemical rationale behind commercial products that catalytically remove ozone from indoor air, using ceramic and metal oxide surfaces. More importantly, the electrophilic nature of ozone also makes it reactive with unsaturated carbon–carbon bonds that are present in a wide range of molecules that partition to surface reservoirs, such as the components of skin oil and cooking oils (squalene, unsaturated triglycerides, cholesterol),175,176,186–190 and terpenoid compounds, especially polar compounds such as terpineol.191–195
Olefinic ozonolysis proceeds via formation of a primary ozonide (see Fig. 8), formed by the π-electrons in a carbon–carbon double bond covalently bonding with unpaired electron density at the terminal oxygen atoms of an ozone molecule.196 The primary ozonide then decomposes to form a Criegee bi-radical intermediate and a carbonyl,197 which frequently is volatile. Gas-phase Criegee intermediates are formed with considerable internal energy which can lead to OH radical formation,77 but it is possible that OH does not form in the condensed phase because the Criegee intermediate internal energy will be rapidly dissipated to the surrounding medium. The stabilized Criegee intermediates isomerize to carboxylic acids, react with carbonyls to form secondary ozonides,177,198 and combine with carboxylic acids to form hydroperoxide esters.199,200 The water content of the surface affects the product distribution because Criegee intermediates can also react with water to form α-hydroxyhydroperoxides.200 This will lower the product yields at high relative humidity of secondary ozonides and hydroperoxide esters and increase the formation of volatile carbonyls relative to their yields under dry conditions.177,201 In the aqueous phase, α-hydroxyhydroperoxides decompose to carbonyls and hydrogen peroxide,202 but whether this occurs on surfaces under sub-saturated conditions is not known. Despite affecting the product distributions, enhanced relative humidity has no effect on the loss rate of ozone with unsaturated oils, such as oleic acid, triolein and squalene.177,187,203 Recently, aspects of this complexity have been added into models of multiphase oxidation processes occurring in the indoor environment, such as the oxidation of unsaturated lipids in skin oil and the formation of associated carbonyls204 and in soiled clothing.205
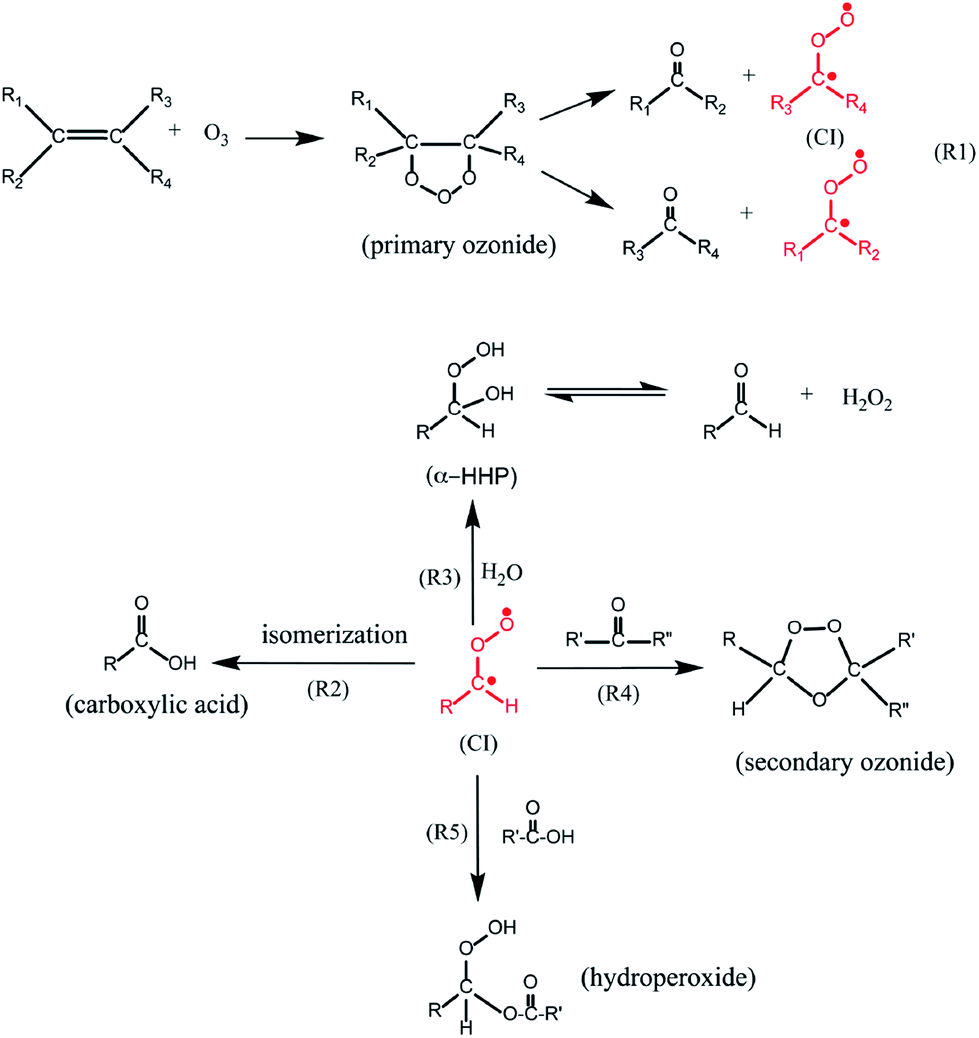 | ||
| Fig. 8 Reaction mechanism for ozonolysis reactions of olefins.187 Reprinted with permission from S. Zhou et al., Environ. Sci. Technol., 2016, 50, 11688–11697. Copyright 2016 American Chemical Society. | ||
Ozone also reacts heterogeneously with many other electron-rich substrates, such as condensed-phase polycyclic aromatic hydrocarbons (PAHs) and nicotine.206–210 These pollutants can arise from incomplete combustion processes, such as candle burning, cooking stove operation, and smoking. Both the PAHs and their oxidation products can be mutagenic and carcinogenic,90 and they have sufficiently low volatility that they largely partition to indoor surface reservoirs and particles. An important environmental fate in outdoor environments is via heterogeneous oxidation with ozone, which proceeds faster for PAHs sorbed to aerosol particles than when those molecules are in the gas phase.209 Few studies have targeted PAH multiphase reactivity under indoor conditions, however one recent indoor study demonstrated that ozone heterogeneously functionalizes benzo[a]pyrene (BaP) into epoxides and epoxy diols.211 The epoxydiol BaP product is highly carcinogenic and known to be formed biochemically in the body's cytochrome P450 enzyme complex. Its environmental consequences when generated by multiphase ozonolysis chemistry have not been evaluated.
The multiphase ozonolysis kinetics of PAHs in organic matrices can only be quantitatively modeled by taking into consideration the effects of phase separation and organic viscosity. For example, the reactivity of BaP in secondary organic aerosol material is enhanced at high relative humidity because the viscosity of the organic matrix is reduced.210 Also, although BaP is fully soluble in cooking oil, its oxygenated ozonolysis reaction products phase separate from the reactants and cooking oil, forming a more viscous reaction medium.212 By preventing the BaP from reacting with incoming ozone molecules, residual BaP is left on the surface even though all the BaP would have reacted away had the solution remained well mixed. Such complex interactions need to be fully understood to arrive at a quantitative description of multiphase reactivity on indoor surfaces and aerosol particles.
It is also known through water treatment studies that ozone is reactive with many other classes of compounds, including proteins and amino acids.213 However, there have not been studies of their heterogeneous reactivity under indoor conditions.
There have been no studies of the heterogeneous reactivity of NO3 radicals which target indoor conditions, although a number of fundamental studies have demonstrated that NO3 is a more selective surface oxidant than OH.220–222 For example, it adds efficiently to carbon–carbon double bonds and it abstracts the hydrogen atom from aldehydic functional groups. Unlike OH, it reacts slowly with saturated compounds, such as alkanes.
There has only been one uptake experiment of genuine indoor films that demonstrated oxidative loss of phthalates and carboxylic acids, with OH concentrations two-to-three orders of magnitude larger than those in the indoor environment.226 Extrapolation to indoor conditions indicated that this process may be an important fate pathway for these condensed-phase molecules on the week-to-month timescale. The heterogeneous uptake of OH was observed to be faster than predicted using standard dry deposition models.226 A recent modeling study of indoor boundary layer chemistry demonstrated that the flux of short-lived species to a wall surface will indeed be enhanced by chemical generation of those species within the fluid dynamical boundary layer adjacent to the surface.227
Although used at very high mixing ratios in hospitals as a disinfecting agent, there are no studies of H2O2 heterogeneous reactions motivated by potential indoor chemistry. However, H2O2 is a well-known oxidant in cloud droplets, reacting with SO2 to form sulfate and with aldehydes to form α-hydroxyhydroperoxides.231 It is unknown whether such reactions occur with the very high H2O2 mixing ratios used in the hospital disinfection scenarios. Lastly, the detailed multiphase chemistry of OClO has not been extensively studied.86
Photochemical degradation can occur via many direct mechanisms. One especially active chromophore is the carbonyl functional group. For example, there was observed to be light-enhanced release of benzaldehyde and other VOCs from a lacquer-coated particle board, probably via Norrish type I reactions that involve carbon–carbon bond breakage from photoinitiators such as 1-phenyl-2-hydroxy-2-methyl-propane-1-one.234 As well, functionalized aromatic compounds absorb in the near UV and visible parts of the spectrum, potentially promoting photochemistry.
There has been a lot of recent research into indirect condensed-phase photochemistry. For example, nitrate photolysis can lead to the formation of condensed-phase OH radicals, as well as the formation of NOx and HONO.232 As well, reactions can proceed via photosensitized processes, such as the photoconversion of a ground singlet state to an excited triplet state of a photosensitizer, such as benzophenone.235 The triplet state can then undergo a suite of reactions, such as by the oxidation of electron-rich species and by electron transfer. For example, NO2− (and HONO) forms on surfaces in the presence of good photosensitizers and reducing agents, as are present in many complex organic substrates.215
One particularly good photooxidation agent widely present indoors is rutile (TiO2), a common whitening agent added to paint.236–240 Under realistic indoor conditions with substrates of white paint on glass in a chamber, there was significant loss of gas-phase NO2 under illumination, but only moderate to no effects were observed for the degradation of a variety of VOCs.239 In another experiment on painted glass surfaces where nitrate had been deposited, common indoor lights sources were shown to photodecompose nitrate into gaseous NOx molecules.238 As well, H2O2 and O3 experience greater loss with light and higher contents of TiO2 in the reaction substrates.241,242 Finally, there is very complex photochemistry that occurs when oxygenated organics, such as gallic acid or oxalic acid, are mixed with iron-containing substrates.243,244 Overall, the degree to which such condensed-phase photosensitized reactions proceed indoors is not known, nor which molecules are the most important photosensitizing agents.
