
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeochemical conditions conducive for retention of trace elements and radionuclides during shale–fluid interactions†
Neha
Mehta
 a and
Benjamin D.
Kocar
*ab
a and
Benjamin D.
Kocar
*ab
aCivil and Environmental Engineering, Massachusetts Institute of Technology, 15 Vassar St, Cambridge, MA 02139, USA
bExponent, Inc., 1055 E. Colorado Blvd, Pasadena, CA 91106, USA. E-mail: bkocar@exponent.com
First published on 3rd September 2019
Abstract
Produced water generated during unconventional oil and gas extractions contains a complex milieu of natural and anthropogenic potentially toxic chemical constituents including arsenic (As), chromium (Cr), and cadmium (Cd), naturally occurring radioactive materials (NORMs) including U and Ra, and a myriad of organic compounds. The human-ecological health risks and challenges associated with the disposal of produced water may be alleviated by understanding geochemical controls on processes responsible for the solubilization of potentially hazardous natural shale constituents to produced water. Here, we investigated, through a series of batch treatments, the leaching behavior of As, Se, Cu, Fe, Ba, Cr, Cd, and radioactive nuclides U, Ra from shale to produced water. Specifically, the effect of four major controls on element mobility was studied: (1) solution pH, (2) ionic strength of the solution, (3) oxic–anoxic conditions, and (4) an additive used in fracking fluid. The mobilization of metals and metalloids from shale was greatest in treatments containing sodium persulfate, an oxidant and a commonly used additive in fracture fluid. In the high ionic strength treatments, dissolved Ba concentrations increased 5-fold compared to low ionic strength treatments. Overall, anoxic conditions superimposed with low pH resulted in the largest increase of dissolved metals and radionuclides such as Ra. Overall, our results suggest that (1) limiting pore water acidification by injection of alkaline fluid in carbonate-low shale and (2) minimizing strong oxidizing conditions in shale formations may result in cost-effective in situ retention of produced water contaminants.
Environmental significanceDisposal and ineffective treatment of large volumes of polluted produced water recovered during hydraulic fracturing poses potential threats to surface and groundwater. To understand these potential impacts, we investigated geochemical controls on mobility and retention of metals and radionuclides to produced water. Our findings could be utilized to inform the synthesis of injection fluids for in situ retardation of produced water pollutants, thereby reducing pollutant loading in produced water above the ground and enable sustainable and safer disposal of produced water. |
1. Introduction
During the last decade, the successful extraction of unconventional oil and gas resources revolutionized the energy landscape in the United States. The key enabler of this revolution is a well stimulation technique called hydraulic fracturing (aka fracking). During fracking, 15–40 million liters of water combined with chemicals and proppants are injected at high pressure into the shale formation to fracture rocks and release trapped hydrocarbons. After the fracturing process, some of the injected fluid returns to the surface in addition to the natural fluid from the formation. Typically, 10–50% of the water used in hydraulic fracturing is recovered.1 At the surface, the recovered water is separated from the extracted hydrocarbon and stored in tanks or pits prior to disposal, treatment, or re-use as injection water.2 These aqueous solutions, hereafter referred to as “produced water” are often chemically complex, containing residual additives, dissolved salts, Naturally Occurring Radioactive Materials (NORMs) and toxic metals and metalloids.3,4 For instance, 133 samples of produced water collected from Marcellus shale gas extraction operations yielded a median radium (Ra) radioactivity of 203 Bq L−1 (combined 226Ra and 228Ra), far above the industrial effluent regulatory limit of 2.2 Bq L−1; elevated dissolved solids (median of 157![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1), and potentially hazardous constituents such as As, Cd, Cu, and Pb.5–8 The constituents of produced water, therefore, represent a potential public and ecological health risk.5,9,10 The potential risks are exacerbated by the large volumes of water produced during the lifetime of a well—as much as 5 million liters are produced by an unconventional Marcellus shale extraction well over its average lifetime of 10 years.2 Managing the storage and transportation of these large volumes is challenging, as illustrated by recorded, large volume spills, inferred infiltration of produced water constituents through impoundment pits and discharge of incompletely treated water to surface waters.10–13
000 mg L−1), and potentially hazardous constituents such as As, Cd, Cu, and Pb.5–8 The constituents of produced water, therefore, represent a potential public and ecological health risk.5,9,10 The potential risks are exacerbated by the large volumes of water produced during the lifetime of a well—as much as 5 million liters are produced by an unconventional Marcellus shale extraction well over its average lifetime of 10 years.2 Managing the storage and transportation of these large volumes is challenging, as illustrated by recorded, large volume spills, inferred infiltration of produced water constituents through impoundment pits and discharge of incompletely treated water to surface waters.10–13
The potential human–ecological health risks and challenges associated with the disposal of produced water may be alleviated by understanding how inorganic constituents are mobilized to produced water. Marcellus shale is enriched in heavy metals like As, Cd, Co, Cr, Hg, Ni, Zn, V, and Ba, radioactive nuclides (e.g. U, 226Ra) and minerals such as calcite, barite, clays, pyrite and silicate minerals.14 During the injection of fracking fluid, long-established solution-mineralogical geochemical conditions that are at (or near) equilibrium are altered, thereby perturbing activities of aqueous constituents. Several previous studies have examined the role of fracking fluid chemistry in element mobilization from Marcellus shale core samples. Tasker et al. characterized the mobility of major elements and organics from Marcellus shale and concluded that metal mobility was strongly dependent on the solution pH.15 The solution pH was shown to be a function of pyrite to carbonate ratio in shale. Harrison et al. reported that shale with low carbonate and high pyrite composition resulted in porewater acidification and thereby increasing the release of major elements and U, Pb, and Ni from shale to porewater.16 In addition to pH, various other solution chemistry parameters could affect the release of trace elements from shale. Wang et al. reported the influence of oxidant concentration, solid:water ratio and pH on the mobility of major elements from Eagle Ford and Bakken shale.17 Phan et al. reported speciation of U, As, and Ba in Marcellus shale using sequential extraction and suggested that U was primarily associated with silicate minerals, and As was primarily associated with organic matter and sulfide minerals.18 Renock et al. focused on the release of Ba and demonstrated that at elevated temperature and under anoxic conditions Ba mobility increases with ionic strength. Recently, experimental studies have begun to focus on reproducing shale–water interactions in the laboratory using synthetic hydraulic fracturing fluids at elevated temperature and pressure batch reactor systems (e.g.ref. 19 and 20).
Prior experimental investigations have focused on examining the mobility of a few selected elements (As, Ba, U) from Marcellus shale under a limited set of solution chemistry conditions. However, the inorganic pollutant load of produced water is derived from a plethora of metals and metalloids. Understanding the role of solution chemistry in the solubilization of all the inorganic contaminants present in the produced water is essential to achieve a reduction in the pollutant load of produced water. Moreover, the relationship between the observed release of elements and the shale mineralogy was established through bulk mineralogical characterization of shale solids. This may obscure the role of microscale heterogeneity in shale mineralogical composition in the release of elements to the solution. Furthermore, little is understood about Ra mobility from shale as a function of solution parameters (e.g.ref. 21). However, it remains unknown how Ra release from shale is affected as a function of pH, fracture fluid additive, etc. Further experimental studies are required to better understand the mobility of Ra and various elements from Marcellus shale under varying solution chemistry conditions. Accordingly, we investigated the effect of solution chemistry parameters on the release of a broad matrix of elements and radionuclides from Marcellus shale to produced water, which has not been demonstrated earlier. Using leaching treatments, we examined the effect of fluid pH, sodium persulfate (additive commonly added in hydraulic fracturing fluid), and ionic strength, under oxic and anoxic conditions on element release from shale. We complemented the experimental study with the comprehensive mineralogical and radiochemical characterization of shale using μ-SXRF, autoradiography, and XRD techniques. We find that injection of oxic and alkaline injection fluid is likely to favor retention of produced water contaminants in situ. We also find that wells utilizing fracturing fluid composed of produced water blended with freshwater are more vulnerable to the formation of radioactive scales, which may decrease well productivity.
2. Materials and methods
2.1. Sample description
The Marcellus shale samples used in this study were obtained from the core repository at the Department of Conservation and Natural Resources, Harrisburg, PA. The cores at the repository were collected as part of the Eastern Gas Shales Project (EGSP) in the late 1970s, to evaluate the gas potential of, and to enhance gas production from the extensive Devonian and Mississippian organic-rich black shales within the Appalachian. From this repository, we obtained 15 rock chips, ranging from approximately 5150 to 5185 feet depth from a Marcellus shale drill core from Mckean county (−78.61242029, 41.868829). Upon receiving the samples, we immediately stored the samples in an anoxic glove bag (95% N2, 5% H2). However, it is possible that there may be some oxidation that would have occurred during sample storage and handling.2.2. μ-SXRF sample preparation
Shale thin section was prepared by embedding the sample in clear resin (EPO-TEK 301-2FL). The embedded sample was placed in a vacuum desiccator for 3 days to cure. After curing, the resin puck was shipped to Spectrum Petrographics for cutting into the petrographic thin section, mounted with superglue to Suprasil 2A Quartz glass, polished with 0.5 μm diamond abrasive to a standard final thickness of 30 μm. Elements in the shale thin section were mapped by μ-SXRF on 2-3 beamline of the Stanford Synchrotron Lightsource (SSRL). The incoming X-ray energy was set to 17200 eV, that is 34 eV above U LIII absorption edge. Measurements were performed using a 1 μm step size, and a dwell time of 50 ms. The μ-XRF spectrum was analyzed using Microtoolkit software (https://www.sams-xrays.com/smak). Correlation plots were created to identify the association of elements with the host matrix. The plots were generated using the Microtoolkit software package. Briefly, the steps involved in generating correlation plots include smoothening the element channel to eliminate the quantization and allow for fractional values and plotting smoothed element channel data on the X-axis and other smoothed element channels on the Y-axis. Pearson's linear correlation coefficients (r) were calculated to identify the positive association of elements with Fe.
