
Vertical transport and sinks of perfluoroalkyl substances in the global open ocean†

Abstract
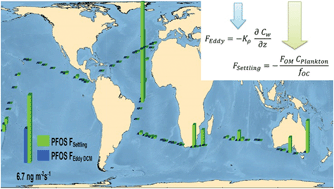
The ubiquitous occurrence of perfluoroalkyl substances (PFAS) in the open ocean has been previously documented, but their vertical transport and oceanic sinks have not been comprehensively characterized and quantified at the oceanic scale. During the Malaspina 2010 circumnavigation expedition, 21 PFAS were measured at the surface and at the deep chlorophyll maximum (DCM) in the Atlantic, Indian and Pacific oceans. In this work, we report an extended data set of PFAS dissolved phase concentrations at the DCM. ∑PFAS at the DCM varied from 130 to 11 000 pg L−1, with a global average value of 500 pg L−1. Perfluorooctanesulfonate (PFOS) abundance contributed 39% of ∑PFAS, followed by perfluorodecanoate (PFDA, 17%), and perfluorohexanoate (PFHxA, 12%). The relative contribution of the remaining compounds was below 10%, with perfluorooctanoate (PFOA) contributing only 5% to PFAS measured at the DCM. Estimates of vertical diffusivity, derived from microstructure turbulence observations in the upper (<300 m) water column, allowed the derivation of PFAS eddy diffusive fluxes from concurrent field measurements of eddy diffusivity and PFAS concentrations. The PFAS concentrations at the DCM predicted from an eddy diffusivity model were lower than field-measured concentrations, suggesting a relevant role of other vertical transport mechanisms. Settling fluxes of organic matter bound PFAS (biological pump), oceanic circulation and potential, yet un-reported, biological transformations are discussed.
- This article is part of the themed collection: PFAS
 Please wait while we load your content...
Please wait while we load your content...