7. Summary
(a) Contrasting the indoor and outdoor environments
This article has demonstrated that there are significant differences in the chemistry that occurs in indoor and outdoor environments, as outlined below. | ||
| Fig. 9 Humans and indoor activities. | ||
Reactive multiphase chemistry occurs in both indoor and outdoor environments. One crucial difference is that this chemistry occurs largely on macroscopic surfaces indoors, such as the many reactions that lead to high deposition velocities for ozone. In the outdoor atmosphere, dry deposition of species like ozone is certainly important but so too is aerosol multiphase chemistry such as N2O5 hydrolysis, halogen recycling, and organic aerosol heterogeneous oxidation. Given the short time for aerosol particle–gas interactions, these reactive aerosol processes are of less importance indoors. Conversely, there is a lot of time – up to months or years – for multiphase chemistry to occur in indoor surface reservoirs.
(b) Future directions
Recent advances have arisen from the application of fast-time-response instrumentation with low detection limits, such as on-line mass spectrometry instruments that sample gases and aerosol particles, and spectroscopic instruments that can monitor reactive intermediates.3,21,29,32,38,56,105,121,252 It is now possible to study how the indoor environment dynamically responds to transient behavior indoors, as during window or door opening, cleaning, cooking, or changing human occupancy. Research questions include: what is the timescale for the lingering chemical effects of humans after they leave an indoor space, or after cooking stops? How quickly does gas–surface partitioning revert to steady state after a short period of enhanced ventilation? What are the time scales over which third-hand smoke impacts the composition of gases and particles in the air? Can a room be flushed clean of contaminants in a reasonable timescale? Can the formation of highly oxygenated molecules that arise through auto-oxidation be observed in real-time, for example after the lights are turned on indoors?105 Overall, we need to better understand how human behavior couples to the indoor environment.253The characterization of the different surface reservoirs and aerosol particles that are present indoors is an exciting challenge. Whereas particle size distributions have been widely reported and analyzed,254 real-time measurements of indoor particle composition have only recently started. However, online aerosol mass spectrometers are limited in their ability to measure the composition of particles below roughly 100 nm diameter, nor do they have much molecular specificity.255 New online analytical methods, such as the extractive electrospray source for aerosol mass spectrometry,256 promise to have major impacts on our understanding of indoor aerosol composition.
Surface reservoirs both in and on building materials and furnishings need to be better characterized. The surface science community has extensively studied the chemical properties of the interfaces of many of these materials (e.g. silica, gypsum, stainless steel, granite) but much less is known about less refractory substances such as wood, upholstery components, and insulation materials. To what degree do these materials outgas and sorb gas-phase molecules, and on what timescales? Moreover, it is likely that the interfaces of all these surface materials are not pristine having been chemically aged by gas and particle deposition soon after construction or placement in an indoor environment. The physical and chemical characteristics of the layers of sorbed chemicals need to be understood, in addition to the properties of the underlying materials. It is notable that no real-time measurements of indoor surface composition have been reported in genuine indoor species. This is needed to follow the dynamics of surface composition change and to assess the semi-volatile species that will not be present if indoor surface samples are taken back to the lab for analysis.
There are many fundamental questions to explore, such as the degree to which the chemical aging that arises indoors drives all surfaces to have the same physical and chemical characteristics. In particular, do all surfaces contain an organic film that masks the interface of the underlying building material to some degree? Even though not all surfaces will age at the same rate, some homogenization of the upper layers of surfaces will simplify the multiphase processes that need to be modeled. Modern surface science analytical techniques permit detailed measurements of specific molecules, functional groups, hygroscopicity, morphology, and homogeneity. Particularly promising are techniques such as direct analysis in real-time mass spectrometry257,258 and atomic force microscopy photothermal infrared spectroscopic analysis259 that provide detailed characterization of surface composition.
With the enormous heterogeneity of indoor spaces, indoor chemistry models have to be tested against measurements in a wide range of environments, to assess the degree to which their predictions are quantitatively accurate and transferable. A modeling consortium has been formed with a hierarchical approach to the development of indoor modeling,4 an approach that is already providing insights into fundamental interactions of gases with building materials152,260 and the heterogeneous chemistry that occurs when ozone interacts with within skin oils and clothing.204,205 This approach is required because detailed processes at the molecular level can not be directly incorporated into models that capture air motions using computational fluid dynamics. Condensed-phase molecular dynamics models can inform the fundamental chemistry that is incorporated into multilayer numerical models that capture both partitioning and reactive processes. As well, box models with detailed gas-phase oxidation chemistry and photochemistry74 can develop parameterizations of key processes for inclusion in computational fluid dynamics simulations.
The multiphase modeling community is moving to couple gas-phase models to multi-layer condensed-phase models.261 As well, predictions of the physical state of condensed-phase substrates will arise. In particular, semi-empirical models arising from the aerosol chemistry community can now predict organic viscosity and the tendency towards inorganic–organic and liquid–liquid phase separation of different chemical mixtures as a function of temperature and relative humidity.262–265 These modeling methods should be directly transferable to studies of indoor surface films. The information needed to drive these models includes the ratio of condensed-phase inorganic salts to organic materials, and the functional groups present in the organics. Knowledge of the thickness of the organic films is important too, to know whether a bulk description is relevant or whether quantum-level interactions with the interface of the underlying building material need to be described.
Coupled modeling and measurement studies will also be needed to further assess the impacts that indoor environments have on outdoor air quality. A recent study illustrated that volatile consumer products, many of which are used indoors, in Los Angeles now match the ozone-forming potential of traffic-related VOC emissions.7 Similar effects may arise in highly congested cities with little biogenic VOC input. It is important to determine the extent to which semivolatile compounds in indoor environments are removed before being emitted to the outdoor environment.
Motivated by our need to understand the extent and mechanisms of human contaminant exposure, the future is rich for the continued application of modern techniques in atmospheric chemistry to study the indoor chemical environment. This work will build upon the pioneering studies conducted over many decades within the indoor chemistry and building science communities. The intent of this review has been to show the highly dynamic nature of the field, illustrating many new opportunities for future research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JA thanks the journal for the invitation to submit this review. His work in the indoor chemistry field has been largely supported by the Chemistry of the Indoor Environment Program of the Alfred P. Sloan Foundation, led by Dr Paula Olsiewski. He also thanks all current and prior members of his research group who have collaborated on indoor chemistry projects, and many members of the indoor chemistry research community for their support and insights. The authors are grateful to Zilin Zhou for crafting Fig. 9.References
- Institute of Medicine, Climate Change, the Indoor Environment, and Health, The National Academies Press, Washington, DC, 2011 Search PubMed.
- C. J. Weschler and W. W. Nazaroff, SVOC exposure indoors: fresh look at dermal pathways, Indoor Air, 2012, 22, 356–377 CrossRef CAS.
- D. K. Farmer, Analytical Challenges and Opportunities For Indoor Air Chemistry Field Studies, Anal. Chem., 2019, 91, 3761–3767 CrossRef CAS.
- M. Shiraiwa, N. Carslaw, D. J. Tobias, M. S. Waring, D. Rim, G. Morrison, P. S. J. Lakey, M. Kruza, M. von Domaros, B. E. Cummings and Y. Won, Modelling consortium for chemistry of indoor environments (MOCCIE): integrating chemical processes from molecular to room scales, Environ. Sci.: Processes Impacts, 2019, 21, 1240–1254 RSC.
- C. Chen, B. Zhao and C. J. Weschler, Indoor Exposure to “Outdoor PM10” Assessing Its Influence on the Relationship Between PM10 and Short-term Mortality in US Cities, Epidemiology, 2012, 23, 870–878 CrossRef.
- J. Xiang, C. J. Weschler, Q. Wang, L. Zhang, J. Mo, R. Ma, J. Zhang and Y. Zhang, Reducing Indoor Levels of “Outdoor PM2.5” in Urban China: Impact on Mortalities, Environ. Sci. Technol., 2019, 53, 3119–3127 CrossRef CAS PubMed.
- B. C. McDonald, J. A. de Gouw, J. B. Gilman, S. H. Jathar, A. Akherati, C. D. Cappa, J. L. Jimenez, J. Lee-Taylor, P. L. Hayes, S. A. McKeen, Y. Y. Cui, S.-W. Kim, D. R. Gentner, G. Isaacman-VanWertz, A. H. Goldstein, R. A. Harley, G. J. Frost, J. M. Roberts, T. B. Ryerson and M. Trainer, Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 2018, 359, 760–764 CrossRef CAS.
- B. Rooney, R. Zhao, Y. Wang, K. H. Bates, A. Pillarisetti, S. Sharma, S. Kundu, T. C. Bond, N. L. Lam, B. Ozaltun, L. Xu, V. Goel, L. T. Fleming, R. Weltman, S. Meinardi, D. R. Blake, S. A. Nizkorodov, R. D. Edwards, A. Yadav, N. K. Arora, K. R. Smith and J. H. Seinfeld, Impacts of household sources on air pollution at village and regional scales in India, Atmos. Chem. Phys., 2019, 19, 7719–7742 CrossRef CAS.
- C. J. Weschler, Changes in indoor pollutants since the 1950s, Atmos. Environ., 2009, 43, 153–169 CrossRef CAS.
- C. J. Weschler, Chemistry in indoor environments: 20 years of research, Indoor Air, 2011, 21, 205–218 CrossRef CAS.
- C. J. Weschler and N. Carslaw, Indoor Chemistry, Environ. Sci. Technol., 2018, 52, 2419–2428 CrossRef CAS.
- W. W. Nazaroff and A. H. Goldstein, Indoor chemistry: research opportunities and challenges, Indoor Air, 2015, 25, 357–361 CrossRef CAS PubMed.
- G. Morrison, Interfacial chemistry in indoor environments, Environ. Sci. Technol., 2008, 42, 3494–3499 CrossRef CAS PubMed.