2.3. Whole rock powder characterization
We chose to prepare a composite powdered shale sample instead of individual rock samples because the amount of each rock sample was not sufficient to yield repeatable experimental measurements. We note that this approach resulted in reducing sample size (n = 1 versus n = 15) for analysis and may limit the generalization of our findings. A composite powdered shale sample was prepared by crushing rock chips (n = 15) with tungsten carbide beads in a benchtop rotary rock tumbler, equipped with a rubber barrel. After crushing, the composite powder sample was sieved to the approximate particle size range of 100–500 μm. To avoid any external metallic contamination, non-metallic sieves (Gilson Nylon sieves) were used. The surface area of the composite sample was measured using the Brunauer–Emmett–Teller (BET) method and ranged between 5 and 7 m2 g−1. Bulk mineralogical characterization and quantification of the major mineral phases in the powdered shale sample was performed on a Panalytical XPert Pro X-ray diffractometer (CMSE, MIT) using a Cu Kα radiation source. The sample was mixed with 9% pure Si internal standard. The sample was run from 4 to 90° 2θ using a step size of 0.01° 2θ. Quantitative analysis of X-ray data was performed using High Score Plus software.For whole-rock element concentrations, approximately 10 mg of composite shale sample was digested using a combination of concentrated HNO3, HCl, H2O2, and HF. The multi-acid digestion procedure was based on a modified version of the USEPA method.22 Details of the digestion procedure can be found in the ESI.† Certified USGS reference material SBC-1 was concurrently digested to ensure quality control and assess the accuracy of the digestion procedure. The contribution of elements in analytical blanks was low, and ranged between 0.001 and 0.1%, depending on the element. Detailed concentrations of elements in the blanks can be found in the ESI (Table S1†). The digestions were performed in triplicate.
2.4. Radioactivity characterization
Long-lived naturally occurring 226Ra in digested shale samples was quantified using a Canberra High Purity Germanium Detector (HPGe), calibrated for energy and efficiency using a mixed multi-nuclide Eckert & Ziegler™ aqueous gamma standard. The method detection limit (MDL) of 226Ra based on Curie's derivation was equal to 0.6 Bq mL−1 for a count time of 1 day. Spatial distribution of U and Th in shale thin section was studied using μ-SXRF (beamline 2-3, Stanford Synchrotron Radiation Light source, SSRL, Stanford). Grain-scale heterogeneity in shale radionuclide distribution was qualitatively analyzed using autoradiography, by exposing a phosphor screen for 7 days to a circular disc of shale and imaging the intensity on a Typhoon™ biomolecular imager.2.5. Sequential extraction
Preliminary association of elements with different shale constituents was quantified using a mixed BCR-Tessier sequential extraction method.23 Traditionally, sequential extraction methods have been a subject of criticism, partly owing to the interpretations of analytical results. Often studies refer to a presumed mineral phase (e.g. bound to carbonates, bound to Fe and Mn oxides) without knowing whether the referred phases are present and are fully or partially responding to the specific chemical treatment. Other problems include non-selectivity of extractants and potential redistribution among phases during extraction. Despite these challenges in interpretation, these extractions do provide first-order qualitative information about the association of metals and metalloids in different solid pools in shale and assist in inferring water–rock interactions. Operationally defined different extracted fractions of elements will be referred to as follows: S1: exchanged fraction, usually includes weakly adsorbed metals, metals that can be released by ion-exchange processes; S2: dilute acid extract fraction, contains metals which are associated with carbonate; S3: oxidized fraction, includes metals bound to organic matter and sulfide that can be easily released under oxidizing conditions; and S4: residual fraction. Six replicates of composite shale were progressively treated with a suite of chemical reagents to selectively extract metals bound in these fractions (Table 1). In all steps, samples were filtered using 0.22 μm polyethersulfone (PES) syringe filters and filtrates were analyzed on ICP-MS and ICP-OES. The concentration of elements in the residual fraction was estimated from the difference between the whole-rock multi-acid digestions and the sum of the sequential fractions (S1–S3). More details of the sequential extraction scheme are provided in the ESI.†| Extracted fraction | Reagents |
|---|---|
| Exchanged (S1) | 16 mL MgCl2 (pH 7), shaken for 1 h at room temperature |
| Dilute acid extract (S2) | 25 mL 1 M CH3COONa (pH 5), shaken for 5 h at room temperature |
| Oxidized (S3) | 40 mL H2O2 (30% w/v), at 85 °C; 20 mL 0.01 N HNO3, shaken for 30 min |
| Residue (S4) | Estimated from the difference between the whole-rock digestions and sum of S1–S3 |
2.6. Batch experiments
The impact of solution chemistry parameters on the release of elements and radionuclides from shale was investigated in a series of batch leaching treatments. The treatments were designed to represent changes in solution chemistry after injection of the hydraulic fracturing fluid into the shale formation. After the injection of oxygenated fracture fluid, the downhole environment transitions from the anoxic to the oxic regime, until all the dissolved oxygen is consumed. The subsurface may remain anoxic until oxidants such as persulfate are introduced to degrade the polymeric gel. We simulated these downhole transitions by performing treatments under both oxic and anoxic conditions. Among other parameters, the pH of the injection water is a master variable affecting the metal release. Prior to the injection of the hydraulic fracturing fluid, often hydrochloric acid is injected into the wells to clean the perforations and enable easier access of injected water to shale.19 Depending on the carbonate content of shale, the pH of the fluid sampled from bore-hole after the injection may range between 2 and 8. The precise data on borehole fluid pH after acid injection are scarce and seldom available. Due to this uncertainty, treatments were run for two scenarios: if the acid were not fully consumed, resulting in pH ∼ 4, and if the majority of the acid were consumed, resulting in pH ∼ 7.Among other solution chemistry parameters, we also investigated the impact of ionic strength of the injection fluid on metal solubilization. With the ongoing shift towards using high salinity water for hydraulic fracturing operations, understanding the impact of high salinity water on the mobilization of metalloids is necessary.24 Traditionally, low TDS water (e.g. surface water, groundwater) was used as an injection fluid in hydraulic fracturing. However, the high-water demand of the fracking process resulted in localized water stresses, forcing operators to use alternative water sources. Simultaneously, the environmental challenges associated with the disposal of large quantities of produced water were growing. Thus, well operators started reusing produced water for subsequent hydraulic fracturing by blending it with freshwater.25 Thomas et al. reported the TDS of blended injection fluid in the range of 30000 mg L−1.26 Thus, treatments were performed under two scenarios: leaching fluid with background electrolyte concentration of 0.5 M NaCl if produced is reused as injection fluid and if freshwater is used as injection water, leaching fluids were prepared with de-ionized water.
In addition to solution parameters, the interaction of chemical additives with shale may also affect the mobility of elements. Fracturing fluids typically comprise 90% water, 9–10% proppants (sand, ceramics) and 0.5–1% chemical additives.2,12 Chemical additives are necessary to achieve a continuous gas flow, but their effect on element mobility is not well understood.27 In order to evaluate these effects, this study focuses on one particular additive-sodium persulfate, widely used in hydraulic fracturing to degrade friction-reducing polymers and guar gels.4,27 Sodium persulfate is an oxidant and extensively used to degrade complex organic compounds in the environment.28,29
The leaching conditions for batch treatments are listed in Table 2. Treatments were named according to the conditions used. For example, E1-ox-DI-pH7 denotes oxic conditions, low ionic strength (de-ionized water), pH buffered at 7. Similarly, E1-anox-Sal-pH7 denotes anoxic conditions, high ionic strength (0.5 M NaCl), and buffered at pH7. The chemical makeup of leaching fluids (LF's) is as follows: LF1, buffered at pH 7 using 0.1 M PIPES solution, under atmospheric conditions; LF2, buffered at pH 4 using a sodium-acetate and acetic acid buffer, under atmospheric conditions; LF3, N2-purged solution buffered at pH 7 using 0.1 M PIPES solution, mixed in an anoxic glove bag (95% N2, 5% H2) to maintain anoxic conditions; LF4 N2-purged solution buffered at pH 4 using a sodium-acetate and acetic acid buffer, mixed in an anoxic glove bag; LF5, N2-purged solution buffered at pH 4 and mixed with 0.5 M sodium persulfate, also in an anoxic glove bag. Depending on the treatment conditions, the ionic strength of the leaching fluids (LF1–5) was either de-ionized water or 0.5 M NaCl. Treatments with LF5 were performed at 85 °C in order to thermally activate sodium persulfate.
