
Aqueous film forming foam and associated perfluoroalkyl substances inhibit methane production and Co-contaminant degradation in an anaerobic microbial community†

Abstract
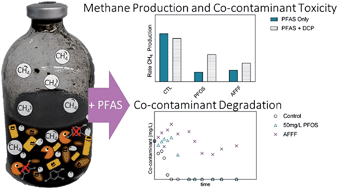
Aqueous film forming foams (AFFF) can contain gram per liter concentrations of per- and polyfluoroalkyl substances (PFAS) and are often released in large quantities directly to the environment as they are used to fight fires. AFFF composition is complex and contains many unknown PFAS in addition to ingredients such as hydrocarbons, solvents, and corrosion inhibitors. While biological effects of single PFAS have been studied, the effects of PFAS-containing mixtures, such as AFFF, are unknown. The effect of PFAS on microorganisms is also not well understood; nevertheless, we rely on microorganisms in locations containing elevated PFAS concentrations to perform certain functions, such as carbon cycling and co-contaminant degradation. This study focused on determining the functional consequences of AFFF and PFAS exposure in a microbial community in both the presence and the absence of a co-contaminant. AFFF, select PFAS, and a PFAS mixture were tested to determine the effect of AFFF on an anaerobic microbial community and the characteristics of the PFAS that drive toxicity in such mixtures. To study this, anaerobic digester communities were exposed to PFAS and a co-contaminant (2,4-dichlorophenol, DCP); methane production, as an indicator of toxicity and the community's ability to cycle carbon, and co-contaminant degradation were monitored. Results showed that PFAS and AFFF can alter the toxicity of DCP, inhibit DCP degradation, decrease the number of methanogens present, and change the microbial community structure. DCP was also able to decrease the toxicity of the PFAS perfluorooctane sulfonate (PFOS), possibly by changing the sorption of PFOS to the microorganisms present. Additionally, it was determined that while PFOS was responsible for AFFF toxicity, no single PFAS or simple PFAS mixture accurately accounted for the inhibition of DCP degradation caused by AFFF exposure.
- This article is part of the themed collection: PFAS
 Please wait while we load your content...
Please wait while we load your content...