- S. Gligorovski and J. P. D. Abbatt, An indoor chemical cocktail, Science, 2018, 359, 631–632 CrossRef PubMed.
- E. Uhde and T. Salthammer, Impact of reaction products from building materials and furnishings on indoor air quality – A review of recent advances in indoor chemistry, Atmos. Environ., 2007, 41, 3111–3128 CrossRef CAS.
- C. Weschler and H. Shields, Potential reactions among indoor pollutants, Atmos. Environ., 1997, 31, 3487–3495 CrossRef.
- A. R. Ravishankara, Heterogeneous and multiphase chemistry in the troposphere, Science, 1997, 276, 1058–1065 CrossRef CAS.
- Z. T. Ai, C. M. Mak and J. L. Niu, Numerical investigation of wind-induced airflow and interunit dispersion characteristics in multistory residential buildings, Indoor Air, 2013, 23, 417–429 CrossRef CAS PubMed.
- D. Murray and D. Burmaster, Residential air exchange rates in the United States – Empirical and estimated parametric distributions by season and climatic region, Risk Anal., 1995, 15, 459–465 CrossRef.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, W. W. Nazaroff and A. H. Goldstein, Detailed investigation of ventilation rates and airflow patterns in a northern California residence, Indoor Air, 2018, 28, 572–584 CrossRef CAS.
- D. B. Collins, R. F. Hems, S. Zhou, M. Alavy, J. A. Siegel and J. P. D. Abbatt, Evidence for Gas–Surface Equilibrium Control of Indoor Nitrous Acid, Environ. Sci. Technol., 2018, 52, 12419–12427 CrossRef CAS.
- A. Manuja, J. Ritchie, K. Buch, Y. Wu, C. M. A. Eichler, J. C. Little and L. C. Marr, Total surface area in indoor environments, Environ. Sci.: Processes Impacts, 2019, 21, 1384–1392 RSC.
- S. Eckhaus, I. Wolock and B. Harris, Porosity of paint films – water vapor adsorption and permeability, Ind. Eng. Chem., 1953, 45, 426–428 CrossRef CAS.
- L. B. Algrim, D. Pagonis, J. A. de Gouw, J. L. Jimenez and P. J. Ziemann, Measurements and Modeling of the Partitioning of Volatile Organic Compounds to Painted Surfaces, Indoor Air, 2019 Search PubMed , submitted.
- A. Gandolfo, V. Gligorovski, V. Bartolomei, S. Tlili, E. Gomez Alvarez, H. Wortham, J. Kleffmann and S. Gligorovski, Spectrally resolved actinic flux and photolysis frequencies of key species within an indoor environment, Build. Environ., 2016, 109, 50–57 CrossRef.
- M. Blocquet, F. Guo, M. Mendez, M. Ward, S. Coudert, S. Batut, C. Hecquet, N. Blond, C. Fittschen and C. Schoemaecker, Impact of the spectral and spatial properties of natural light on indoor gas-phase chemistry: Experimental and modeling study, Indoor Air, 2018, 28, 426–440 CrossRef CAS PubMed.
- S. F. Kowal, S. R. Allen and T. F. Kahan, Wavelength-Resolved Photon Fluxes of Indoor Light Sources: Implications for HOx Production, Environ. Sci. Technol., 2017, 51, 10423–10430 CrossRef CAS PubMed.
- C. J. Weschler, Roles of the human occupant in indoor chemistry, Indoor Air, 2016, 26, 6–24 CrossRef CAS PubMed.
- S. Liu, R. Li, R. J. Wild, C. Warneke, J. A. de Gouw, S. S. Brown, S. L. Miller, J. C. Luongo, J. L. Jimenez and P. J. Ziemann, Contribution of human-related sources to indoor volatile organic compounds in a university classroom, Indoor Air, 2016, 26, 925–938 CrossRef CAS PubMed.
- X. Tang, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Siloxanes Are the Most Abundant Volatile Organic Compound Emitted from Engineering Students in a Classroom, Environ. Sci. Technol. Lett., 2015, 2, 303–307 CrossRef CAS.
- J. Williams, C. Stönner, J. Wicker, N. Krauter, B. Derstroff, E. Bourtsoukidis, T. Klüpfel and S. Kramer, Cinema audiences reproducibly vary the chemical composition of air during films, by broadcasting scene specific emissions on breath, Sci. Rep., 2016, 6, 25464 CrossRef CAS PubMed.
- D. Pagonis, D. J. Price, L. B. Algrim, D. A. Day, A. V. Handschy, H. Stark, S. L. Miller, J. de Gouw, J. L. Jimenez and P. J. Ziemann, Time-Resolved Measurements of Indoor Chemical Emissions, Deposition, and Reactions in a University Art Museum, Environ. Sci. Technol., 2019, 53, 4794–4802 CrossRef CAS.
- C. Yu and D. Crump, A review of the emission of VOCs from polymeric materials used in buildings, Build. Environ., 1998, 33, 357–374 CrossRef.
- H. Destaillats, R. L. Maddalena, B. C. Singer, A. T. Hodgson and T. E. McKone, Indoor pollutants emitted by office equipment: A review of reported data and information needs, Atmos. Environ., 2008, 42, 1371–1388 CrossRef CAS.
- T. Salthammer, S. Mentese and R. Marutzky, Formaldehyde in the Indoor Environment, Chem. Rev., 2010, 110, 2536–2572 CrossRef CAS PubMed.
- F. Haghighat and L. D. Bellis, Material emission rates: Literature review, and the impact of indoor air temperature and relative humidity, Build. Environ., 1998, 33, 261–277 CrossRef.
- C.-C. Lin and R. L. Corsi, Texanol® ester alcohol emissions from latex paints: Temporal variations and multi-component recoveries, Atmos. Environ., 2007, 41, 3225–3234 CrossRef CAS.
- Y. Liu, P. K. Misztal, J. Xiong, Y. Tian, C. Arata, R. J. Weber, W. W. Nazaroff and A. H. Goldstein, Characterizing sources and emissions of volatile organic compounds in a northern California residence using space- and time-resolved measurements, Indoor Air, 2019, 29, 630–644 CAS.
- K. L. Abdullahi, J. M. Delgado-Saborit and R. M. Harrison, Emissions and indoor concentrations of particulate matter and its specific chemical components from cooking: A review, Atmos. Environ., 2013, 71, 260–294 CrossRef CAS.
- F. Klein, N. J. Farren, C. Bozzetti, K. R. Daellenbach, D. Kilic, N. K. Kumar, S. M. Pieber, J. G. Slowik, R. N. Tuthill, J. F. Hamilton, U. Baltensperger, A. S. H. Prevot and I. El Haddad, Indoor terpene emissions from cooking with herbs and pepper and their secondary organic aerosol production potential, Sci. Rep., 2016, 6, 36623 CrossRef CAS.
- Y. Zhao and B. Zhao, Emissions of air pollutants from Chinese cooking: A literature review, Build. Simul., 2018, 11, 977–995 CrossRef.
- K. E. R. Dawe, T. C. Furlani, S. F. Kowal, T. F. Kahan, T. C. VandenBoer and C. J. Young, Formation and emission of hydrogen chloride in indoor air, Indoor Air, 2019, 29, 70–78 CrossRef CAS PubMed.
- W. W. Nazaroff and C. J. Weschler, Cleaning products and air fresheners: exposure to primary and secondary air pollutants, Atmos. Environ., 2004, 38, 2841–2865 CrossRef CAS.
- D. A. Olson and R. L. Corsi, In-home formation and emissions of trihalomethanes: The role of residential dishwashers, J. Exposure Anal. Environ. Epidemiol., 2004, 14, 109–119 CrossRef CAS PubMed.
- J. P. S. Wong, N. Carslaw, R. Zhao, S. Zhou and J. P. D. Abbatt, Observations and impacts of bleach washing on indoor chlorine chemistry, Indoor Air, 2017, 27, 1082–1090 CrossRef CAS PubMed.
- M. Odabasi, T. Elbir, Y. Dumanoglu and S. C. Sofuoglu, Halogenated volatile organic compounds in chlorine-bleach-containing household products and implications for their use, Atmos. Environ., 2014, 92, 376–383 CrossRef CAS.
- R. H. Dunstan, D. L. Sparkes, B. J. Dascombe, M. M. Macdonald, C. A. Evans, C. J. Stevens, M. J. Crompton, J. Gottfries, J. Franks, G. Murphy, R. Wood and T. K. Roberts, Sweat Facilitated Amino Acid Losses in Male Athletes during Exercise at 32–34 degrees C, PLoS One, 2016, 11, e0167844 CrossRef PubMed.
- J. Fenske and S. Paulson, Human breath emissions of VOCs, J. Air Waste Manage. Assoc., 1999, 49, 594–598 CrossRef CAS PubMed.
- F. M. Schmidt, O. Vaittinen, M. Metsala, M. Lehto, C. Forsblom, P.-H. Groop and L. Halonen, Ammonia in breath and emitted from skin, J. Breath Res., 2013, 7, 017019 CrossRef PubMed.
- C. Stoenner, A. Edtbauer and J. Williams, Real-world volatile organic compound emission rates from seated adults and children for use in indoor air studies, Indoor Air, 2018, 28, 164–172 CrossRef CAS PubMed.
- X. Tang, P. K. Misztal, W. W. Nazaroff and A. H. Goldstein, Volatile Organic Compound Emissions from Humans Indoors, Environ. Sci. Technol., 2016, 50, 12686–12694 CrossRef CAS PubMed.
- P. K. Misztal, D. S. Lymperopoulou, R. I. Adams, R. A. Scott, S. E. Lindow, T. Bruns, J. W. Taylor, J. Uehling, G. Bonito, R. Vilgalys and A. H. Goldstein, Emission Factors of Microbial Volatile Organic Compounds from Environmental Bacteria and Fungi, Environ. Sci. Technol., 2018, 52, 8272–8282 CrossRef CAS PubMed.
- T. Schettler, Human exposure to phthalates via consumer products, Int. J. Androl., 2006, 29, 134–139 CrossRef CAS PubMed.
- J. R. Girman, M. G. Apte, G. W. Traynor, J. R. Allen and C. D. Hollowell, Pollutant emission rates from indoor combustion appliances and sidestream cigarette smoke, Environ. Int., 1982, 8, 213–221 CrossRef CAS.