| Treatment | Buffer | pH | Oxic/anoxic | Ionic strength | T (°C) | Additive | LF type |
|---|---|---|---|---|---|---|---|
| E1-ox-DI-pH7 | 0.1 M PIPES | 7 | Oxic | De-ionized water | Ambient | — | LF1 |
| E2-ox-DI-pH4 | Sodium acetate and acetic acid | 4 | Oxic | De-ionized water | Ambient | — | LF2 |
| E3-anox-DI-pH7 | 0.1 M PIPES | 7 | Anoxic | De-ionized water | Ambient | — | LF3 |
| E4-anox-DI-pH4 | Sodium acetate and acetic acid | 4 | Anoxic | De-ionized water | Ambient | — | LF4 |
| E5-anox-DI-pH4 | Sodium acetate and acetic acid | 4 | Anoxic | De-ionized water | 85 °C | 0.5 M sodium persulfate | LF5 |
| E1-ox-Sal-pH7 | 0.1 M PIPES | 7 | Oxic | 0.5 M NaCl | Ambient | — | LF1 |
| E2-ox-Sal-pH4 | Sodium acetate and acetic acid | 4 | Oxic | 0.5 M NaCl | Ambient | — | LF2 |
| E3-anox-Sal-pH7 | 0.1 M PIPES | 7 | Anoxic | 0.5 M NaCl | Ambient | — | LF3 |
| E4-anox-Sal-pH4 | Sodium acetate and acetic acid | 4 | Anoxic | 0.5 M NaCl | Ambient | — | LF4 |
| E5-anox-Sal-pH4 | Sodium acetate and acetic acid | 4 | Anoxic | 0.5 M NaCl | 85 °C | 0.5 M sodium persulfate | LF5 |
In each treatment, 0.5 gram of solid was shaken with 10 mL leaching fluid (liquid-to-solid ratio of 20:1) in acid washed falcon tubes and placed on a rotary shaker (at 100 rpm) for 24 hours. Thereafter, samples were centrifuged, and the supernatants were filtered using 0.22 μm (PES) syringe filters and acidified using 2 μL concentrated HNO3. Filtrates were analyzed for Cr, Mn, Fe, Co, Ni, Cu As, Se, Sr, Cd, Ba, Pb and U using ICP-MS, and 226Ra in solution was measured using a gamma spectroscopy system. Each test was performed in six replicates. Blank controls were performed that did not contain sample powders. Concentrations of metal leached from shale were calculated by subtracting concentrations in fluids reacted with shale from fluids reacted with no shale (blanks). We note that the leaching treatments (except E5) were performed under ambient pressure and temperature conditions. The use of ambient pressure rather than reservoir pressure is expected to result in minor differences between experimental and field conditions. The types of minerals that dissolve and their relative rates of dissolution are likely not strongly impacted by differences in pressures, but the temperature will likely influence reaction rates to a greater extent. Therefore, our results represent a conservative estimate of metals and radionuclide release from shale to porewater.
2.7. Element concentration in solution
Trace metal concentrations in leachates, multi-acid digestates and in sequentially extracted fractions were measured using both Agilent 7900 ICP-MS and Agilent 5100 ICP-OES at the MIT CEHS Bioimaging and Chemical Analysis Core Facility and at the MIT CMSE facility. The elements of interest to our study occurred in the samples in a range of concentrations, and neither instrument alone is suitable for measuring all these elements. Therefore, Al, Ca and K were measured on ICP-OES, while the rest of the elements were measured on ICP-MS. We calculated the detection limit as suggested in EPA method 200.8. The method describes detection limit (MDL) as the value at three standard deviations of the 7 replicate runs of 3% (w/v) HNO3. A typical detection limit was 6.6 μg L−1 in the solution for Al, 56.4 μg L−1 in the solution for Ca, and 22.06 μg L−1 for K in solution.The ICP-MS instrument was calibrated with a dilution of the Agilent Environmental Calibration Standard Mix at 3% nitric acid for all samples. The trace elements of our interest suffer polyatomic interferences during the generation of ions from the plasma and/or the sample. The interferences result in the generation of ions which carry a mass-to-charge ratio that is identical to that of analyte ions. Some examples of such interferences relevant to this study are: 40Ar35Cl+ on As (m/z = 75) and ArAr+ on Se (m/z = 80). Therefore, trace metal concentration in all samples, including whole rock assays, sequential extracts, and produced waters, was measured by ICP-MS under kinetic energy discrimination (KED) mode using helium as the collision gas, based on the US EPA 200.8 method and an Agilent application note (Tetsushi Sakai and Ed McCurdy, 2014). This has been demonstrated to successfully remove polyatomic interferences 40Ar35Cl+, 40Ca35Cl+, and 37Cl2H, ArO+, ArAr+ on the m/z ratio of metals such as Fe, Se, and As.
Samples, calibration standards, and blanks were prepared in acid-washed falcon tubes. All samples were diluted with 3% HNO3 (trace metal grade) to ensure the stability of elements. Use of HCl was avoided in the preparation of samples for ICP-MS, due to the formation of Cl based interferences on elements such as As, Se, and Cr. All samples were spiked with an internal standard before analysis on ICP-MS, consisting of 20 ppb of lithium (Li), scandium (Sc), yttrium (Y), germanium (Ge), bismuth (Bi), terbium (Tb), and indium (In). At the beginning of the analysis, 3–5 blanks of milliQ water acidified to 3% nitric acid were run to clear out any residual contamination from previous runs. Additionally, one analytical blank was run between every five samples to ensure no crossover between samples and that there was no build-up of contamination within the machine. The ICP-MS method detection limit was calculated in the same way as for ICP-OES. The concentration of elements in the control blanks was negligible and can be found in the ESI (Table S1†).
3. Results and discussion
3.1. Bulk chemistry and mineralogy of shale
Based on the XRD measurement, the presence of calcite, quartz, pyrite and clay minerals was confirmed in the shale. Quantitative analysis using an internal standard method showed that the composite shale sample had a mineralogical composition of 35% quartz, 39% illite, 4% pyrite, 3% calcite, 3% chlorite, and 1% smectite. The approximate error in measurement was ±5 wt%. Although our study focused on one composite shale sample, the mineralogical composition presented here was consistent with the previously reported studies (e.g.ref. 16 and 30). The total concentrations of elements in the shale and certified reference material (SBC-1) are summarized in Table 3. Element recoveries in total digestion procedures ranged within 70–130%, deemed acceptable as prescribed in EPA method 200.8. The element recoveries were calculated based on SBC-1. Appreciable concentrations of trace elements such as As, Cu, Se, Cd, U, Ba, Co, and Pb were measured in whole-rock digestates (Table 3). The concentrations of major elements in shale were Al (7.6% wt), Ca (2.1% wt), K (3.7% wt) and Fe (3.4% wt).| Alb, mg kg−1 | Cab, mg kg−1 | Kb, mg kg−1 | Fe, mg kg−1 | Sr, mg kg−1 | Ba, mg kg−1 | Mn, mg kg−1 | |
|---|---|---|---|---|---|---|---|
| a ND: not detected. X: the residual concentration was negative as the sum of elemental data from sequential extraction steps exceeded the total element concentration. b Measured on ICP-OES and rest of the elements were measured on ICP-MS. c The concentrations are reported as mean ± standard deviation from triplicate measurements. d Reference material used in this analysis is USGS SBC-1, which is representative of marine shale of the lower Conemaugh Group, Glenshaw Formation. e Calculated by subtracting total of concentrations in extracted fractions from the total concentration in whole rock. f Calculated by dividing the measured elemental concentration by the corresponding certified concentration in SBC-1. | |||||||
| Total | 76533 ± 1445 |
21613 ± 509 |
37400 ± 925 |
34753 ± 776 |
237 ± 5 | 647 ± 17 | 310 ± 5 |
| Exchangeable | 10.1 ± 0.3 | 12731 ± 790 |
7755 ± 581 | 0.19 ± 0.05 | 74 ± 6 | 21 ± 1 | 2.44 ± 0.03 |
| Acid soluble | 1979 ± 35 | 19363 ± 1071 |
6636 ± 774 | 1481 ± 39 | 21 ± 6 | 65 ± 2 | 12.0 ± 0.2 |
| Oxidized | 1834 ± 234 | ND | ND | 7470 ± 651 | 205 ± 18 | 73 ± 34 | 201 ± 3 |
| Residuee | 72711 ± 1464 |
X | 23008 ± 1369 |
25802 ± 1014 |
X | 488 ± 38 | 94 ± 5.9 |
|
|||||||
| Data quality control: SBC-1 | |||||||
| Measured | 82057 ± 9038 |
14284 ± 1538 |
29324 ± 1762 |
66250 ± 622 |
167 ± 10.4 | 722 ± 37 | 919 ± 1 |
| Certified | 111197 ± 212 |
21063 ± 71 |
28635 ± 83 |
67914 ± 140 |
178 ± 1.4 | 788 ± 8 | 1162 ± 8 |
| Element recoveryf | 74% | 87% | 102% | 98% | 94% | 92% | 79% |
| Cr, mg kg−1 | Co, mg kg−1 | Ni, mg kg−1 | Cu, mg kg−1 | As, mg kg−1 | Se, mg kg−1 | Cd, mg kg−1 | Pb, mg kg−1 | U, mg kg−1 | |
|---|---|---|---|---|---|---|---|---|---|
| Total | 92.8 ± 1.2 | 323 ± 4 | 162 ± 1 | 160 ± 2 | 27 ± 0.2 | 3.36 ± 0.14 | 0.67 ± 0.01 | 42.19 ± 0.47 | 14.8 ± 0.1 |
| Exchangeable | 0.17 ± 0.01 | 45.2 ± 2.1 | 1.31 ± 0.01 | 0.18 ± 0.01 | 0.011 ± 0.001 | 0.09 ± 0.01 | 0.015 ± 0.001 | ND | 0.25 ± 0.01 |
| Acid soluble | 1.32 ± 0.03 | 37.6 ± 2.5 | 4.5 ± 2.65 | 3.01 ± 0.07 | 0.17 ± 0.00 | 0.17 ± 0.02 | 0.01 ± 0.00 | 0.68 ± 0.06 | 0.29 ± 0.01 |
| Oxidized | 5.00 ± 0.37 | 82.1 ± 1.2 | 42.29 ± 7.41 | 81 ± 9 | 0.80 ± 0.07 | 2.66 ± 0.39 | 0.1 ± 0.02 | 9.3 ± 2.8 | 1.0 ± 0.15 |
| Residuee | 86 ± 1.3 | 159 ± 5 | 114 ± 8.0 | 76 ± 9.6 | 26 ± 0.2 | 0.44 ± 0.41 | 0.50 ± 0.02 | 32 ± 2.8 | 13 ± 0.2 |
|
|||||||||
| Data quality control: SBC-1 | |||||||||
| Measured | 101 ± 1 | 20 ± 0.9 | 89 ± 0.9 | 32 ± 0.9 | 25 ± 0.9 | 1.5 ± 0.9 | 0.48 ± 0.9 | 29 ± 1 | 5.3 ± 0.9 |
| Certified | 109 ± 1 | 23 ± 0.3 | 83 ± 0.8 | 31 ± 0.6 | 26 ± 0.7 | 1.2 ± 0.5 | 0.40 ± 0.02 | 35 ± 0.3 | 5.8 ± 0.1 |
| Element recoveryf | 93% | 87% | 107% | 103% | 96% | 125% | 120% | 83% | 91% |
Fig. 1 shows μ-SXRF elemental maps in the shale thin section. Previous studies have identified iron-bearing minerals as a major source of trace metals.16,31 Therefore, we plotted correlation plots of Fe with other elements to identify which elements were associated with iron solids. The elements which were positively associated with Fe are Cu (r = 0.687), Ni (r = 0.738), Mn (r = 0.847), Cr (r = 0.716), PbAs (r = 0.734), and S (r = 0.687) and their correlation plots with Fe are shown in Fig. 2. Regions exhibiting a positive correlation between iron and sulfur indicate two trends of our data, which might represent two different forms of Fe–S solids. Trends with high S and low Fe (marked as trend 1 in Fig. 2) likely correspond to grains of pyrite. The trend with high Fe, low S (marked as trend 2 in Fig. 2) may correspond to numerous Fe(II)-bearing solids; however, it was unclear whether there were differences in the trace metal concentrations in these areas relative to the high S and low Fe. This positive association of As, Pb, Cu, Ni, S, and Mn associated with Fe bearing solids suggests that the solubility of iron minerals will strongly affect the mobility of these trace metals in shale.
