
Closing the gap – inclusion of ultrashort-chain perfluoroalkyl carboxylic acids in the total oxidizable precursor (TOP) assay protocol†
 ‡a
Karsten
Nödler,
a
Marco
Scheurer,
a
Oliver
Happel,
a
Christian
Zwiener
b
and
Frank Thomas
Lange
*a
‡a
Karsten
Nödler,
a
Marco
Scheurer,
a
Oliver
Happel,
a
Christian
Zwiener
b
and
Frank Thomas
Lange
*a
Abstract
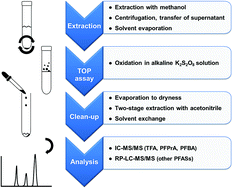
An improved protocol of the total oxidizable precursor (TOP) assay was developed for precursors to C2–C14 perfluoroalkyl carboxylic acids (PFCAs) and C4–C8 and C10 perfluoroalkyl sulfonic acids (PFSAs). The proposed protocol was tested and validated for contaminated soil samples. The perfluoroalkyl acids (PFAAs) present in the soil extract solutions after oxidation with persulfate were separated from the inorganic salts by vacuum-assisted drying of the digestion solution followed by solid–liquid extraction of the PFAAs with acetonitrile from the dry residue. Ion chromatography (for C2–C4 PFCAs) and reversed phase liquid chromatography (for all other PFASs), both coupled to tandem mass spectrometry, were used for quantification. High procedural recoveries of PFAAs between 68% and 123% with RSDs between 0.2% and 25% (n = 3) were achieved. The method was validated using selected polyfluoroalkyl phosphoric acid esters (PAPs) and bis-[2-(N-ethyl perfluorooctane-1-sulfonamido)ethyl] phosphoric acid ester (diSAmPAP) as model precursors in pure solutions and in the presence of soil matrix. The oxidation led to characteristic and reproducible PFCA patterns (in the case of PAPs) or PFOA (in the case of diSAmPAP) with total reaction yields between 92 ± 4% and 123 ± 13% (n = 3). The impact of the soil matrix on transformation yields was negligible. In a soil core from a PFAS-polluted agricultural site, precursors were concentrated in the upper 40 cm with long-chain precursors being prevalent. After oxidative digestion, the total molar PFAA-concentrations increased by factors of 1.6 to 5.0. More than 40 cm below ground precursors of TFAA, PFPrA and PFBA accounted for ∼50% of the reaction products, underlining the importance of their inclusion in mass balances based on the TOP assay.
- This article is part of the themed collection: PFAS
 Please wait while we load your content...
Please wait while we load your content...