- T. Salthammer, T. Schripp, S. Wientzek and M. Wensing, Impact of operating wood-burning fireplace ovens on indoor air quality, Chemosphere, 2014, 103, 205–211 CrossRef CAS PubMed.
- R. F. Hems, C. Wang, D. B. Collins, S. Zhou, N. Borduas-Dedekind, J. A. Siegel and J. P. D. Abbatt, Sources of isocyanic acid (HNCO) indoors: a focus on cigarette smoke, Environ. Sci.: Processes Impacts, 2019, 21, 1334–1341 RSC.
- C. Spicer, D. Kenny, G. Ward and I. Billick, Transformations, lifetimes and sources of NO2, HONO and HNO3 in indoor environments, J. Air Waste Manage. Assoc., 1993, 43, 1479–1485 CrossRef CAS PubMed.
- Z. Vecera and P. Dasgupta, Indoor nitrous acid levels – production of nitrous acid from open flame sources, Int. J. Environ. Anal. Chem., 1994, 56, 311–316 CrossRef CAS.
- S. Zhou, C. J. Young, T. C. VandenBoer, S. F. Kowal and T. F. Kahan, Time-Resolved Measurements of Nitric Oxide, Nitrogen Dioxide, and Nitrous Acid in an Occupied New York Home, Environ. Sci. Technol., 2018, 52, 8355–8364 CrossRef CAS PubMed.
- H. Zhao, E. T. Gall and B. Stephens, Measuring the Building Envelope Penetration Factor for Ambient Nitrogen Oxides, Environ. Sci. Technol., 2019, 53, 9695–9704 CrossRef CAS PubMed.
- J. Pitts, T. Wallington, H. Biermann and A. Winer, Identification and measurement of nitrous acid in an indoor environment, Atmos. Environ., 1985, 19, 763–767 CrossRef CAS.
- S. Brown, M. Sim, M. Abramson and C. Gray, Concentrations of volatile organic compounds in indoor air – a review, Indoor Air, 1994, 4, 123–134 CrossRef CAS.
- H.-J. Su, C.-J. Chao, H.-Y. Chang and P.-C. Wu, The effects of evaporating essential oils on indoor air quality, Atmos. Environ., 2007, 41, 1230–1236 CrossRef CAS.
- U. Schlink, M. Rehwagen, M. Damm, M. Richter, M. Borte and O. Herbarth, Seasonal cycle of indoor-VOCs: comparison of apartments and cities, Atmos. Environ., 2004, 38, 1181–1190 CrossRef CAS.
- C. J. Young, S. Zhou, J. A. Siegel and T. F. Kahan, Illuminating the dark side of indoor oxidants, Environ. Sci.: Processes Impacts, 2019, 21, 1229–1239 RSC.
- S. Gligorovski and C. J. Weschler, The Oxidative Capacity of Indoor Atmospheres, Environ. Sci. Technol., 2013, 47, 13905–13906 CrossRef CAS PubMed.
- C. Weschler, Ozone in indoor environments: Concentration and chemistry, Indoor Air, 2000, 10, 269–288 CrossRef CAS PubMed.
- M. S. Waring and J. R. Wells, Volatile organic compound conversion by ozone, hydroxyl radicals, and nitrate radicals in residential indoor air: Magnitudes and impacts of oxidant sources, Atmos. Environ., 2015, 106, 382–391 CrossRef CAS PubMed.
- J. Zhang and P. Lioy, Ozone in residential air – concentrations, I/O ratios, indoor chemistry and exposures, Indoor Air, 1994, 4, 95–105 CrossRef CAS.
- B. Stephens, E. T. Gall and J. A. Siegel, Measuring the Penetration of Ambient Ozone into Residential Buildings, Environ. Sci. Technol., 2012, 46, 929–936 CrossRef CAS PubMed.
- N. Britigan, A. Alshawa and S. Nizkorodov, Quantification of ozone levels in indoor environments generated by ionization and ozonolysis air purifiers, J. Air Waste Manage. Assoc., 2006, 56, 601–610 CrossRef CAS PubMed.
- M. S. Waring and J. A. Siegel, The effect of an ion generator on indoor air quality in a residential room, Indoor Air, 2011, 21, 267–276 CrossRef CAS PubMed.
- K. Leovic, L. Sheldon, D. Whitaker, R. Hetes, J. Calcagni and J. Baskir, Measurement of indoor air emissions from dry-process photocopy machines, J. Air Waste Manage. Assoc., 1996, 46, 821–829 CrossRef CAS PubMed.
- N. Carslaw, A new detailed chemical model for indoor air pollution, Atmos. Environ., 2007, 41, 1164–1179 CrossRef CAS.
- G. Sarwar, R. Corsi, Y. Kimura, D. Allen and C. Weschler, Hydroxyl radicals in indoor environments, Atmos. Environ., 2002, 36, 3973–3988 CrossRef CAS.
- C. Weschler and H. Shields, Production of the hydroxyl radical in indoor air, Environ. Sci. Technol., 1996, 30, 3250–3258 CrossRef CAS.
- J. Kroll, N. Donahue, V. Cee, K. Demerjian and J. Anderson, Gas-phase ozonolysis of alkenes: Formation of OH from anti carbonyl oxides, J. Am. Chem. Soc., 2002, 124, 8518–8519 CrossRef CAS PubMed.
- E. Gomez Alvarez, D. Amedro, C. Afif, S. Gligorovski, C. Schoemaecker, C. Fittschen, J.-F. Doussin and H. Wortham, Unexpectedly high indoor hydroxyl radical concentrations associated with nitrous acid, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13294–13299 CrossRef PubMed.
- V. Bartolomei, E. Gomez Alvarez, J. Wittmer, S. Tlili, R. Strekowski, B. Temime-Roussel, E. Quivet, H. Wortham, C. Zetzsch, J. Kleffmann and S. Gligorovski, Combustion Processes as a Source of High Levels of Indoor Hydroxyl Radicals through the Photolysis of Nitrous Acid, Environ. Sci. Technol., 2015, 49, 6599–6607 CrossRef CAS PubMed.
- C. Weschler and H. Shields, Measurements of the hydroxyl radical in a manipulated but realistic indoor environment, Environ. Sci. Technol., 1997, 31, 3719–3722 CrossRef CAS.
- N. Carslaw, L. Fletcher, D. Heard, T. Ingham and H. Walker, Significant OH production under surface cleaning and air cleaning conditions: Impact on indoor air quality, Indoor Air, 2017, 27, 1091–1100 CrossRef CAS PubMed.
- C. Weschler, M. Brauer and P. Koutrakis, Indoor air ad nitrogen dioxide – a potential pathway to the generation of nitrate radicals, dinitrogen pentaoxide and nitric acid indoors, Environ. Sci. Technol., 1992, 26, 179–184 CrossRef CAS.
- C. Arata, K. J. Zarzana, P. K. Misztal, Y. Liu, S. S. Brown, W. W. Nazaroff and A. H. Goldstein, Measurement of NO3 and N2O5 in a Residential Kitchen, Environ. Sci. Technol. Lett., 2018, 5, 595–599 CrossRef CAS.
- J. K. Nojgaard, Indoor measurements of the sum of the nitrate radical, NO3, and nitrogen pentoxide, N2O5 in Denmark, Chemosphere, 2010, 79, 898–904 CrossRef PubMed.
- D. J. Weber, H. Kanamori and W. A. Rutala, ‘No touch’ technologies for environmental decontamination: focus on ultraviolet devices and hydrogen peroxide systems, Curr. Opin. Infect. Dis., 2016, 29, 424–431 CrossRef CAS PubMed.
- H. Hubbard, D. Poppendieck and R. L. Corsi, Chlorine Dioxide Reactions with Indoor Materials during Building Disinfection: Surface Uptake, Environ. Sci. Technol., 2009, 43, 1329–1335 CrossRef CAS PubMed.
- M. Odabasi, Halogenated volatile organic compounds from the use of chlorine-bleach-containing household products, Environ. Sci. Technol., 2008, 42, 1445–1451 CrossRef CAS PubMed.
- N. Borduas, J. G. Murphy, C. Wang, G. da Silva and J. P. D. Abbatt, Gas Phase Oxidation of Nicotine by OH Radicals: Kinetics, Mechanisms, and Formation of HNCO, Environ. Sci. Technol. Lett., 2016, 3, 327–331 CrossRef CAS.
- R. Atkinson, A. Winer and J. Pitts, Rate constants for the gas-phase reactions of O3 with the natural hydrocarbons isoprene and alpha-pinene and beta-pinene, Atmos. Environ., 1982, 16, 1017–1020 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the lower and upper atmosphere, Academic Press, New York, 2000 Search PubMed.
- H.-L. Huang, T.-J. Tsai, N.-Y. Hsu, C.-C. Lee, P.-C. Wu and H.-J. Su, Effects of essential oils on the formation of formaldehyde and secondary organic aerosols in an aromatherapy environment, Build. Environ., 2012, 57, 120–125 CrossRef.
- G. Sarwar, D. Olson, R. Corsi and C. Weschler, Indoor fine particles: The role of terpene emissions from consumer products, J. Air Waste Manage. Assoc., 2004, 54, 367–377 CrossRef CAS PubMed.
- H. Destaillats, M. M. Lunden, B. C. Singer, B. K. Coleman, A. T. Hodgson, C. J. Weschler and W. W. Nazaroff, Indoor secondary pollutants from household product emissions in the presence of ozone: A bench-scale chamber study, Environ. Sci. Technol., 2006, 40, 4421–4428 CrossRef CAS PubMed.
- Z. Fan, C. Weschler, I. Han and J. Zhang, Co-formation of hydroperoxides and ultra-fine particles during the reactions of ozone with a complex VOC mixture under simulated indoor conditions, Atmos. Environ., 2005, 39, 5171–5182 CrossRef CAS.
- C. Wang and M. S. Waring, Secondary organic aerosol formation initiated from reactions between ozone and surface-sorbed squalene, Atmos. Environ., 2014, 84, 222–229 CrossRef CAS.
- A. M. Avery, M. S. Waring and P. F. DeCarlo, Human occupant contribution to secondary aerosol mass in the indoor environment, Environ. Sci.: Processes Impacts, 2019, 21, 1301–1312 RSC.