 | ||
| Fig. 1 μ-SXRF maps of metals within shale. The scale bar in these maps is 20 μm. Regions in blue indicate low concentrations of elements relative to regions in red containing higher concentrations. | ||
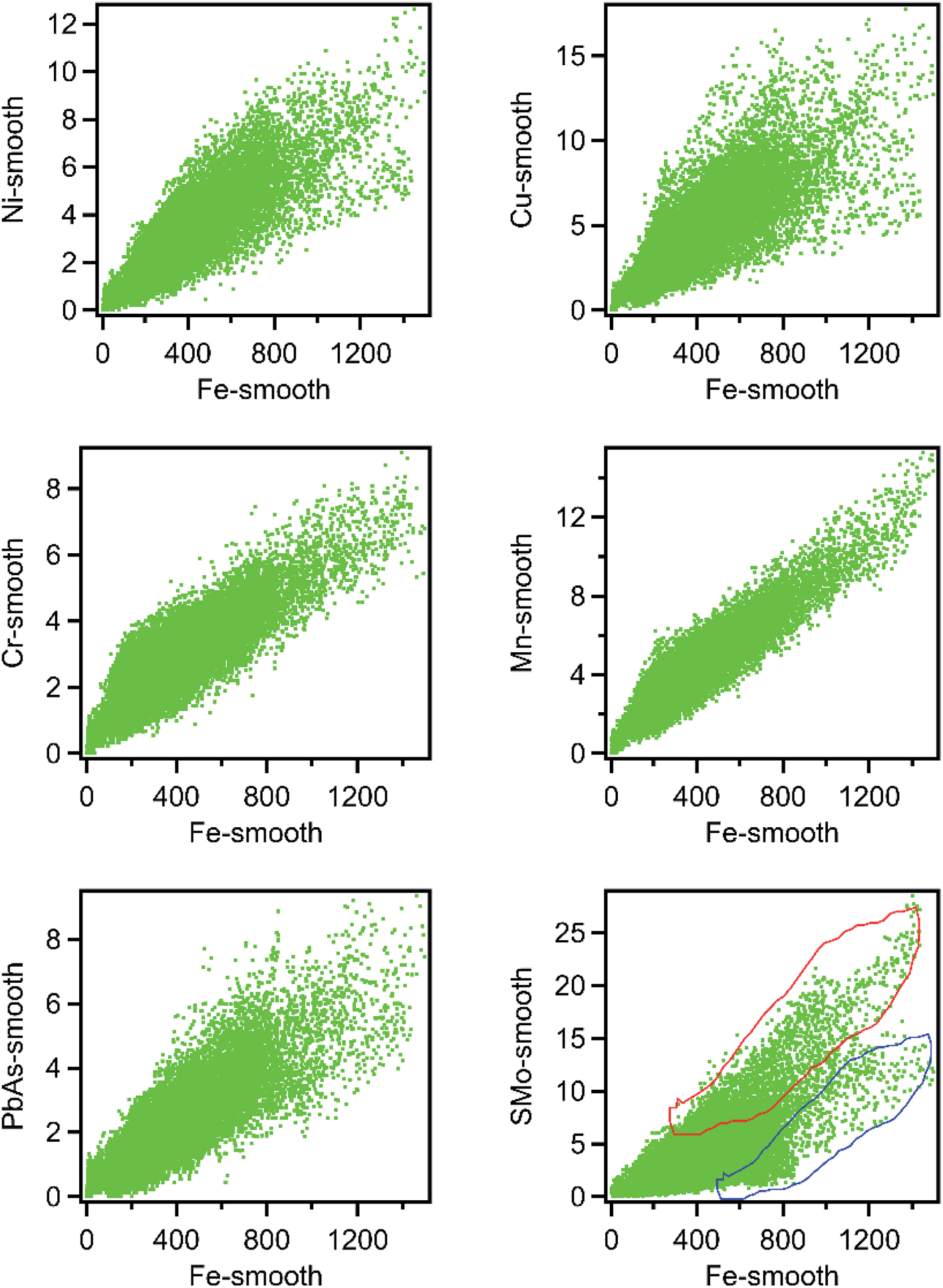 | ||
| Fig. 2 Correlation plots of Ni, Cu, Cr, PbAs, SMo, and Mn with Fe in shale thin section. The two trends in Fe–S minerals are marked with a red polygon (trend 1) and a blue polygon (trend 2). | ||
3.2. Sequential extractions
The behavior of different elements to the sequential extraction solutions varied based on chemical affinities and mineral phase(s) extracted by the solution (Fig. 3). Of the alkaline earth elements, Ca, Ba, and Sr showed large variability in their distribution across extracted fractions. Calcium is held primarily in dilute acid fractions, likely in carbonate phases. For reasons unknown, the sum of dilute acid and exchangeable fraction for Ca and Sr exceeded the total Ca and Sr concentration measured in shale. There is a small percentage of Ba (10%), Co (12%) and Sr (5%) also associated with carbonate minerals as inferred by the dilute acid extract. Nearly 70% of Ba is extracted in the residual fraction, whereas Sr is primarily either in the exchanged fraction or oxidized fraction. This suggests that a large fraction of Ba is tied up in the form of barite, which is not significantly dissolved by the extraction fluids used here, consistent with previously reported Ba speciation in the shale.30 On the other hand, Sr and Ca appear to have a strong affinity for charged surfaces and are more readily released to solution in comparison to Ba.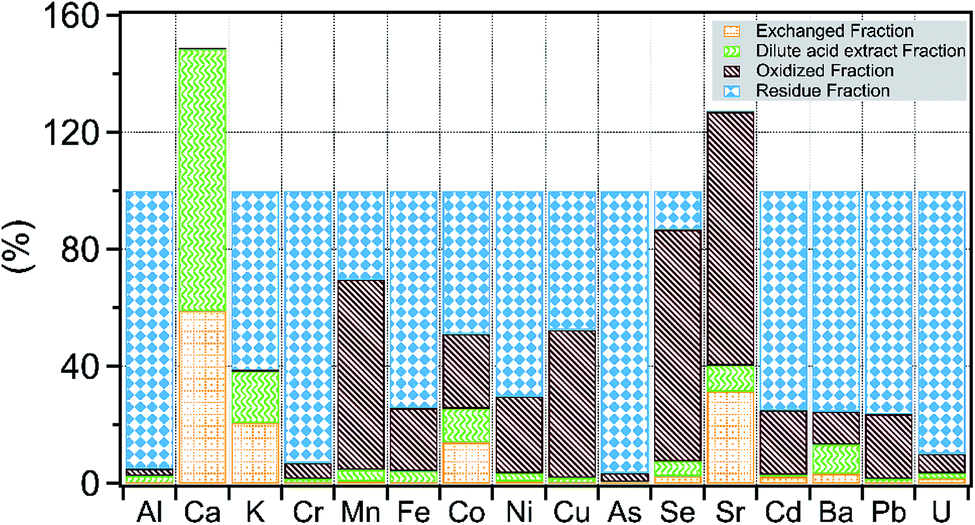 | ||
| Fig. 3 Distribution of major and trace elements in sequentially extracted fractions of composite shale sample. | ||
The remaining elements were distributed in either an oxidized fraction or residual fraction. The highest residual fraction was found for Cr, As and U (an average of 93%) followed by Fe, Ni, Ba, Cd and Pb (average of 74%). The oxidized fraction was highest for Sr (86%), and Se (80%) followed by Mn (65%). Based on the sequential leaching trends, it appears likely that organic matter and iron cycling reactions are the main scavengers and sources of Cr, Cu, As, Se, Cd, and U in shale.
3.3. Effect of pH, oxic–anoxic conditions and iron cycling reactions
The impact of solution chemistry parameters on the release of metals under each treatment is shown in Fig. 4–6 and Table S2.† At circumneutral pH, anoxic conditions increased dissolved Cd, Cu, and U, whereas they reduced dissolved As and Se by 50% and 35% respectively (Fig. 4A). There was no measurable mobilization of Fe and Cr at circumneutral pH under oxic or anoxic conditions in low ionic strength experiments (Table S2†). Among divalent ions, at circumneutral pH, anoxic conditions resulted in an increase of Sr concentration by 30% in comparison to oxic conditions (Table S2†), whereas, Ba concentration in leachates remained unaffected by oxic or anoxic conditions. Fig. 4B and C show the effect of pH on low ionic strength treatments. Low pH under oxic conditions resulted in high concentrations of As, Se, Cd, Ba and U in leachates (Fig. 4B). Anoxic conditions superimposed with low pH increased dissolved concentrations of all the selected metals. Moreover, Fe and Cr exhibited the highest relative change in concentration upon a decrease in pH; dissolved concentration of Fe and Cr increased by 100-fold to 481 ± 66 μg g−1 and to 0.44 ± 0.03 μg g−1 respectively in E4-anox-DI-pH4.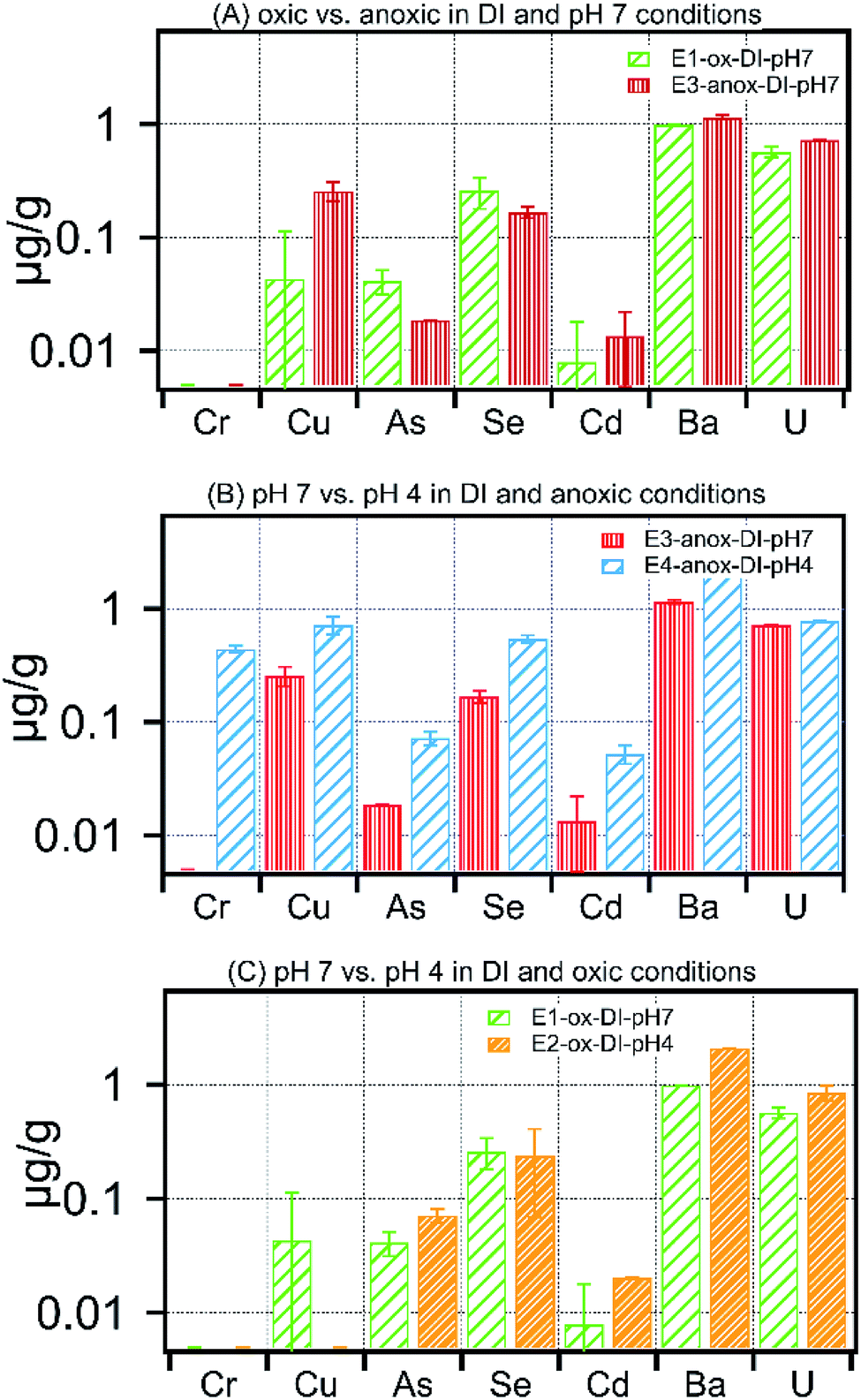 | ||
| Fig. 4 Concentration of metals released in (A) oxic vs. anoxic in DI water and pH 7 treatment, (B) pH 7 vs. pH 4 in DI water and anoxic treatment, (C) pH 7 vs. pH 4 in DI water and oxic treatments. | ||
The solution chemistry in a shale–water system is affected by a myriad of interactions, but here we will focus on redox transformations of Fe-bearing solids present in shale such as pyrite, and iron in phyllosilicate minerals, and dissolution of carbonate minerals. Both these mineral matrixes also contain the majority of elements within them as evident in sequential extractions (Fig. 3). In the presence of dissolved O2, pyrite undergoes oxidative dissolution in accordance with eqn (1) and (2), releasing metals contained within to solution.32,33 These reactions may occur during the initial stage of the hydraulic fracturing process when an oxic fluid is injected into a shale formation.20 This explains the observed increase in metal concentration in leachates of oxic treatment.
| FeS2 + 3.5O2 + H2O = Fe2+ + 2SO42− + 2H+ | (1) |
| FeS2 + 3.75O2 + 0.5H2O = Fe3+ + 2SO42− + H+ | (2) |
Simultaneously, pyrite oxidation generates acidity (eqn (1)) which decreases pore water pH. If abundant carbonate minerals are present in shale, acidity generated during the oxidative dissolution of pyrite could be neutralized by the dissolution of calcite and other carbonate minerals (e.g. dolomite). Each mole of pyrite requires 4 moles of calcite to achieve complete neutralization.14 Here, Marcellus shale has a pyrite-to-calcite molar ratio of 1.3 (Table 3), which means that there's not sufficient calcite to achieve complete neutralization. Compared to shale formations containing abundant carbonate-bearing minerals (e.g. calcite, and dolomite), pore water acidification may impart greater control on metalloid mobilization in Marcellus shale. For example, lower total metal concentrations were reported to be leached from high carbonate shale in comparison to those with low mineral carbonate content.34
Leaching of carbonate-poor Marcellus shale with low pH fluids would further favor an increase in pore water metal concentration as evident in Fig. 4B and C. The total metal concentration within the low pH (4.0) leaching fluid equilibrated with Marcellus shale is substantially higher than the same material equilibrated with pH 7.0 leaching fluid; arsenic, for example, is 40% lower at pH 7.0 (E1-ox-DI-pH7) than pH 4.0 (E2-ox-DI-pH4). The current paradigm of fracturing a well involves the injection of HCl prior to injection of the hydraulic fracturing fluid.19 Depending on the residual acid, and pyrite-to-carbonate ratio, the pH of the borehole fluid could range anywhere between 2 and 8.19 For carbonate-low Marcellus shale, use of injection fluid buffered at 7 or likely higher may provide additional buffering capacity against porewater acidification and residual acid, resulting in reduced pollutant loading in produced water. To realize this modification at a hydraulic fracturing site, it might require appropriate addition of scale inhibitors and iron control agents (e.g. phosphonic acid, thioglycolic acid)27 to suppress mineral scale formation triggered by the alkaline pH of the injection fluid.
We note that the pH of the leachates was not measured. Based on the previous studies monitoring pH of the unbuffered solution in contact with Marcellus shale, the pH of leachates drifts approximately by ∼2 units from the initial solution pH due to the interplay between the acid generating component (pyrite dissolution) and acid buffering component (calcite dissolution).16,20 The change in solution pH reported in previous studies is not sufficient to destroy the buffering capacity of the leaching fluids associated with our treatments. Therefore, we choose to not measure the final pH of the solution.
Oxidative dissolution of pyrite is commonly accompanied by precipitation of secondary Fe(III)-(oxy)hydroxide phase(s).35 A recent study using synchrotron-based spectroscopic techniques demonstrated that various poorly crystalline Fe(III)-bearing phases and mixed valence Fe-bearing solids including ferrihydrite, lepidocrocite, and goethite, precipitated in shale after their treatment with oxygenated synthetic fracture fluid.31 Depending on the pH and redox conditions, these newly formed Fe(III)-bearing secondary phases are excellent sorbents of metals such as As, Cu, Cr, Ni etc.36 For example, Cr(III) readily combines with amorphous iron-(oxy)-hydroxides to form a sparingly soluble solid solution (CrxFe(1−x)(OH)3), thereby removing Cr from solution.36 Hence, it is conceivable that similar processes may occur in batch treatments. Low (below detection) aqueous Cr concentrations in oxic treatments support the occurrence of these processes (Fig. 4C).
3.4. Effect of ionic strength on metal release
Clay minerals impart an overarching control on the retention and release of metals within shales. These minerals commonly include 2:1 layered clays such as illite, and smectite that are dominant in Marcellus shale,14 and which act as repositories for exchangeable cations, including Ba, and Sr.37,38 These exchangeable cations associated with phyllosilicate clay minerals are readily displaced in the presence of high ionic strength solutions and explains the overall increased metal concentration in leachates of high ionic strength treatments (Fig. 5). The effect of increased ionic strength on the release of Ba and Sr is of particular interest. Measured Ba concentration in high ionic strength fluids ranged between 5 and 6 μg g−1 across E1-Sal to E4-Sal treatments, which is 5-fold higher compared to low ionic strength treatments (Fig. 4 and 5). The solution chemistry had no significant effect on Ba release in high and low ionic strength treatments. Similarly, compared to low ionic strength treatments, the Sr concentration was 1.5 times more in high ionic strength treatment (Table S2†), with the highest concentration recorded in E4-anox-Sal-pH4 (Fig. 5D–F and Table S2†). The high concentration of Ba and Sr in solution could result in the formation of minerals such as barite (BaSO4), and celestite (SrSO4), which are common components of scale. The increase in Ba and Sr concentrations in leachates of high ionic strength treatments implies that in the presence of sulfate ions, the solution may rapidly reach oversaturation with respect to BaSO4 and SrSO4, resulting in precipitation of barite and celestite. Common sources of sulfate in the shale–water system include persulfate decomposition and oxidation of sulfide minerals preset in shale. Barite and celestite precipitation could contribute to the formation of scale, and thereby limit and reduce well productivity by plugging the formation matrix. In comparison to Ba and Sr, the effect of ionic strength on the mobility of U, Se, As, Cr, Cu, and Cd was negligible, consistent with their sequential extraction data.
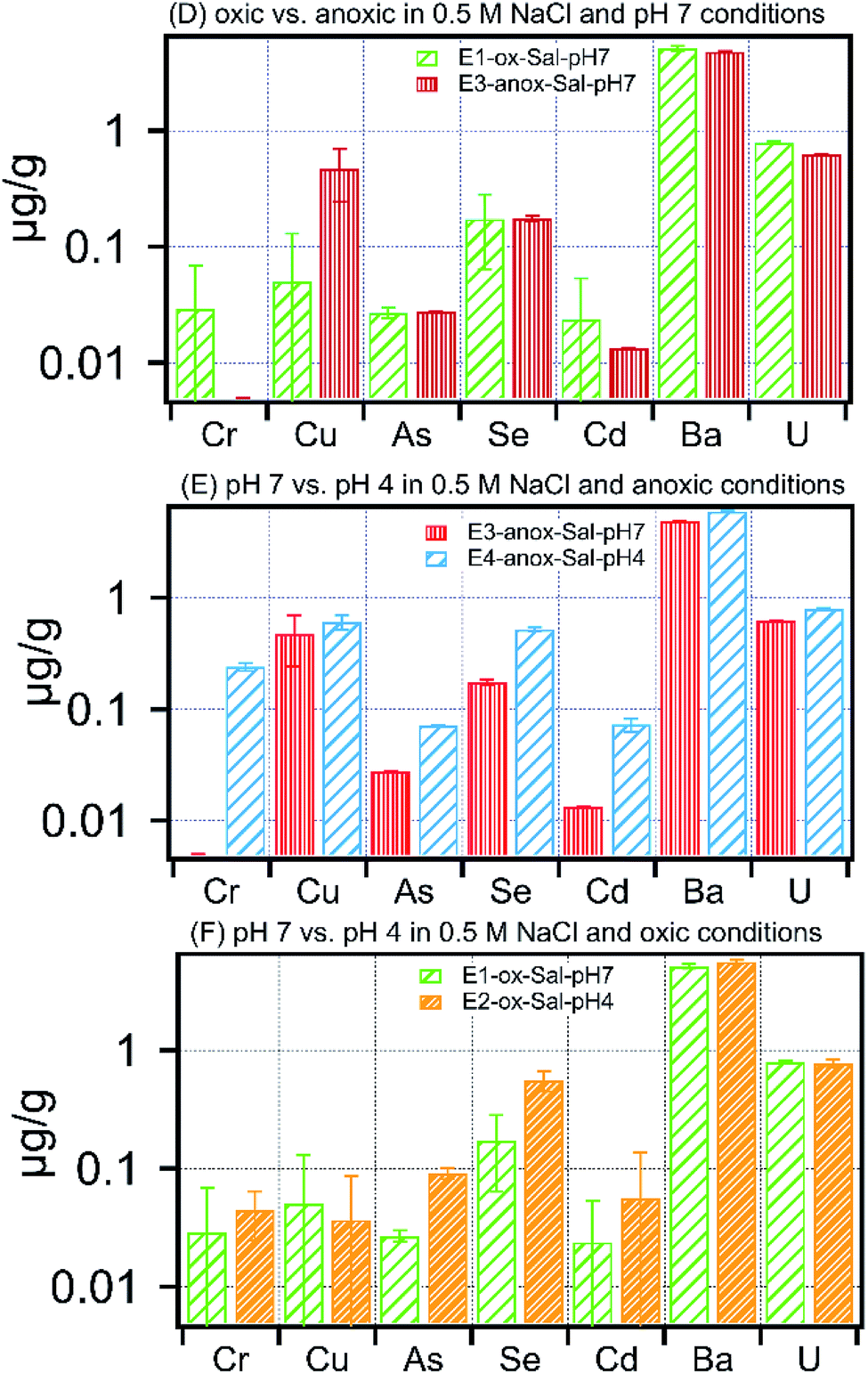 | ||
| Fig. 5 Concentration of metals released in (A) oxic vs. anoxic in 0.5 M NaCl and pH 7 treatment, (B) pH 7 vs. pH 4 in 0.5 M NaCl and anoxic treatment, (C) pH 7 vs. pH 4 in 0.5 M NaCl and oxic treatments. | ||
3.5. Effect of oxidant on metal release
With the exception of Ba, the concentrations of other elements in leachates of E5 treatment (containing persulfate) was greatest across all batch treatments (Fig. 6 and Table S2†). Highly reactive species such as SO4−˙ and HO˙ are generated upon decomposition of sodium persulfate.29,39 These species are effective in degrading complex organic compounds and oxidize sulfide minerals present in shale.39 Consequently, such degradation of shale organic matter and sulfide minerals may mobilize metals and other contaminants complexed within these matrixes to solution,40 consistent with the measured high concentrations of metals (except Ba) in the leachate of the E5 treatment. Previous studies (e.g.ref. 28) have shown that the solution pH is drastically reduced upon sodium persulfate oxidation. Low pH conditions are not only favorable to maintain trace metals in solution but also suppress precipitation of iron-(oxy)-hydroxides formed during oxidation of pyrite by persulfate, resulting in the observed high Fe concentration in E5 treatment. Among other products of persulfate decomposition is sulfate anion, which may induce the formation of barite and explain the observed low concentrations of Ba measured in E5 treatment. Aqueous sulfate concentrations could also result in the formation of soluble uranium–sulfate complexes (such as UO2SO04 and UO2(SO4)2+),41,42 and likely result in the measured high mobility of U in E5 treatment (Fig. 6). Additionally, oxidation of uraninite may also result in a high concentration of U in E5 treatment.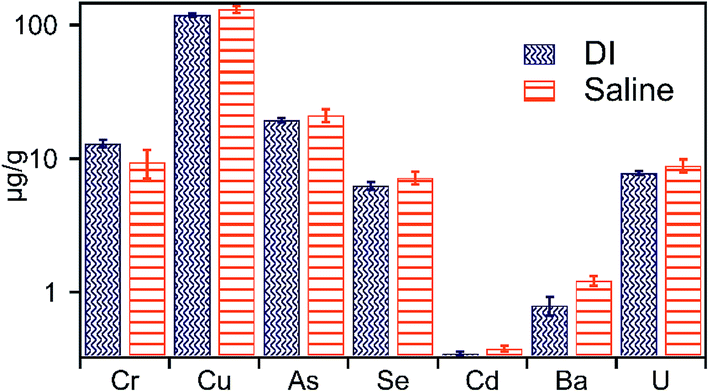 | ||
| Fig. 6 Concentration of metals liberated in treatment containing 0.5 M sodium persulfate, an oxidative, and commonly used fracture fluid additive. | ||
3.6. Bulk radioactivity and radium mobilization
Black shale of marine origin such as Marcellus shale often contains an appreciable U content of about 20 ppm in comparison to other sedimentary rocks (average U content of 3–4 ppm).43 Here, shale had a U content of 15 ppm as measured on ICP-MS (Table 3). The bulk distribution of radionuclides within shale appeared uniformly dispersed as shown in the autoradiograph (Fig. 7). The total activity of 226Ra (a decay product of 238U), in shale, was measured to be 200 ± 2.5 Bq kg−1. The 226Ra activity measured here is consistent with the previously reported 226Ra activity in Marcellus shale (50–500 Bq kg−1).44 | ||
| Fig. 7 Autoradiographs of shale, illustrating spatial bulk radionuclide distribution within the shale studied here. The area of analysis for the sample spans ∼10 × 10 cm. | ||
The radioactivity of produced water is often elevated, primarily due to high concentrations of Ra isotopes. Generally, produced water contains total dissolved Ra (226Ra and 228Ra) ranging between 37 and 555 Bq L−1, whereas dissolved U concentration is typically low, ranging between 0.084 μg L−1 and 3.26 μg L−1.5,18 For comparison, the U.S. EPA maximum contaminant level for drinking water is 0.2 Bq L−1 total dissolved Ra and 30 μg L−1 dissolved U. Here, sequential extraction data show that approximately 60% of the total 226Ra was extracted in the exchanged fraction, likely adsorbed to clay minerals, organic matter and Fe-solids38 (Fig. 8). Nearly 40% of total Ra was present within the dilute acid fraction and oxidized fraction combined, suggesting that carbonates, organic matter, and sulfide minerals are important host matrices of Ra in shale (Fig. 8).
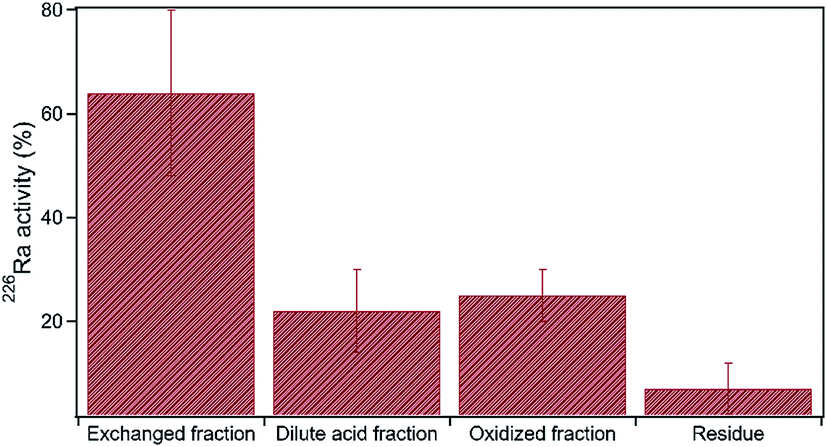 | ||
| Fig. 8 Sequential extraction data of 226Ra in shale. The y-axis shows the percentage of total metal extracted within each extraction step. Error bars denote the standard deviation, n = 3. | ||
In the batch leaching treatments, 226Ra activity in solution varied anywhere between 0 and 3 Bq kg−1 (Fig. 9). Compared to oxic and low pH treatments, increased dissolved activities of 226Ra were measured under anoxic and low pH conditions (Fig. 9). At high pH (=7), no difference was observed in Ra activity in oxic and anoxic treatments. Adsorption of Ra is also influenced by ionic strength. For example, several studies have shown increased mobilization of 226Ra from clays such as kaolinite and montmorillonite in the presence of a high concentration of NaCl.45 Here, an increase in the ionic strength of the leaching fluid had no pronounced effect on Ra activity in leachates, except for E1 treatment. Compared to low ionic strength treatment, high Ra activity was measured in leachates of E1-ox-Sal-pH7 (Fig. 9). Furthermore, in treatments containing sodium persulfate, (E5), negligible 226Ra activity was detected irrespective of solution ionic strength. Simultaneously Ba concentration in the leachate of E5 treatment was low (Fig. 6). Although no XRD analysis was performed on the solid residue from E5 treatments, based on the low Ba solubility in leachates of E5 treatment and previously reported incorporation of Ra in barite,46 we conclude that Ra is most likely scavenged from E5 leachates due to incorporation into barite. Combining sequential extraction data and batch leaching treatments, it is apparent that the majority of Ra in shale is concentrated on exchangeable sites in minerals such as clay and Fe-solids and is readily mobilized to solution irrespective of ionic strength.
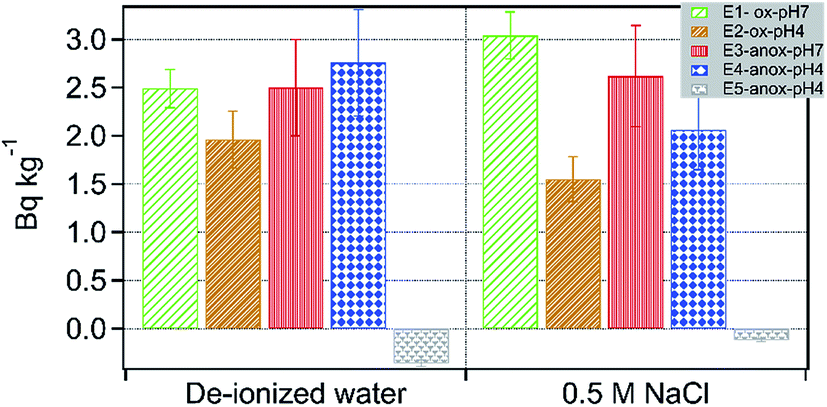 | ||
| Fig. 9 Specific activity of 226Ra released in various batch treatments. | ||
In addition to geochemical controls on Ra mobility in pore water, the physical process of alpha-recoil is an equally important mechanism for supplying Ra to pore water, especially in the context of hydraulic fracturing.44,47 Alpha recoil is a physical process of displacement of a radioactive decay product from its initial position in a crystal lattice of a mineral grain because of energy gained during alpha-decay. The distance traversed by the recoiled Ra atoms (referred to as recoil length) is approximately 30 nm (in quartz). Owing to the small magnitude of recoil range in comparison to typical grain diameters (ranging from mm to μm), only 226Ra atoms produced by the decay of U atoms located within a distance less than 226Ra recoil range from grain surface will have a non-zero probability of escaping the pore–grain boundary. The recoiled Ra atoms produced in the solid at a distance greater than their recoil range remain embedded in the grain. The fracture network generated during hydraulic fracturing substantially increases the surface area of the shale intercepted by the injected fluid exposing a larger number of Ra atoms closer to the grain–water boundary, which otherwise would remain embedded in the solid unable to escape the grain–water boundary. Hence, it is conceivable that the elevated supply of Ra to produced water is a direct consequence of physical structure modification induced in shale by hydraulic fracturing.
4. Conclusions
Management of large volumes of polluted produced water is one of the key environmental challenges being faced by the unconventional oil and gas industry. Various above surface wastewater treatment technologies are effective in reducing contaminant concentration in produced water but are limited in their application due to the high treatment cost.48 Alternatively, immobilizing contaminants in situ could provide a unique approach for reducing pollutant loading in produced water and thereby reduce environmental concerns associated with produced water disposal. To inform such approaches, this study focuses on understanding geochemical controls on processes responsible for mobilizing hazardous pollutants to produced water.Our findings show that anoxic conditions superimposed with acidic pH increased dissolved concentrations of all the selected metals. At circumneutral pH, anoxic conditions significantly increased the aqueous concentrations of Cd, Cu, and U, whereas they decreased As and Se dissolved concentrations by 50% and 35% respectively. Therefore, injection of oxic and alkaline injection fluid is likely to favor retention of produced water contaminants in situ. Radium mobility was enhanced under low pH and anoxic conditions, implying that maintaining alkaline pH conditions in situ may reduce the radioactivity of produced water. Our data also suggest that elevated concentration of sulfate ion is conducive for Ra retardation in situ due to incorporation into barite solids, but, at the same time, may impact well productivity by contributing to scale formation. Thus, controlling injected solution pH (and buffering) may serve as a more favorable strategy for limiting produced water radioactivity and not compromising well productivity. A lesser studied control on Ra mobility is the effect of physical alteration of shale during hydraulic fracturing on recoil supply of Ra. The controls on Ra recoil supply are independent of solution geochemistry and instead depend on the spatial distribution of the parent nuclide in aquifer solids, the microstructure of porous medium, and decay kinetics.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank John Neubaum (DCNR-Bureau of Topographic and Geological Survey, Harrisburg, Pennsylvania) for providing Marcellus shale cores, Charlie Settens (MIT, Cambridge) for his assistance with X-ray Diffraction analysis, and Bogdan I Fedeles (CEHS, MIT) for his help in using autoradiography, and SSRL scientists for their help with acquiring synchrotron-based data. This work was supported by the MIT energy initiative (MITEI). We would also like to thank the anonymous reviewers for their valuable comments and insight, which helped improve the paper.References
- R. D. Vidic, S. L. Brantley, J. M. Vandenbossche, D. Yoxtheimer and J. D. Abad, Impact of Shale Gas Development on Regional Water Quality, Science, 2013, 340, 1235009–1235018 CrossRef CAS PubMed.
- U.S. EPA (U.S. Environmental Protection Agency), Hydraulic Fracturing for Oil and Gas: Impacts from the Hydraulic Fracturing Water Cycle on Drinking Water Resources in the United States. Executive Summary, EPA/600/R-16/236ES, Office of Research and Development, Washington, DC, 2016 Search PubMed.
- I. Ferrer and E. M. Thurman, Chemical constituents and analytical approaches for hydraulic fracturing waters, Trends Environ. Anal. Chem., 2015, 5, 18–25 CrossRef CAS.
- N. Mehta and F. O. Sullivan, in Food, Energy, and Water, ed. S. Ahuja, Elsevier, Boston, 1st edn, 2015, pp. 217–241 Search PubMed.
- E. L. Rowan, M. A. Engle, C. S. Kirby and T. F. Kraemer, Radium Content of Oil and Gas Field Produced Waters in the Northern Appalachian Basin (USA): Summary of Data, U.S. Geological Survey Scientific Investigations Report 2011-5135, Reston, VA, 2011 Search PubMed.
- L. O. Haluszczak, A. W. Rose and L. R. Kump, Geochemical evaluation of flowback brine from Marcellus gas wells in Pennsylvania, USA, Appl. Geochem., 2013, 28, 55–61 CrossRef CAS.
- H. Thomas, Sampling and Analysis of Water Streams Associated with the Development of Marcellus Shale Gas, Gas Technology Institute, Des Plaines, IL, 2009 Search PubMed.
- E. C. Chapman, R. C. Capo, B. W. Stewart, C. S. Kirby, R. W. Hammack, K. T. Schroeder and H. M. Edenborn, Geochemical and Strontium Isotope Characterization of Produced Waters from Marcellus Shale Natural Gas Extraction, Environ. Sci. Technol., 2012, 46, 3545–3553 CrossRef CAS.
- B. C. Gordalla, U. Ewers and F. H. Frimmel, Hydraulic fracturing: A toxicological threat for groundwater and drinking-water?, Environ. Earth Sci., 2013, 70, 3875–3893 CrossRef CAS.
- N. R. Warner, C. A. Cidney, R. B. Jackson and A. Vengosh, Impacts of Shale Gas Wastewater Disposal on Water Quality in Western PA, Environ. Sci. Technol., 2013, 1–14 Search PubMed.
- R. B. Jackson, A. Vengosh, J. W. Carey, R. J. Davies, T. H. Darrah, F. O'Sullivan and G. Pétron, The Environmental Costs and Benefits of Fracking, Annu. Rev. Environ. Resour., 2014, 39, 327–362 CrossRef.
- K. B. Gregory, R. D. Vidic and D. A. Dzombak, Water Management Challenges Associated with the Production of Shale Gas by Hydraulic Fracturing, Elements, 2011, 7, 181–186 CrossRef.
- S. Entrekin, M. Evans-White, B. Johnson and E. Hagenbuch, Rapid expansion of natural gas development poses a threat to surface waters, Front. Ecol. Environ., 2011, 9, 503–511 CrossRef.
- J. A. Chermak and M. E. Schreiber, Mineralogy and trace element geochemistry of gas shales in the United States: Environmental implications, Int. J. Coal Geol., 2014, 126, 32–44 CrossRef CAS.
- T. L. Tasker, P. K. Piotrowski, F. L. Dorman and W. D. Burgos, Metal Associations in Marcellus Shale and Fate of Synthetic Hydraulic Fracturing Fluids Reacted at High Pressure and Temperature, Environ. Eng. Sci., 2016, 33, 753–765 CrossRef CAS.
- A. L. Harrison, A. D. Jew, M. K. Dustin, D. L. Thomas, C. M. Joe-Wong, J. R. Bargar, N. Johnson, G. E. Brown and K. Maher, Element release and reaction-induced porosity alteration during shale-hydraulic fracturing fluid interactions, Appl. Geochem., 2017, 82, 47–62 CrossRef CAS.
- L. Wang, J. D. Fortner and D. E. Giammar, Impact of Water Chemistry on Element Mobilization from Eagle Ford Shale, Environ. Eng. Sci., 2015, 32, 310–320 CrossRef.
- T. T. Phan, R. C. Capo, B. W. Stewart, J. R. Graney, J. D. Johnson, S. Sharma and J. Toro, Trace metal distribution and mobility in drill cuttings and produced waters from Marcellus Shale gas extraction: Uranium, arsenic, barium, Appl. Geochem., 2015, 60, 89–103 CrossRef CAS.
- A. N. P. Vankeuren, J. A. Hakala, K. Jarvis, J. E. Moore, W. Virginia, U. States, A. N. Paukert Vankeuren, J. A. Hakala, K. Jarvis and J. E. Moore, Mineral Reactions in Shale Gas Reservoirs: Barite Scale Formation from Reusing Produced Water As Hydraulic Fracturing Fluid, Environ. Sci. Technol., 2017, 51, 9391–9402 CrossRef PubMed.
- V. Marcon, C. Joseph, K. E. Carter, S. W. Hedges, C. L. Lopano, G. D. Guthrie and J. A. Hakala, Experimental insights into geochemical changes in hydraulically fractured Marcellus Shale, Appl. Geochem., 2017, 76, 36–50 CrossRef CAS.
- J. D. Landis, M. Sharma and D. Renock, Rapid desorption of radium isotopes from black shale during hydraulic fracturing. 2. A model reconciling radium extraction with Marcellus wastewater production, Chem. Geol., 2018, 500, 194–206 CrossRef CAS.
- United States Environmental Protection Agency Method 3052, Microwave Assisted Acid Digestion of Siliceous and Organically Based Matrices, available at https://www.epa.gov/sites/production/files/2015-12/documents/3052.pdf, 1996.
- V. Cappuyns, R. Swennen and M. Niclaes, Application of the BCR sequential extraction scheme to dredged pond sediments contaminated by Pb–Zn mining: A combined geochemical and mineralogical approach, J. Geochem. Explor., 2007, 93, 78–90 CrossRef CAS.
- R. A. Lebas, T. W. Shahan, P. Lord and D. Luna, in SPE Hydraulic Fracturing Technology Conference, Society of Petroleum Engineers, Woodlands, Texas, USA, 2013 Search PubMed.
- D. S. Alessi, A. Zolfaghari, S. Kletke, J. Gehman, D. M. Allen and G. G. Goss, Comparative analysis of hydraulic fracturing wastewater practices in unconventional shale development: Water sourcing, treatment and disposal practices, Can. Water Resour. J., 2017, 42, 105–121 CrossRef.
- H. Thomas, Sampling and Analysis of Water Streams Associated with the Development of Marcellus Shale Gas, Final report prepared for the Marcellus Shale Coalition, 2009 Search PubMed.
- W. T. Stringfellow, J. K. Domen, M. K. Camarillo, W. L. Sandelin and S. Borglin, Physical, chemical, and biological characteristics of compounds used in hydraulic fracturing, J. Hazard. Mater., 2014, 275, 37–54 CrossRef CAS PubMed.
- K. C. Huang, Z. Zhao, G. E. Hoag, A. Dahmani and P. A. Block, Degradation of volatile organic compounds with thermally activated persulfate oxidation, Chemosphere, 2005, 61, 551–560 CrossRef CAS PubMed.
- D. Zhao, X. Liao, X. Yan and S. G. Huling, Effect and mechanism of persulfate activated by different methods for PAHs removal in soil, J. Hazard. Mater., 2013, 254–255, 228–235 CrossRef CAS.
- D. Renock, J. D. Landis and M. Sharma, Reductive weathering of black shale and release of barium during hydraulic fracturing, Appl. Geochem., 2016, 65, 73–86 CrossRef CAS.
- A. D. Jew, M. K. Dustin, A. L. Harrison, C. M. Joe-Wong, D. L. Thomas, K. Maher, G. E. Brown and J. R. Bargar, Impact of Organics and Carbonates on the Oxidation and Precipitation of Iron during Hydraulic Fracturing of Shale, Energy Fuels, 2017, 31, 3643–3658 CrossRef CAS.
- W. Salomons, Environmental impact of metals derived from mining activities: Processes, predictions, prevention, J. Geochem. Explor., 1995, 52, 5–23 CrossRef CAS.
- D. D. Rimstidt and D. J. Vaughan, Pyrite oxidation: A state-of-the-art assessment of the reaction mechanism, Geochim. Cosmochim. Acta, 2003, 67, 873–880 CrossRef.
- A. S. Jeng, Weathering of Some Norwegian Alum Shales, II. Laboratory Simulations to Study the Influence of Aging, Acidification and Liming on Heavy Metal Release, Acta Agric. Scand., Sect. B, 1992, 42, 76–87 CAS.
- D. K. Nordstrom, in Acid Sulfate Weathering, ed. J. A. Kittrick, D. S. Fanning and L. R. Hossner, SSSA Spec. Publ. 10. SSSA, Madison, WI, 1982, pp. 37–56 Search PubMed.
- A. Kappler and K. L. Straub, Geomicrobiological Cycling of Iron, Rev. Mineral. Geochem., 2005, 59, 85–108 CrossRef CAS.
- G. Atun and E. Bascetin, Adsorption of barium on kaolinite, illite and montmorillonite at various ionic strengths, Radiochim. Acta, 2003, 91, 223–228 CAS.
- T. Missana, E. Colàs, F. Grandia, J. Olmeda, M. Mingarro, M. García-Gutiérrez, I. Munier, J. C. Robinet and M. Grivé, Sorption of radium onto early cretaceous clays (Gault and Plicatules Fm). Implications for a repository of low-level, long-lived radioactive waste, Appl. Geochem., 2017, 86, 36–48 CrossRef CAS.
- K. C. Huang, R. A. Couttenye and G. E. Hoag, Kinetics of Heat-Assisted Persulfate Oxidation of Methyl tert-Butyl Ether (MTBE), Chemosphere, 2002, 11, 447–448 Search PubMed.
- M. Wengel, E. Kothe, C. M. Schmidt, K. Heide and G. Gleixner, Degradation of organic matter from black shales and charcoal by the wood-rotting fungus Schizophyllum commune and release of DOC and heavy metals in the aqueous phase, Sci. Total Environ., 2006, 367, 383–393 CrossRef CAS PubMed.
- D. Gorman-lewis, P. C. Burns and J. B. Fein, Review of uranyl mineral solubility measurements, J. Chem. Thermodyn., 2008, 40, 335–352 CrossRef CAS.
- C. M. Zammit, J. Brugger, G. Southam and F. Reith, In situ recovery of uranium – The microbial influence, Hydrometallurgy, 2014, 150, 236–244 CrossRef CAS.
- J. S. Leventhal, Comparison of organic geochemistry and metal enrichment in two black shales: Cambrian Alum Shale of Sweden and Devonian Chattanooga Shale of United States, Miner. Deposita, 1991, 26, 104–112 CAS.
- E. S. Eitrheim, D. May, T. Z. Forbes and A. W. Nelson, Disequilibrium of Naturally Occurring Radioactive Materials (NORM) in Drill Cuttings from a Horizontal Drilling Operation, Environ. Sci. Technol. Lett., 2016, 3, 425–429 CrossRef CAS.
- M. Chen and B. Kocar, Radium Sorption to Iron (hydr)oxides, Pyrite, and Montmorillonite: Implications for Ra Mobility, Environ. Sci. Technol., 2018, 52, 4023–4030 CrossRef CAS PubMed.
- T. Zhang, K. Gregory, R. W. Hammack and R. D. Vidic, Co-precipitation of radium with barium and strontium sulfate and its impact on the fate of radium during treatment of produced water from unconventional gas extraction, Environ. Sci. Technol., 2014, 48, 4596–4603 CrossRef CAS PubMed.
- N. Mehta and B. Kocar, Deciphering and Predicting Microscale Controls on Radon Production in Soils, Sediments and Rock, Soil Syst., 2018, 2, 1–14 CrossRef.
- T. Liden, I. C. Santos, Z. L. Hildenbrand and K. A. Schug, Treatment modalities for the reuse of produced waste from oil and gas development, Sci. Total Environ., 2018, 643, 107–118 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00244h |
| This journal is © The Royal Society of Chemistry 2019 |