- T. Liu, Z. Li, M. Chan and C. K. Chan, Formation of secondary organic aerosols from gas-phase emissions of heated cooking oils, Atmos. Chem. Phys., 2017, 17, 7333–7344 CrossRef CAS.
- T. Liu, Q. Liu, Z. Li, L. Huo, M. Chan, X. Li, Z. Zhou and C. K. Chan, Emission of volatile organic compounds and production of secondary organic aerosol from stir-frying spices, Sci. Total Environ., 2017, 599–600, 1614–1621 CrossRef CAS PubMed.
- T. Liu, Z. Wang, D. D. Huang, X. Wang and C. K. Chan, Significant Production of Secondary Organic Aerosol from Emissions of Heated Cooking Oils, Environ. Sci. Technol. Lett., 2018, 5, 32–37 CrossRef CAS.
- C. Wang, D. B. Collins, R. F. Hems, N. Borduas, M. Antinolo and J. P. D. Abbatt, Exploring Conditions for Ultrafine Particle Formation from Oxidation of Cigarette Smoke in Indoor Environments, Environ. Sci. Technol., 2018, 52, 4623–4631 CrossRef CAS PubMed.
- M. S. Waring, Secondary organic aerosol in residences: predicting its fraction of fine particle mass and determinants of formation strength, Indoor Air, 2014, 24, 376–389 CrossRef CAS PubMed.
- G. Sarwar, R. Corsi, D. Allen and C. Weschler, The significance of secondary organic aerosol formation and growth in buildings: experimental and computational evidence, Atmos. Environ., 2003, 37, 1365–1381 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I. H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petaja, A. Wahner, V. M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–480 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- D. Pagonis, L. B. Algrim, D. J. Price, D. A. Day, A. V. Handschy, H. Stark, S. L. Miller, J. A. de Gouw, J. L. Jimenez and P. J. Ziemann, Autoxidation of Limonene Emitted in a University Art Museum, Environ. Sci. Technol. Lett., 2019, 6, 520–524 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- C. Wang, D. B. Collins and J. P. D. Abbatt, Indoor Illumination of Terpenes and Bleach Emissions Leads to Particle Formation and Growth, Environ. Sci. Technol., 2019, 53, 11792–11800 CrossRef CAS PubMed.
- J. Li, T. Lin, S.-H. Pan, Y. Xu, X. Liu, C. Zhang and X.-D. Li, Carbonaceous matter and PBDEs on indoor/outdoor glass window surfaces in Guangzhou and Hong Kong, South China, Atmos. Environ., 2010, 44, 3254–3260 CrossRef CAS.
- Q. T. Liu, R. Chen, B. E. McCarry, M. L. Diamond and B. Bahavar, Characterization of polar organic compounds in the organic film on indoor and outdoor glass windows, Environ. Sci. Technol., 2003, 37, 2340–2349 CrossRef CAS PubMed.
- L. A. Wallace, W. R. Ott, C. J. Weschler and A. C. K. Lai, Desorption of SVOCs from Heated Surfaces in the Form of Ultrafine Particles, Environ. Sci. Technol., 2017, 51, 1140–1146 CrossRef CAS PubMed.
- C. J. Weschler and W. W. Nazaroff, Growth of organic films on indoor surfaces, Indoor Air, 2017, 27, 1101–1112 CrossRef CAS PubMed.
- C. M. A. Eichler, J. Cao, G. Isaacman-VanWertz and J. C. Little, Modeling the formation and growth of organic films on indoor surfaces, Indoor Air, 2019, 29, 17–29 CrossRef CAS PubMed.
- S. M. Duncan, K. G. Sexton and B. J. Turpin, Oxygenated VOCs, aqueous chemistry, and potential impacts on residential indoor air composition, Indoor Air, 2018, 28, 198–212 CrossRef CAS PubMed.
- H. Schwartz-Narbonne and D. J. Donaldson, Water uptake by indoor surface films, Sci. Rep., 2019, 9, 11089 CrossRef PubMed.
- W. Hertl and M. Hair, Adsorption of water on silica, Nature, 1969, 223, 1150–1151 CrossRef CAS.
- R. Razouk and A. Salem, The adsorption of water on glass surfaces, J. Phys. Colloid Chem., 1948, 52, 1208–1227 CrossRef CAS.
- H. A. Al-Abadleh and V. H. Grassian, FT-IR Study of Water Adsorption on Aluminum Oxide Surfaces, Langmuir, 2003, 19, 341–347 CrossRef CAS.
- R. Razouk, A. Salem and R. Mikhail, The adsorption of water vapor on dehydrated gypsum, J. Phys. Chem., 1960, 64, 1350–1355 CrossRef CAS.
- R. Jeffries, The sorption of water by cellulose and eight other textile polymers, J. Text. Inst., Trans., 1960, 51, T339–T340 CrossRef CAS.
- A. van der Zanden and E. Goossens, The measurement of the sorption isotherm of water in paint films, Chem. Eng. Process., 2004, 43, 739–743 CrossRef CAS.
- L. Ampollini, E. F. Katz, S. Bourne, Y. Tian, A. Novoselac, A. H. Goldstein, G. Lucic, M. S. Waring and P. F. DeCarlo, Observations and Contributions of Real-Time Indoor Ammonia Concentrations during HOMEChem, Environ. Sci. Technol., 2019, 53, 8591–8598 CrossRef CAS PubMed.
- N. Nicolaides, Skin lipids – Their biochemical uniqueness, Science, 1974, 186, 19–26 CrossRef CAS PubMed.
- D. T. Downing and J. S. Strauss, Synthesis and composition of surface lipids of human skins, J. Invest. Dermatol., 1974, 62, 228–244 CrossRef CAS.
- D. T. Downing, A. M. Stranieri and J. S. Strauss, The Effect of Accumulated Lipids on Measurements of Sebum Secretion in Human Skin, J. Invest. Dermatol., 1982, 79, 226–228 CrossRef CAS PubMed.
- L. M. Milstone, Epidermal desquamation, J. Dermatol. Sci., 2004, 36, 131–140 CrossRef PubMed.
- C. J. Weschler, S. Langer, A. Fischer, G. Beko, J. Toftum and G. Clausen, Squalene and Cholesterol in Dust from Danish Homes and Daycare Centers, Environ. Sci. Technol., 2011, 45, 3872–3879 CrossRef CAS PubMed.
- E. Géhin, O. Ramalho and S. Kirchner, Size distribution and emission rate measurement of fine and ultrafine particle from indoor human activities, Atmos. Environ., 2008, 42, 8341–8352 CrossRef.
- A. Manoukian, E. Quivet, B. Temime-Roussel, M. Nicolas, F. Maupetit and H. Wortham, Emission characteristics of air pollutants from incense and candle burning in indoor atmospheres, Environ. Sci. Pollut. Res., 2013, 20, 4659–4670 CrossRef CAS PubMed.
- T. Hussein, T. Glytsos, J. Ondráček, P. Dohányosová, V. Ždímal, K. Hämeri, M. Lazaridis, J. Smolík and M. Kulmala, Particle size characterization and emission rates during indoor activities in a house, Atmos. Environ., 2006, 40, 4285–4307 CrossRef CAS.
- D. K. Farmer, M. E. Vance, J. P. D. Abbatt, A. Abeleira, M. R. Alves, C. Arata, E. Boedicker, S. Bourne, F. Cardoso-Saldaña, R. Corsi, P. F. DeCarlo, A. H. Goldstein, V. H. Grassian, L. Hildebrandt Ruiz, J. L. Jimenez, T. F. Kahan, E. F. Katz, J. M. Mattila, W. W. Nazaroff, A. Novoselac, R. E. O'Brien, V. W. Or, S. Patel, S. Sankhyan, P. S. Stevens, Y. Tian, M. Wade, C. Wang, S. Zhou and Y. Zhou, Overview of HOMEChem: House Observations of Microbial and Environmental Chemistry, Environ. Sci.: Processes Impacts, 2019, 21, 1280–1300 RSC.
- J. Qian, J. Peccia and A. R. Ferro, Walking-induced particle resuspension in indoor environments, Atmos. Environ., 2014, 89, 464–481 CrossRef CAS.
- B. E. Boor, J. A. Siegel and A. Novoselac, Wind Tunnel Study on Aerodynamic Particle Resuspension from Monolayer and Multilayer Deposits on Linoleum Flooring and Galvanized Sheet Metal, Aerosol Sci. Technol., 2013, 47, 848–857 CrossRef CAS.
- C. Monn, A. Fuchs, D. Högger, M. Junker, D. Kogelschatz, N. Roth and H.-U. Wanner, Particulate matter less than 10 μm (PM10) and fine particles less than 2.5 μm (PM2.5): relationships between indoor, outdoor and personal concentrations, Sci. Total Environ., 1997, 208, 15–21 CrossRef CAS PubMed.
- C. Long, H. Suh, P. Catalano and P. Koutrakis, Using time- and size-resolved particulate data to quantify indoor penetration and deposition behavior, Environ. Sci. Technol., 2001, 35, 2089–2099 CrossRef CAS PubMed.
- B. Stephens and J. A. Siegel, Penetration of ambient submicron particles into single-family residences and associations with building characteristics, Indoor Air, 2012, 22, 501–513 CrossRef CAS PubMed.
- B. A. Tichenor, Z. Guo, J. E. Dunn, L. E. Sparks and M. A. Mason, The Interaction of Vapour Phase Organic Compounds with Indoor Sinks, Indoor Air, 1991, 1, 23–35 CrossRef.
- D. Won, R. Corsi and M. Rynes, Sorptive interactions between VOCs and indoor materials, Indoor Air, 2001, 11, 246–256 CrossRef CAS PubMed.
- R. Jorgensen and O. Bjorseth, Sorption behaviour of volatile organic compounds on material surfaces – The influence of combinations of compounds and materials compared to sorption of single compounds on single materials, Environ. Int., 1999, 25, 17–27 CrossRef CAS.
- R. Jorgensen, O. Bjorseth and B. Malvik, Chamber testing of adsorption of volatile organic compounds (VOCs) on material surfaces, Indoor Air, 1999, 9, 2–9 CrossRef CAS PubMed.
- Y. Wu, C. M. A. Eichler, W. Leng, S. S. Cox, L. C. Marr and J. C. Little, Adsorption of Phthalates on Impervious Indoor Surfaces, Environ. Sci. Technol., 2017, 51, 2907–2913 CrossRef CAS PubMed.
- G. Morrison, H. Li, S. Mishra and M. Buechlein, Airborne phthalate partitioning to cotton clothing, Atmos. Environ., 2015, 115, 149–152 CrossRef CAS.
- B. Singer, K. Revzan, T. Hotchi, A. Hodgson and N. Brown, Sorption of organic gases in a furnished room, Atmos. Environ., 2004, 38, 2483–2494 CrossRef CAS.
- B. C. Singer, A. T. Hodgson, T. Hotchi, K. Y. Ming, R. G. Sextro, E. E. Wood and N. J. Brown, Sorption of organic gases in residential rooms, Atmos. Environ., 2007, 41, 3251–3265 CrossRef CAS.
- K. Kristensen, D. M. Lunderberg, Y. Liu, P. K. Misztal, Y. Tian, C. Arata, W. W. Nazaroff and A. H. Goldstein, Sources and dynamics of semivolatile organic compounds in a single-family residence in northern California, Indoor Air, 2019, 29, 645–655 CAS.
- C. Wang, D. B. Collins, C. Arata, A. H. Goldstein, J. M. Mattila, D. K. Farmer, L. Ampollini, P. F. DeCarlo, A. Novoselac, M. E. Vance, W. W. Nazaroff and J. P. D. Abbatt, Surface reservoirs dominate dynamic gas–surface partitioning of many indoor air constituents, Sci. Adv., 2019 Search PubMed , accepted - technical hold.
- P. F. DeCarlo, A. M. Avery and M. S. Waring, Thirdhand smoke uptake to aerosol particles in the indoor environment, Sci. Adv., 2018, 4, eaap8368 CrossRef PubMed.
- G. E. Matt, P. J. E. Quintana, H. Destaillats, L. A. Gundel, M. Sleiman, B. C. Singer, P. Jacob III, N. Benowitz, J. P. Winickoff, V. Rehan, P. Talbot, S. Schick, J. Samet, Y. Wang, B. Hang, M. Martins-Green, J. F. Pankow and M. F. Hovell, Thirdhand Tobacco Smoke: Emerging Evidence and Arguments for a Multidisciplinary Research Agenda, Environ. Health Perspect., 2011, 119, 1218–1226 CrossRef CAS PubMed.
- P. Jacob III, N. L. Benowitz, H. Destaillats, L. Gundel, B. Hang, M. Martins-Green, G. E. Matt, P. J. E. Quintana, J. M. Samet, S. F. Schick, P. Talbot, N. J. Aquilina, M. F. Hovell, J.-H. Mao and T. P. Whitehead, Thirdhand Smoke: New Evidence, Challenges, and Future Directions, Chem. Res. Toxicol., 2017, 30, 270–294 Search PubMed.
- S. M. Duncan, S. Tomaz, G. Morrison, M. Webb, J. Atkin, J. D. Surratt and B. J. Turpin, Dynamics of Residential Water-Soluble Organic Gases: Insights into Sources and Sinks, Environ. Sci. Technol., 2019, 53, 1812–1821 CrossRef CAS PubMed.
- D. Valeri and A. J. A. Meirelles, Viscosities of fatty acids, triglycerides, and their binary mixtures, J. Am. Oil Chem. Soc., 1997, 74, 1221–1226 CrossRef CAS.
- T. Koop, J. Bookhold, M. Shiraiwa and U. Poschl, Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC.
- Y. Fang, P. S. J. Lakey, S. Riahi, A. T. McDonald, M. Shrestha, D. J. Tobias, M. Shiraiwa and V. H. Grassian, A molecular picture of surface interactions of organic compounds on prevalent indoor surfaces: limonene adsorption on SiO2, Chem. Sci., 2019, 10, 2906–2914 RSC.
- R. L. Corsi and C.-C. Lin, Emissions of 2,2,4-Trimethyl-1,3-Pentanediol Monoisobutyrate (TMPD-MIB) from Latex Paint: A Critical Review, Crit. Rev. Environ. Sci. Technol., 2009, 39, 1052–1080 CrossRef CAS.
- R. Meininghaus, L. Gunnarsen and H. N. Knudsen, Diffusion and Sorption of Volatile Organic Compounds in Building Materials-Impact on Indoor Air Quality, Environ. Sci. Technol., 2000, 34, 3101–3108 CrossRef CAS.
- M. Wensing, E. Uhde and T. Salthammer, Plastics additives in the indoor environment – flame retardants and plasticizers, Sci. Total Environ., 2005, 339, 19–40 CrossRef CAS PubMed.
- K. Chin, A. Laguerre, P. Ramasubramanian, D. Pleshakov, B. Stephens and E. T. Gall, Emerging investigator series: primary emissions, ozone reactivity, and byproduct emissions from building insulation materials, Environ. Sci.: Processes Impacts, 2019, 21, 1255–1267 RSC.
- C. Weschler, Indoor/outdoor connections exemplified by processes that depend on an organic compound's saturation vapor pressure, Atmos. Environ., 2003, 37, 5455–5465 CrossRef CAS.
- D. B. Collins, C. Wang and J. P. D. Abbatt, Selective Uptake of Third-Hand Tobacco Smoke Components to Inorganic and Organic Aerosol Particles, Environ. Sci. Technol., 2018, 52, 13195–13201 CrossRef CAS PubMed.
- A. M. Avery, M. S. Waring and P. F. DeCarlo, Seasonal variation in aerosol composition and concentration upon transport from the outdoor to indoor environment, Environ. Sci.: Processes Impacts, 2019, 21, 528–547 RSC.
- C. J. Weschler and W. W. Nazaroff, Semivolatile organic compounds in indoor environments, Atmos. Environ., 2008, 42, 9018–9040 CrossRef CAS.
- F. Wania, Y. D. Lei, C. Wang, J. P. D. Abbatt and K.-U. Goss, Using the chemical equilibrium partitioning space to explore factors influencing the phase distribution of compounds involved in secondary organic aerosol formation, Atmos. Chem. Phys., 2015, 15, 3395–3412 CrossRef CAS.
- R. Sander, Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 2019 Search PubMed.
- N. Borduas, B. Place, G. R. Wentworth, J. P. D. Abbatt and J. G. Murphy, Solubility and reactivity of HNCO in water: insights into HNCO's fate in the atmosphere, Atmos. Chem. Phys., 2016, 16, 703–714 CrossRef CAS.
- H. S. S. Ip, X. H. H. Huang and J. Z. Yu, Effective Henry's law constants of glyoxal, glyoxylic acid, and glycolic acid, Geophys. Res. Lett., 2009, 36, L01802 CrossRef.
- US EPA, Estimation Programs Interface Suite, United States Environmental Protection Agency, Washington, DC, 2012, vol. 4.11 Search PubMed.
- G. Morrison and W. Nazaroff, The rate of ozone uptake on carpet: mathematical modeling, Atmos. Environ., 2002, 36, 1749–1756 CrossRef CAS.
- T. Grontoft and M. Raychaudhuri, Compilation of tables of surface deposition velocities for O3, NO2 and SO2 to a range of indoor surfaces, Atmos. Environ., 2004, 38, 533–544 CrossRef CAS.
- G. C. Morrison and W. W. Nazaroff, The rate of ozone uptake on carpets: Experimental studies, Environ. Sci. Technol., 2000, 34, 4963–4968 CrossRef CAS.
- O. A. Abbass, D. J. Sailor and E. T. Gall, Effect of fiber material on ozone removal and carbonyl production from carpets, Atmos. Environ., 2017, 148, 42–48 CrossRef CAS.
- B. K. Coleman, H. Destaillats, A. T. Hodgson and W. W. Nazaroff, Ozone consumption and volatile byproduct formation from surface reactions with aircraft cabin materials and clothing fabrics, Atmos. Environ., 2008, 42, 642–654 CrossRef CAS.
- A. C. Rai, B. Guo, C. H. Lin, J. Zhang, J. Pei and Q. Chen, Ozone reaction with clothing and its initiated VOC emissions in an environmental chamber, Indoor Air, 2014, 24, 49–58 CrossRef CAS.
- E. Gall, E. Darling, J. A. Siegel, G. C. Morrison and R. L. Corsi, Evaluation of three common green building materials for ozone removal, and primary and secondary emissions of aldehydes, Atmos. Environ., 2013, 77, 910–918 CrossRef CAS.
- L. S. Pandrangi and G. C. Morrison, Ozone interactions with human hair: Ozone uptake rates and product formation, Atmos. Environ., 2008, 42, 5079–5089 CrossRef CAS.
- A. Wisthaler and C. J. Weschler, Reactions of ozone with human skin lipids: Sources of carbonyls, dicarbonyls, and hydroxycarbonyls in indoor air, Proc. Natl. Acad. Sci. U. S. A., 2009, 107, 6568–6575 CrossRef PubMed.
- S. Zhou, M. W. Forbes, Y. Katrib and J. P. D. Abbatt, Rapid Oxidation of Skin Oil by Ozone, Environ. Sci. Technol. Lett., 2016, 3, 170–174 CrossRef CAS.
- Z. Zhou, S. Zhou and J. P. D. Abbatt, Kinetics and Condensed-phase Products in Multiphase Ozonolysis of an Unsaturated Triglyceride, Environ. Sci. Technol., 2019, 53, 12467–12475 CrossRef CAS.
- G. Morrison, W. Nazaroff, J. Cano-Ruiz, A. Hodgson and M. Modera, Indoor air quality impacts of ventilation ducts: Ozone removal and emissions of volatile organic compounds, J. Air Waste Manage. Assoc., 1998, 48, 941–952 CrossRef CAS.
- P. Zhao, J. A. Siegel and R. L. Corsi, Ozone removal by HVAC filters, Atmos. Environ., 2007, 41, 3151–3160 CrossRef CAS.
- M. Hyttinen, P. Pasanen, J. Salo, M. Bjorkroth, M. Vartiainen and P. Kalliokoski, Reactions of ozone on ventilation filters, Indoor Built Environ., 2003, 12, 151–158 CrossRef CAS.
- D. G. Poppendieck, H. F. Hubbard, C. J. Weschler and R. L. Corsi, Formation and emissions of carbonyls during and following gas-phase ozonation of indoor materials, Atmos. Environ., 2007, 41, 7614–7626 CrossRef CAS.
- H. Wang and G. Morrison, Ozone-surface reactions in five homes: surface reaction probabilities, aldehyde yields, and trends, Indoor Air, 2010, 20, 224–234 CrossRef CAS.
- D. Rim, E. T. Gall, R. L. Maddalena and W. W. Nazaroff, Ozone reaction with interior building materials: Influence of diurnal ozone variation, temperature and humidity, Atmos. Environ., 2016, 125, 15–23 CrossRef CAS.
- E. T. Gall, R. L. Corsi and J. A. Siegel, Barriers and opportunities for passive removal of indoor ozone, Atmos. Environ., 2011, 45, 3338–3341 CrossRef CAS.
- F. Hanisch and J. N. Crowley, Ozone decomposition on Saharan dust: an experimental investigation, Atmos. Chem. Phys., 2003, 3, 119–130 CrossRef CAS.
- J. R. Wells, G. C. Morrison and B. K. Coleman, Kinetics and reaction products of ozone and surface-bound squalene, J. ASTM Int., 2008, 5, 1–12 CrossRef.
- S. Zhou, M. W. Forbes and J. P. D. Abbatt, Kinetics and Products from Heterogeneous Oxidation of Squalene with Ozone, Environ. Sci. Technol., 2016, 50, 11688–11697 CrossRef CAS PubMed.
- L. Petrick and Y. Dubowski, Heterogeneous oxidation of squalene film by ozone under various indoor conditions, Indoor Air, 2009, 19, 381–391 CrossRef CAS.
- D. Fu, C. B. Leng, J. Kelley, G. Zeng, Y. H. Zhang and Y. Liu, ATR-IR Study of Ozone Initiated Heterogeneous Oxidation of Squalene in an Indoor Environment, Environ. Sci. Technol., 2013, 47, 10611–10618 CrossRef CAS.
- J. Gumulka and L. L. Smith, Ozonation of cholesterol, J. Am. Chem. Soc., 1983, 105, 1972–1979 CrossRef CAS.
- J. Fick, L. Pommer, A. Astrand, R. Ostin, C. Nilsson and B. Andersson, Ozonolysis of monoterpenes in mechanical ventilation systems, Atmos. Environ., 2005, 39, 6315–6325 CrossRef CAS.
- S. Shu and G. C. Morrison, Rate and reaction probability of the surface reaction between ozone and dihydromyrcenol measured in a bench scale reactor and a room-sized chamber, Atmos. Environ., 2012, 47, 421–427 CrossRef CAS.
- S. Shu and G. C. Morrison, Surface Reaction Rate and Probability of Ozone and Alpha-Terpineol on Glass, Polyvinyl Chloride, and Latex Paint Surfaces, Environ. Sci. Technol., 2011, 45, 4285–4292 CrossRef CAS.
- M. Springs, J. R. Wells and G. C. Morrison, Reaction rates of ozone and terpenes adsorbed to model indoor surfaces, Indoor Air, 2011, 21, 319–327 CrossRef CAS.
- M. S. Waring and J. A. Siegel, Indoor Secondary Organic Aerosol Formation Initiated from Reactions between Ozone and Surface-Sorbed D-Limonene, Environ. Sci. Technol., 2013, 47, 6341–6348 CrossRef CAS.
- W.-T. Chan and I. P. Hamilton, Mechanisms for the ozonolysis of ethene and propene: Reliability of quantum chemical predictions, J. Chem. Phys., 2003, 118, 1688–1701 CrossRef CAS.
- R. Criegee, Mechanism of ozonolysis, Angew. Chem., Int. Ed. Engl., 1975, 14, 745–752 CrossRef.
- C. Lai, B. Finlayson-Pitts and W. Willis, Formation of secondary ozonides from the reaction of an unsaturated phosphatidylcholine with ozone, Chem. Res. Toxicol., 1990, 3, 517–523 Search PubMed.
- J. Qiu, S. Ishizuka, K. Tonokura, A. J. Colussi and S. Enami, Reactivity of Monoterpene Criegee Intermediates at Gas-Liquid Interfaces, J. Phys. Chem. A, 2018, 122, 7910–7917 CrossRef CAS PubMed.
- S. Zhou, S. Joudan, M. W. Forbes, Z. Zhou and J. P. D. Abbatt, Reaction of Condensed-Phase Criegee Intermediates with Carboxylic Acids and Perfluoroalkyl Carboxylic Acids, Environ. Sci. Technol. Lett., 2019, 6, 243–250 CrossRef CAS.
- N. Heine, F. A. Houle and K. R. Wilson, Connecting the Elementary Reaction Pathways of Criegee Intermediates to the Chemical Erosion of Squalene Interfaces during Ozonolysis, Environ. Sci. Technol., 2017, 51, 13740–13748 CrossRef CAS PubMed.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska, X. M. Kuang, J. Chen, S. E. Paulson and J. H. Seinfeld, Rapid Aqueous-Phase Hydrolysis of Ester Hydroperoxides Arising from Criegee Intermediates and Organic Acids, J. Phys. Chem. A, 2018, 122, 5190–5201 CrossRef CAS PubMed.
- T. Thornberry and J. P. D. Abbatt, Heterogeneous reaction of ozone with liquid unsaturated fatty acids: detailed kinetics and gas-phase product studies, Phys. Chem. Chem. Phys., 2004, 6, 84–93 RSC.
- P. S. J. Lakey, A. Wisthaler, T. Berkemeier, T. Mikoviny, U. Poeschl and M. Shiraiwa, Chemical kinetics of multiphase reactions between ozone and human skin lipids: Implications for indoor air quality and health effects, Indoor Air, 2017, 27, 816–828 CrossRef CAS.
- P. S. J. Lakey, G. C. Morrison, Y. Won, K. M. Parry, M. von Domaros, D. J. Tobias, D. Rim and M. Shiraiwa, The impact of clothing on ozone and squalene ozonolysis products in indoor environments, Commun. Chem., 2019, 2, 56 CrossRef.
- H. Destaillats, B. C. Singer, S. K. Lee and L. A. Gundel, Effect of ozone on nicotine desorption from model surfaces: Evidence for heterogeneous chemistry, Environ. Sci. Technol., 2006, 40, 1799–1805 CrossRef CAS.
- B. T. Mmereki and D. J. Donaldson, Direct observation of the kinetics of an atmospherically important reaction at the air-aqueous interface, J. Phys. Chem. A, 2003, 107, 11038–11042 CrossRef CAS.
- L. Petrick, H. Destaillats, I. Zouev, S. Sabach and Y. Dubowski, Sorption, desorption, and surface oxidative fate of nicotine, Phys. Chem. Chem. Phys., 2010, 12, 10356–10364 RSC.
- U. Poschl, T. Letzel, C. Schauer and R. Niessner, Interaction of ozone and water vapor with spark discharge soot aerosol particles coated with benzo[a]pyrene: O3 and H2O adsorption, benzo[a]pyrene degradation, and atmospheric implications, J. Phys. Chem. A, 2001, 105, 4029–4041 CrossRef.
- S. M. Zhou, M. Shiraiwa, R. D. McWhinney, U. Poschl and J. P. D. Abbatt, Kinetic limitations in gas-particle reactions arising from slow diffusion in secondary organic aerosol, Faraday Discuss., 2013, 165, 391–406 RSC.
- S. Zhou, L. W. Y. Yeung, M. W. Forbes, S. Mabury and J. P. D. Abbatt, Epoxide formation from heterogeneous oxidation of benzo[a] pyrene with gas-phase ozone and indoor air, Environ. Sci.: Processes Impacts, 2017, 19, 1292–1299 RSC.
- S. Zhou, B. C. H. Hwang, P. S. J. Lakey, A. Zuend, J. P. D. Abbatt and M. Shiraiwa, Multiphase reactivity of polycyclic aromatic hydrocarbons is driven by phase separation and diffusion limitations, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11658–11663 CAS.
- V. K. Sharma and N. J. D. Graham, Oxidation of Amino Acids, Peptides and Proteins by Ozone: A Review, Ozone: Sci. Eng., 2010, 32, 81–90 CrossRef CAS.
- B. Finlayson-Pitts, L. Wingen, A. Sumner, D. Syomin and K. Ramazan, The heterogeneous hydrolysis of NO2 in laboratory systems and in outdoor and indoor atmospheres: An integrated mechanism, Phys. Chem. Chem. Phys., 2003, 5, 223–242 RSC.
- C. George, R. S. Strekowski, J. Kleffmann, K. Stemmler and M. Ammann, Photoenhanced uptake of gaseous NO2 on solid organic compounds: a photochemical source of HONO?, Faraday Discuss., 2005, 130, 195–210 RSC.
- D. G. Aubin and J. P. D. Abbatt, Interaction of NO2 with hydrocarbon soot: Focus on HONO yield, surface modification, and mechanism, J. Phys. Chem. A, 2007, 111, 6263–6273 CrossRef CAS PubMed.
- F. Arens, L. Gutzwiller, H. W. Gäggeler and M. Ammann, The reaction of NO2 with solid anthrarobin (1,2,10-trihydroxy-anthracene), Phys. Chem. Chem. Phys., 2002, 4, 3684–3690 RSC.
- J. Liu, S. Li, J. Zeng, M. Mekic, Z. Yu, W. Zhou, G. Loisel, A. Gandolfo, W. Song, X. Wang, Z. Zhou, H. Herrmann, X. Li and S. Gligorovski, Assessing indoor gas phase oxidation capacity through real-time measurements of HONO and NOx in Guangzhou, China, Environ. Sci.: Processes Impacts, 2019, 21, 1393–1402 RSC.
- M. Sleiman, L. A. Gundel, J. F. Pankow, P. Jacob III, B. C. Singer and H. Destaillats, Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6576–6581 CrossRef CAS PubMed.
- I. J. George and J. P. D. Abbatt, Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2010, 2, 713–722 CrossRef CAS PubMed.
- S. Gross, R. Iannone, S. Xiao and A. K. Bertram, Reactive uptake studies of NO3 and N2O5 on alkenoic acid, alkanoate, and polyalcohol substrates to probe nighttime aerosol chemistry, Phys. Chem. Chem. Phys., 2009, 11, 7792–7803 RSC.
- Y. Rudich, R. K. Talukdar and A. R. Ravishankara, Reactive uptake of NO3 on pure water and ionic solutions, J. Geophys. Res.: Atmos., 1996, 101, 21023–21031 CrossRef CAS.
- A. K. Bertram, A. V. Ivanov, M. Hunter, L. T. Molina and M. J. Molina, The reaction probability of OH on organic surfaces of tropospheric interest, J. Phys. Chem. A, 2001, 105, 9415–9421 CrossRef CAS.
- I. J. George, A. Vlasenko, J. G. Slowik, K. Broekhuizen and J. P. D. Abbatt, Heterogeneous oxidation of saturated organic aerosols by hydroxyl radicals: uptake kinetics, condensed-phase products, and particle size change, Atmos. Chem. Phys., 2007, 7, 4187–4201 CrossRef CAS.
- J. H. Kroll, J. D. Smith, D. L. Che, S. H. Kessler, D. R. Worsnop and K. R. Wilson, Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol, Phys. Chem. Chem. Phys., 2009, 11, 8005–8014 RSC.
- R. Alwarda, S. Zhou and J. P. D. Abbatt, Heterogeneous oxidation of indoor surfaces by gas-phase hydroxyl radicals, Indoor Air, 2018, 28, 655–664 CrossRef CAS PubMed.
- G. C. Morrison, P. S. J. Lakey, J. P. D. Abbatt and M. Shiraiwa, Indoor boundary layer chemistry, Indoor Air, 2019, 29, 956–967 CrossRef CAS PubMed.
- M. Deborde and U. von Gunten, Reactions of chlorine with inorganic and organic compounds during water treatment – Kinetics and mechanisms: A critical review, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
- H. Schwartz-Narbonne, C. Wang, S. Zhou, J. P. D. Abbatt and J. Faust, Heterogeneous Chlorination of Squalene and Oleic Acid, Environ. Sci. Technol., 2019, 53, 1217–1224 CrossRef CAS PubMed.
- D. M. Popolan-Vaida, C.-L. Liu, T. Nah, K. R. Wilson and S. R. Leone, Reaction of Chlorine Molecules with Unsaturated Submicron Organic Particles, Z. Phys. Chem., 2015, 229, 1521–1540 CAS.
- R. Zhao, A. K. Y. Lee, R. Soong, A. J. Simpson and J. P. D. Abbatt, Formation of aqueous-phase alpha-hydroxyhydroperoxides (alpha-HHP): potential atmospheric impacts, Atmos. Chem. Phys., 2013, 13, 5857–5872 CrossRef.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS PubMed.
- P. Sotero and R. Arce, Major products in the photochemistry of perylene adsorbed in models of atmospheric particulate matter, J. Photochem. Photobiol., A, 2008, 199, 14–22 CrossRef CAS.
- T. Salthammer, A. Bednarek, F. Fuhrmann, R. Funaki and S. Tanabe, Formation of organic indoor air pollutants by UV-curing chemistry, J. Photochem. Photobiol., A, 2002, 152, 1–9 CrossRef CAS.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- A. Gandolfo, V. Bartolomei, E. Gomez Alvarez, S. Tlili, S. Gligorovski, J. Kleffmann and H. Wortham, The effectiveness of indoor photocatalytic paints on NOx and HONO levels, Appl. Catal., B, 2015, 166, 84–90 CrossRef.
- A. Gandolfo, L. Rouyer, H. Wortham and S. Gligorovski, The influence of wall temperature on NO2 removal and HONO levels released by indoor photocatalytic paints, Appl. Catal., B, 2017, 209, 429–436 CrossRef CAS.
- H. Schwartz-Narbonne, S. H. Jones and D. J. Donaldson, Indoor Lighting Releases Gas Phase Nitrogen Oxides from Indoor Painted Surfaces, Environ. Sci. Technol. Lett., 2019, 6, 92–97 CrossRef CAS.
- T. Salthammer and F. Fuhrmann, Photocatalytic surface reactions on indoor wall paint, Environ. Sci. Technol., 2007, 41, 6573–6578 CrossRef CAS.
- H. Chen, C. E. Nanayakkara and V. H. Grassian, Titanium Dioxide Photocatalysis in Atmospheric Chemistry, Chem. Rev., 2012, 112, 5919–5948 CrossRef CAS.
- A. Boonstra and C. Mutsaers, Adsorption of hydrogen peroxide on surface of titanium dioxide, J. Phys. Chem., 1975, 79, 1940–1943 CrossRef CAS.
- H. Chen, C. O. Stanier, M. A. Young and V. H. Grassian, A Kinetic Study of Ozone Decomposition on Illuminated Oxide Surfaces, J. Phys. Chem. A, 2011, 115, 11979–11987 CrossRef CAS.
- G. R. Wentworth and H. A. Al-Abadleh, DRIFTS studies on the photosensitized transformation of gallic acid by iron(III) chloride as a model for HULIS in atmospheric aerosols, Phys. Chem. Chem. Phys., 2011, 13, 6507–6516 RSC.
- S. A. Styler and D. J. Donaldson, Heterogeneous Photochemistry of Oxalic Acid on Mauritanian Sand and Icelandic Volcanic Ash, Environ. Sci. Technol., 2012, 46, 8756–8763 CrossRef CAS.
- S. Chino, S. Kato, J. Seo and Y. Ataka, Study on emission of decomposed chemicals of esters contained in PVC flooring and adhesive, Build. Environ., 2009, 44, 1337–1342 CrossRef.
- A. Sjoberg and O. Ramnas, An experimental parametric study of VOC from flooring systems exposed to alkaline solutions, Indoor Air, 2007, 17, 450–457 CrossRef CAS.
- A. Bope, S. R. Haines, B. Hegarty, C. J. Weschler, J. Peccia and K. C. Dannemiller, Degradation of phthalate esters in floor dust at elevated relative humidity, Environ. Sci.: Processes Impacts, 2019, 21, 1268–1279 RSC.
- P. Wolkoff and S. K. Kjaergaard, The dichotomy of relative humidity on indoor air quality, Environ. Int., 2007, 33, 850–857 CrossRef CAS.
- M. Kruza and N. Carslaw, How do breath and skin emissions impact indoor air chemistry?, Indoor Air, 2019, 29, 369–379 CrossRef CAS.
- D. Licina, G. C. Morrison, G. Beko, C. J. Weschler and W. W. Nazaroff, Clothing-Mediated Exposures to Chemicals and Particles, Environ. Sci. Technol., 2019, 53, 5559–5575 CrossRef CAS.
- G. C. Morrison, C. J. Weschler, G. Bekö, H. M. Koch, T. Salthammer, T. Schripp, J. Toftum and G. Clausen, Role of clothing in both accelerating and impeding dermal absorption of airborne SVOCs, J. Exposure Sci. Environ. Epidemiol., 2015, 26, 113 CrossRef.
- S. Liu, S. L. Thompson, H. Stark, P. J. Ziemann and J. L. Jimenez, Gas-Phase Carboxylic Acids in a University Classroom: Abundance, Variability, and Sources, Environ. Sci. Technol., 2017, 51, 5454–5463 CrossRef CAS.
- D. Yan, W. O'Brien, T. Hong, X. Feng, H. B. Gunay, F. Tahmasebi and A. Mahdavi, Occupant behavior modeling for building performance simulation: Current state and future challenges, Energy Build., 2015, 107, 264–278 CrossRef.
- W. Nazaroff, Indoor particle dynamics, Indoor Air, 2004, 14, 175–183 CrossRef.
- J. T. Jayne, D. C. Leard, X. F. Zhang, P. Davidovits, K. A. Smith, C. E. Kolb and D. R. Worsnop, Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 2000, 33, 49–70 CrossRef CAS.
- L. Qi, M. Chen, G. Stefenelli, V. Pospisilova, Y. Tong, A. Bertrand, C. Hueglin, X. Ge, U. Baltensperger, A. S. H. Prevot and J. G. Slowik, Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 2019, 19, 8037–8062 CrossRef CAS.
- R. B. Cody, J. A. Laramee and H. D. Durst, Versatile new ion source for the analysis of materials in open air under ambient conditions, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS.
- S. M. Zhou, M. W. Forbes and J. P. D. Abbatt, Application of Direct Analysis in Real Time-Mass Spectrometry (DART-MS) to the Study of Gas–Surface Heterogeneous Reactions: Focus on Ozone and PAHs, Anal. Chem., 2015, 87, 4733–4740 CrossRef CAS.
- V. W. Or, M. R. Alves, M. Wade, S. Schwab, R. L. Corsi and V. H. Grassian, Crystal Clear? Microspectroscopic Imaging and Physicochemical Characterization of Indoor Depositions on Window Glass, Environ. Sci. Technol. Lett., 2018, 5, 514–519 CrossRef CAS.
- Y. Fang, S. Riahi, A. T. McDonald, M. Shrestha, D. J. Tobias and V. H. Grassian, What Is the Driving Force behind the Adsorption of Hydrophobic Molecules on Hydrophilic Surfaces?, J. Phys. Chem. Lett., 2019, 10, 468–473 CrossRef CAS.
- M. Shiraiwa, C. Pfrang and U. Poeschl, Kinetic multi-layer model of aerosol surface and bulk chemistry (KM-SUB): the influence of interfacial transport and bulk diffusion on the oxidation of oleic acid by ozone, Atmos. Chem. Phys., 2010, 10, 3673–3691 CrossRef CAS.
- A. Zuend and J. H. Seinfeld, Modeling the gas-particle partitioning of secondary organic aerosol: the importance of liquid-liquid phase separation, Atmos. Chem. Phys., 2012, 12, 3857–3882 CrossRef CAS.
- M. Shiraiwa, A. Zuend, A. K. Bertram and J. H. Seinfeld, Gas-particle partitioning of atmospheric aerosols: interplay of physical state, non-ideal mixing and morphology, Phys. Chem. Chem. Phys., 2013, 15, 11441–11453 RSC.
- W.-S. DeRieux, Y. Li, P. Lin, J. Laskin, A. Laskin, A. K. Bertram, S. A. Nizkorodov and M. Shiraiwa, Predicting the glass transition temperature and viscosity of secondary organic material using molecular composition, Atmos. Chem. Phys., 2018, 18, 6331–6351 CrossRef.
- A. Zuend, C. Marcolli, T. Peter and J. H. Seinfeld, Computation of liquid–liquid equilibria and phase stabilities: implications for RH-dependent gas/particle partitioning of organic–inorganic aerosols, Atmos. Chem. Phys., 2010, 10, 7795–7820 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |