
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlobal transport of perfluoroalkyl acids via sea spray aerosol†
J. H.
Johansson‡
 *a,
M. E.
Salter‡
ab,
J. C.
Acosta Navarro
ab,
C.
Leck
bc,
E. D.
Nilsson
ab and
I. T.
Cousins
a
*a,
M. E.
Salter‡
ab,
J. C.
Acosta Navarro
ab,
C.
Leck
bc,
E. D.
Nilsson
ab and
I. T.
Cousins
a
aDepartment of Environmental Science and Analytical Chemistry, Stockholm University, 11418 Stockholm, Sweden. E-mail: jana.johansson@aces.su.se
bBolin Centre for Climate Research, 11418 Stockholm, Sweden
cDepartment of Meteorology, Stockholm University, 11418 Stockholm, Sweden
First published on 19th March 2019
Abstract
Perfluoroalkyl acids (PFAAs) are persistent organic pollutants found throughout the world's oceans. Previous research suggests that long-range atmospheric transport of these substances may be substantial. However, it remains unclear what the main sources of PFAAs to the atmosphere are. We have used a laboratory sea spray chamber to study water-to-air transfer of 11 PFAAs via sea spray aerosol (SSA). We observed significant enrichment of all PFAAs relative to sodium in the SSA generated. The highest enrichment was observed in aerosols with aerodynamic diameter < 1.6 μm, which had aerosol PFAA concentrations up to ∼62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times higher than the PFAA water concentrations in the chamber. In surface microlayer samples collected from the sea spray chamber, the enrichment of the substances investigated was orders of magnitude smaller than the enrichment observed in the aerosols. In experiments with mixtures of structural isomers, a lower contribution of branched PFAA isomers was observed in the surface microlayer compared to the bulk water. However, no clear trend was observed in the comparison of structural isomers in SSA and bulk water. Using the measured enrichment factors of perfluorooctanoic acid and perfluorooctane sulfonic acid versus sodium we have estimated global annual emissions of these substances to the atmosphere via SSA as well as their global annual deposition to land areas. Our experiments suggest that SSA may currently be an important source of these substances to the atmosphere and, over certain areas, to terrestrial environments.
000 times higher than the PFAA water concentrations in the chamber. In surface microlayer samples collected from the sea spray chamber, the enrichment of the substances investigated was orders of magnitude smaller than the enrichment observed in the aerosols. In experiments with mixtures of structural isomers, a lower contribution of branched PFAA isomers was observed in the surface microlayer compared to the bulk water. However, no clear trend was observed in the comparison of structural isomers in SSA and bulk water. Using the measured enrichment factors of perfluorooctanoic acid and perfluorooctane sulfonic acid versus sodium we have estimated global annual emissions of these substances to the atmosphere via SSA as well as their global annual deposition to land areas. Our experiments suggest that SSA may currently be an important source of these substances to the atmosphere and, over certain areas, to terrestrial environments.
Environmental significanceThe oceans are currently considered to be the ultimate sink for perfluoroalkyl acids (PFAAs). However, our experiments suggest that the ocean may act as a significant source of PFAAs to the atmosphere. The experiments demonstrate that PFAAs are highly enriched in sea spray aerosols (SSAs) smaller than 1.6 μm in aerodynamic diameter, a size which facilitates long-range atmospheric transport. Since PFAAs do not environmentally degrade, PFAAs present in SSA will be a continuous source to terrestrial environments long after anthropogenic emissions of PFAAs cease. |
1 Introduction
Perfluoroalkyl acids (PFAAs) are a class of anthropogenic surfactants which have been manufactured since the 1950s.1–3 These substances are made up of a fully fluorinated carbon chain linked to an acid group.2 The perfluorinated carbon chain provides oleophobic and hydrophobic properties, as well as high stability.4 These properties make PFAAs valuable chemicals on which many industry sectors rely. Examples of their applications include use as surfactants in firefighting foams1,3,5 and as processing aids in the production of fluoropolymers.1 Recent estimates of the cumulative global emissions of PFAAs are at least 46000 tonnes with a large fraction of this released directly to environmental water.1,5 As a result, PFAAs are present in rivers downstream of manufacturing facilities6 and throughout the world's oceans.7–13
PFAAs have been observed in both humans and biota worldwide.14–17 Particular concern has been raised regarding perfluoroalkane sulfonic acids (PFSAs) and perfluoroalkyl carboxylic acids (PFCAs), as these substance classes include compounds identified as persistent,18 bioaccumulative19 and toxic.20 A number of PFAAs are subject to regulation under REACH,21 and perfluorooctane sulfonic acid (PFOS) is listed under the Stockholm Convention.22
Observations of PFAAs in air23–27 and precipitation,23,28–32 as well as remote inland environments,33–36 suggest that long-range atmospheric transport may be substantial. Several pathways have been proposed to explain the origin of PFAAs observed in air and precipitation, including direct releases of PFAAs to air from manufacturing sources37,38 and atmospheric formation of PFAAs through the degradation of volatile precursor substances.39–43 Although water-to-air transfer of PFAAs from the global oceans via sea spray aerosol (SSA) has previously been discussed in the scientific literature,44–46 the magnitude of this pathway on a global scale has not been properly quantified. Further, since PFAAs are predominantly present in the oceans in their involatile and highly water soluble anionic form,47–50 efficient transport of these substances to the atmosphere via volatilization is considered unlikely. As such, it is assumed that PFAAs that enter the global surface oceans will remain there until they are ultimately transported into the deep oceans.51–53
SSAs are droplets of seawater that are ejected into the atmosphere when bubbles burst on the ocean surface. The bubbles responsible are mainly formed when air is entrained into the ocean by breaking waves which result from the interaction of wind with the ocean surface. The formation of SSAs by bubble bursting is thought to take place via two different mechanisms. First, the film cap of each bubble fragments into numerous so-called film droplets. This type of droplet is thought to make up the majority of SSAs smaller than 1 μm in diameter.54,55 The cavity created as the film cap bursts then collapses, forming a jet of water. This water jet subsequently disintegrates into a small number of so-called jet droplets56,57 which are large in comparison to the film droplets and are thought to comprise the majority of SSA larger than 1 μm in diameter.55 Production of SSAs is a complex process governed by factors such as wind speed, salinity, air temperature, water temperature and the presence of surfactants.58–61
SSA consists of a complex mixture of sea salt and organic matter.62–65 Bubbles are thought to scavenge surface-active organic matter as they travel through the bulk ocean66 and the air–sea interface.67 Previous studies have consistently shown that the organic mass fraction of SSAs increases with decreasing particle size.65,68–73 Critically, formation of both smaller film droplets and larger jet droplets is known to be sensitive to bubble size.60 As such, it is an essential requirement of any laboratory system that is designed to produce nascent SSAs with relevant physical and chemical characteristics that it reproduces the numbers and sizes of bubbles entrained by breaking waves in the open ocean.74
Previous studies have demonstrated strong enrichment of PFAAs in laboratory generated aerosols relative to their bulk water concentration44,45,75 using a number of different methods to produce bubbles. McMurdo et al.45 utilised a piezoelectric crystal ultrasonic aerosol generating device to generate aerosols much larger (∼50 μm) than those typically produced over the ocean (<10 μm). Although the underlying mechanism of droplet formation by ultrasonic nebulisation is not fully understood,76 it is clear that this process is very different to the process of air entrainment in the open ocean and any bubbles present will be very different in size to those found in oceanic breaking waves. A further limitation of the McMurdo et al.45 study is that aerosol enrichment factors were only derived for one PFAA, namely perfluorooctanoic acid (PFOA). Reth et al.44 investigated aerosols produced via bubble bursting following air entrainment using a plunging jet, a technique that is considered more reflective of the process of nascent SSA generation,77 and extended the target substances to a range of PFCAs and PFSAs. Unfortunately, although these experiments were useful in revealing that PFAA-enrichment in aerosols is dependent on the perfluoroalkyl chain length, they were performed using tap water, which is likely to produce bubble-bursting aerosols that are very different, both in size and chemical composition, to aerosols produced by bubble bursting in seawater. Furthermore, the production of SSAs is strongly influenced by seawater salinity.78–81 Ebersbach et al.75 generated aerosols from wastewater by entraining air using a diffuser/frit. Experiments using such an approach are non-ideal in that the bubble sizes generated depend on the exact frit used and do not reproduce the bubble size distributions found in oceanic breaking waves. In summary, none of the previous studies produced data that enabled quantification of the environmental relevance of the water-to air-transport pathway of PFAAs via SSA on a global scale – that is size-resolved aerosol enrichment factors relative to an SSA tracer compound included in global circulation models, such as sodium (Na+).
Our study improves upon these initial laboratory experiments by using artificial seawater in a sea spray simulation chamber which produces a bubble size distribution similar to that found in breaking waves.82 Size-resolved samples of the produced aerosols are obtained through the use of a low pressure impactor (LPI) connected downstream of the sea spray chamber. Sampling the aerosols in this way enabled us to determine (i) whether PFAAs are enriched in SSA under conditions which accurately reflect the process of SSA formation and (ii) whether PFAAs aerosolized as SSA have a size that facilitates long-range atmospheric transport. Another important advance over previous studies is the concurrent measurement of Na+, an important tracer of SSA, which enabled the generation of aerosol enrichment factors and subsequent estimation of the magnitude of SSA-mediated ocean-to-atmosphere transfer of PFAAs using a global circulation model. Due to a lack of field data for model parameterization, model output is only generated for the two most well-studied substances, PFOA and perfluorooctane sulfonic acid (PFOS). Further experiments were performed to study the enrichment of structural PFAA isomers, as patterns of these are interesting for source elucidation of PFAAs in the atmosphere.
2 Materials and methods
In the following, the methods are described in brief. Full details of the sampling, extraction and instrumental analysis can be found in the ESI.†2.1 Sea spray simulation chamber
All experiments were performed using a sea spray generator developed by Salter et al.82 Here, nascent SSAs were generated in the laboratory using a plunging jet (Fig. S1†). Using this setup, artificial seawater was circulated continuously at 1.7 L min−1 from the bottom of a chamber, 47 cm in diameter and 100 cm in height, through a stainless steel nozzle with an inner diameter of 4.3 mm held in a vertical position 30 cm above the air–water interface. Within this chamber the seawater was filled to a depth of 60 cm, leaving a headspace of 40 cm (100 L seawater). All surfaces below the water level on the inside of the stainless steel tank are coated with polytetrafluoroethylene. All tubing in contact with sample water was made of silicone. Dry particle-free sweep-air entered the chamber at 32 L min−1 after passing through an ultrafilter (Type H cartridge, MSA) and an activated carbon filter (Ultrafilter, AG-AK).2.2 Experiments
Two experiments were conducted using artificial seawater spiked with native target compounds. The artificial seawater was prepared by rehydration of Sigma Aldrich sea salt to an absolute salinity of 35 g kg−1 using low-organic-carbon standard deionized water (MilliQ, >18.2 MΩ cm), hereafter referred to as DIW. All experiments were performed at the same salinity and temperature (15 °C). Experiment A was performed with a mix of linear compounds: perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA), PFOA, perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluoroundecanoic acid (PFUnDA), perfluorododecanoic acid (PFDoDA), perfluorotridecanoic acid (PFTriDA), perfluorotetradecanoic acid (PFTeDA), perfluorobutane sulfonic acid (PFBS), perfluorohexane sulfonic acid (PFHxS), PFOS and perfluorodecane sulfonic acid (PFDS). For experiment B the seawater was spiked with technical standards of PFOA, PFHxS and PFOS (TPFOA, brPFHxS and TPFOS). The technical standards are characterized mixtures of branched and linear isomers. The nominal concentration of each substance was approximately 10 ng L−1 in experiment A. In experiment B the bulk water was spiked to 52, 45 and 49 ng L−1 with TPFOA, brPFHxS and TPFOS respectively. The choice of the PFAA concentration used in experiment A was a compromise between the wish to use an environmentally relevant concentration (see Fig. S2†) and that to ensure quantifiable levels in all sample types. The target concentrations of the substances included in experiment B were increased to ensure quantifiable levels of each individual isomer. The experiments were initiated following 1 hour of system equilibration with the plunging jet switched on. Aerosols were then sampled for approximately 6 hours. Each experiment was replicated three times. The seawater was not replaced between each replicate run but the concentration of PFAAs in the seawater was monitored over the period of the three replicates.2.3 Aerosol sampling
To determine the mass of Na+, PFCAs and PFSAs in particles generated using the sea spray chamber we used a 13-stage (30 L min−1) low pressure impactor (LPI, Dekati). Immediately upstream of the impactor the relative humidity (RH) was measured using a Vaisala model HMT333 probe. During all measurements, the sample flowing to the LPI as well as the LPI itself were heated by placing a heating jacket around the sample line and the LPI. This ensured that the relative humidity at the inlet of the LPI was always below ∼40% (see Table S1†). The LPI had 50% cut-off diameters of 0.029, 0.060, 0.104, 0.165, 0.253, 0.391, 0.634, 0.990, 1.60, 2.45, 3.96, 6.57, and 10.16 μm aerodynamic diameter. No back-up filter was used to sample particles smaller than 0.029 μm in aerodynamic diameter. Polycarbonate collection foils (Whatman Nuclepore Track-Etch Membrane; 800203) were used as the collection substrates. The outlet pressure on the LPI was continuously monitored using an analog pressure sensor.Following removal from the impactor, LPI substrates were placed in a polypropylene centrifuge tube with 10 mL of DIW and extracted in an ultrasonic bath for 60 min. LPI substrates from stages 1 to 7 were pooled, in order to achieve quantifiable concentrations in the final extracts, while substrates from stages 8 to 13 were extracted individually. All handling of filter substrates was performed inside a glove box.
Four dynamic handling blanks were prepared during the experimental period to account for potential contamination of the LPI filter substrates during handling and transport to and from the sea spray chamber. For each dynamic handling blank, the impactor was loaded with substrates and transported to the sea spray chamber where it was left for 60 min. The impactor was returned to the laboratory and the filters were handled and analysed in the same manner as the samples.
2.4 Bulk water sampling
Bulk water was sampled in 50 mL polypropylene centrifuge tubes, at the start and end of each replicate experiment. While the jet was still turned on, bulk water was sampled in triplicate through a tap located on the side of the chamber approximately halfway between the water surface and the bottom of the chamber.2.5 Surface microlayer sampling
Samples of the surface microlayer (SML) within the sea spray simulation chamber were collected in triplicate following cessation of the final aerosol sampling period for each experiment. SML samples were collected using a glass plate as per the methods of Harvey.83 Here, a clean hydrophilic glass plate (rinsed with ethanol and DIW) was immersed into the seawater sample and withdrawn at a controlled rate so that the thin surface layer of the seawater is retained.2.6 Determination of Na+ and PFAAs
Aliquots (1 mL) of all aerosol samples were subsampled to determine the concentration of Na+ by chemically suppressed ion chromatography (IC; Dionex ICS-2000) using CG16/CS16 columns.Bulk seawater (50 mL) and SML (15 mL) samples as well as the remaining aliquots of the aerosol samples (9 mL) were spiked with an isotope-labeled internal standard and concentrated on Oasis weak-anion exchange (WAX) solid phase extraction (SPE) cartridges (6 cm−3, 150 mg, 30 μm) using a previously published method.84
The final extracts were analyzed for PFAA content using ultra performance liquid chromatography coupled to tandem mass spectrometry, as further described in the ESI.†
Linear analytes were quantified using the internal standard method. All analytes had matching stable isotope-labelled internal standards except PFBS, PFDS, PFTriDA and PFTeDA, for which quantification was performed using 18O2-PFHxS (PFBS), 13C4-PFOS (PFDS) and 13C4-PFOA (PFTriDA and PFTeDA). The distribution between the linear and the sum of branched isomers was determined by comparing their respective peak areas in the precursor/product ion transitions 413/369, 399/80 and 499/80 for PFOA, PFHxS and PFOS respectively. To investigate fractionation of structural isomers, the intensity of isomer-specific product ions was monitored, according to a strategy previously described by Benskin et al.85
2.7 Quality assurance
In addition to dynamic handling blanks, each batch of samples was extracted along with a blank prepared from a non-used polycarbonate membrane and 10 mL DIW. All blank samples contained high background contamination of PFBA. As such, this analyte was omitted from the study. Apart from PFBA, no other analyte was observed above its respective instrumental quantification limit in the blank samples and hence no subtraction of these concentrations from the measured values for the samples was conducted. To test the accuracy and precision of the method, unused membranes were spiked with 8 ng of each linear target analyte and extracted in DIW according to the procedure described above. Poor accuracy and high relative standard deviation (RSD) were observed for PFDS, PFTriDA and PFTeDA (Table S3†). This was likely due to their high surface activity, causing losses during sampling and storage. In addition, the quantification of these substances was not performed relative to an identical isotope-labelled internal standard and should therefore only be viewed as semi-quantitative. For these reasons enrichment factors of PFDS, PFTriDA and PFTeDA are not reported herein. To test the performance of the isomer analysis, unused membranes were spiked with 20, 25 and 20 ng of TPFOA, brPFHxS and TPFOS, respectively. Good precision (RSD 1.0–12%) and accuracy (93–118%) was observed for the sum of branched isomers as well as ratios of individual structural isomers. The mean recoveries of the internal standards relative to 13C8-PFOA are given in Table S4.†2.8 Calculation of enrichment factors
The measured data were used to determine the enrichment factors of each of the PFAAs in the aerosol (aerosol EFs), relative to their bulk water concentrations, as a function of particle size. They were calculated using a classical approach where the aerosol EF is defined as the ratio of the concentration of substance X in the particle to that in the bulk seawater and the concentration of substance X is normalized to the concentration of one of the major constituents of seawater, generally Na+:86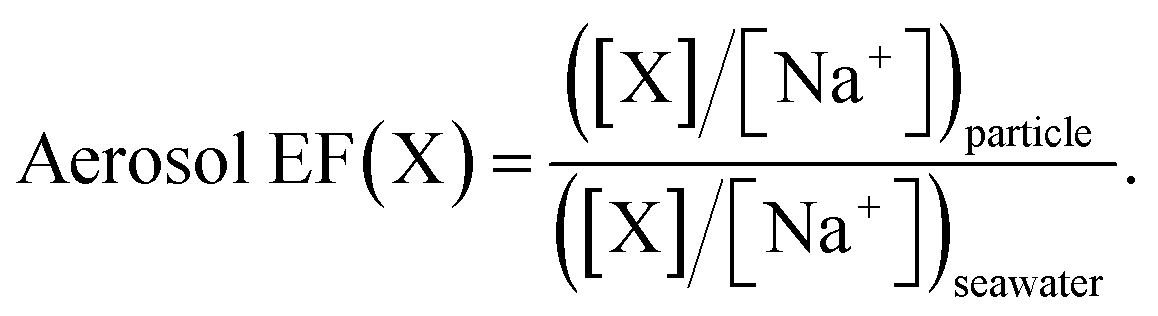 | (1) |
The reported aerosol EFs were calculated from the average concentration of substance X in triplicate LPI samples representing a specific aerosol size range and the average concentration of the same substance in the bulk water during the course of the complete experiment (n = 6). Such enrichment factors are used under the assumption that the mass fraction of Na+ is the same in both the seawater and the nascent aerosol produced from it.
2.9 Global modelling
A Norwegian Earth System Model (NorESM)87,88 was used to determine the magnitude of the transport of PFOA and PFOS to the atmosphere via SSA as well as the magnitude of the deposition of these substances to terrestrial environments via SSA transport. To estimate SSA emissions, NorESM uses the inorganic SSA source function developed by Salter et al.89 This source function simulates the number of SSAs produced from a unit area of ocean in a unit of time as a function of particle size. The source function consists of three log-normal modes (modal diameters: 0.095 μm, 0.6 μm and 1.5 μm) and depends on two environmental parameters thought to be most important for SSA generation, wind speed and seawater temperature. Importantly, the source function was developed using the same sea spray simulation chamber utilized in the current study and when compared to a wide range of SSA source functions in the literature, it estimates an annual global flux of inorganic SSA close to the median value.89Annual average PFOA and PFOS emissions via SSA were modeled by rearranging eqn (1) using the mass emissions of Na+via SSA in NorESM, and relevant seawater PFOA and PFOS concentrations and the measured PFOA and PFOS aerosol enrichment factors are presented in Fig. 2. The relevant mean enrichment factor for both PFOA and PFOS was selected for each of the modes in the source function (0.095 μm: stages 1–7 of the LPI, 0.6 μm: stages 1–7 of the LPI, 1.5 μm: stage 8 of the LPI). With regard to relevant seawater concentrations of PFOA and PFOS, a series of studies8–12,90–93 have measured the open ocean surface water concentrations of these substances and box and whisker diagrams summarising these measurements are presented in Fig. S2c and S2d.† For these calculations we have used the median value of reported open ocean PFOA and PFOS concentrations, 34 pg L−1 and 20 pg L−1, respectively.
Since our approach to calculating PFOA and PFOS emissions (rearranging eqn (1)) assumes that the absolute magnitude of these emissions scales linearly with the relevant enrichment factor and the seawater concentration of the substance, as well as the mass of Na+ emitted as SSAs, it is also possible to use ranges of each of these parameters to determine a best estimate of global annual PFOA and PFOS emissions along with upper and lower bounds. We have utilised the standard deviations of the calculated enrichment factors presented as error bars in Fig. 2 for the lower and upper bounds of the enrichment factors along with the mean values as a best estimate. To generate a best estimate and upper and lower bounds of seawater PFOA and PFOS concentrations we have used the median and 25th and 75th percentiles of the data presented in Figs. S2c and S2d.† We have also included the uncertainty in SSA emissions by utilising a review of SSA emissions computed by 12 chemical transport and general circulation models participating in the AeroCom aerosol model intercomparison.94,95 A summary of these 12 estimates is presented in Fig. S2b† and we have used the median and 25th and 75th percentiles for our best estimate and lower and upper bounds, respectively. For these calculations we assume that the mass of sea salt in the best estimate and upper and lower bounds is distributed across particle size in the same manner as in the parameterisation of Salter et al.89 so that we can apply appropriate enrichment factors. We also assume that the fraction of sea salt deposited to terrestrial regions is the same for these estimates as calculated by NorESM (total deposition).
3 Results and discussion
3.1 Laboratory experiments
The RSD of the triplicate bulk seawater samples taken at the start and end of each aerosol sampling period was below 10% for all substances (Fig. S3†). The concentrations of the least surface active substances were stable over the course of the experiment, while a 40% decrease in concentration was observed for PFDoDA between the start of experiment A1 and the termination of experiment A3 (Fig. S3†). Clear discrepancies existed between the target concentrations and the measured seawater concentrations. Notably, this discrepancy increased with increasing PFAA chain length, which is a proxy for the surface activity of the substance.96–98 Agreement between target and measured concentrations in seawater was within 20% for some short-chain homologues, namely perfluorobutane sulfonic acid (PFBS), perfluoropentanoic acid (PFPeA) and perfluorohexanoic acid (PFHxA), whereas a 69% discrepancy was observed for perfluorododecanoic acid (PFDoDA), the most surface active substance. Similar behavior was observed by Reth et al.,44 who suggested that it was the result of sorptive losses to the chamber walls and partitioning to the air–water interface. Observation of concentrations above the spiked target can be attributed to background levels of PFAAs in the tap water used to prepare artificial seawater for the experiments. In experiment B, the target concentrations of the individual PFAAs studied were approximately 5 times higher than their corresponding target concentrations in experiment A. However, the target concentration for the sum of all PFAAs was 150 ng L−1 in both experiments. In experiment B, the discrepancy between target and measured concentrations was 53, 29 and 48% for PFHxS, PFOA and PFOS, respectively (sum of branched and linear isomers). In experiment A, the discrepancy was 11, 8 and 23% for PFHxS, PFOA and PFOS, respectively (only linear isomers). The larger discrepancy observed in experiment B might be explained by a lower influence of background PFAA levels in the artificial seawater.Low surface microlayer enrichment factors (SML EFs; defined as the ratio of the concentration in the SML to the concentration in the bulk seawater) were observed for PFCAs with up to 9 carbon chain lengths (SML EF: 1.1–2.9) and PFSAs with up to 6 carbon chain lengths (SML EF: 1.1, 1.2), while more substantial enrichment was observed for longer chain substances (SML EF: 13–47) (Fig. 1). Notably, SML EFs increased with increasing homologue chain length (Fig. 1). The SML EFs observed in experiments A and B agreed well for PFOA (1.4 and 2.1, respectively) and PFHxS (1.2 and 1.6, respectively). However, the SML EFs for PFOS were more than three times higher in experiment B (43) than in experiment A (13). The difference in the observed SML EF for PFOS in the two experiments is likely due to the well-known challenges in achieving repeatability of SML sampling.
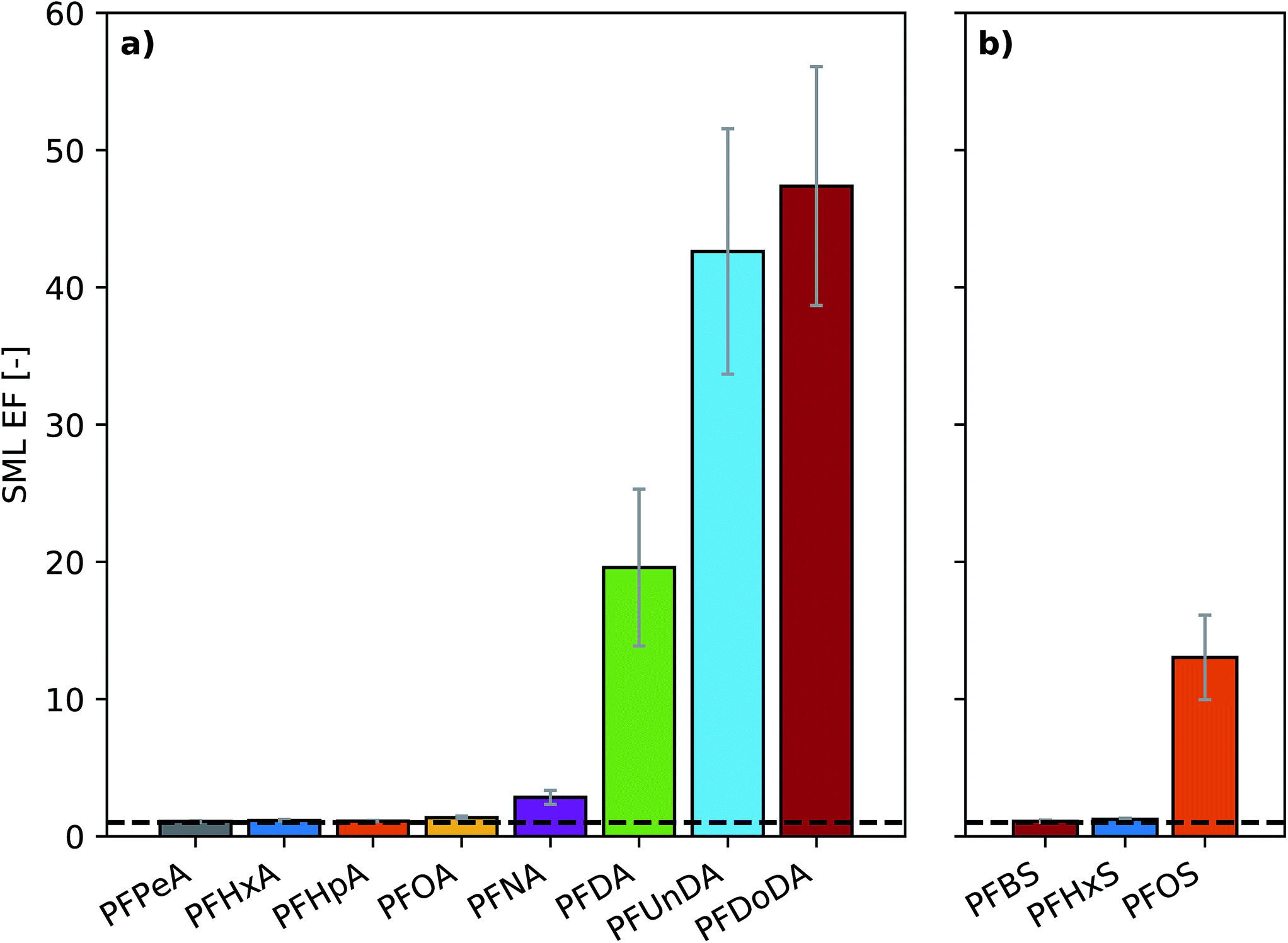 | ||
| Fig. 1 The SML enrichment of (a) perfluoroalkyl carboxylic acids (PFCAs) and (b) perfluoroalkane sulfonic acids (PFSAs) in experiment A. Here error bars represent 1 standard deviation and the dashed line represents an enrichment factor of 1. | ||
With the exception of PFPeA in the two smallest stages of the LPI (i.e. in aerosols <1.60 μm), quantifiable masses of the spiked PFAAs were present across all stages of the LPI (Fig. S4 and S5†). Similar to the trend observed in the SML EFs (Fig. 1), the aerosol EFs increased with homologue chain length (Fig. 2). This suggests that similar processes are enriching PFAAs in the aerosols and the sea surface microlayer. The observed aerosol enrichment factors increased with decreasing particle size and exhibited maxima in the two lowest LPI stages. In experiment A, PFOA and PFOS aerosol enrichment factors increased from ∼1800 to ∼17100 and ∼200 to ∼62100, respectively, between the largest aerosol particles (>10.16 μm) and the smallest aerosol particles (0.029–0.99 μm). The aerosol EFs observed in experiments A and B were generally within 15% agreement, although differences between 19 and 35% were observed for five aerosol EFs. Notably, the aerosol EFs were orders of magnitude larger than the corresponding SML EFs for each substance.
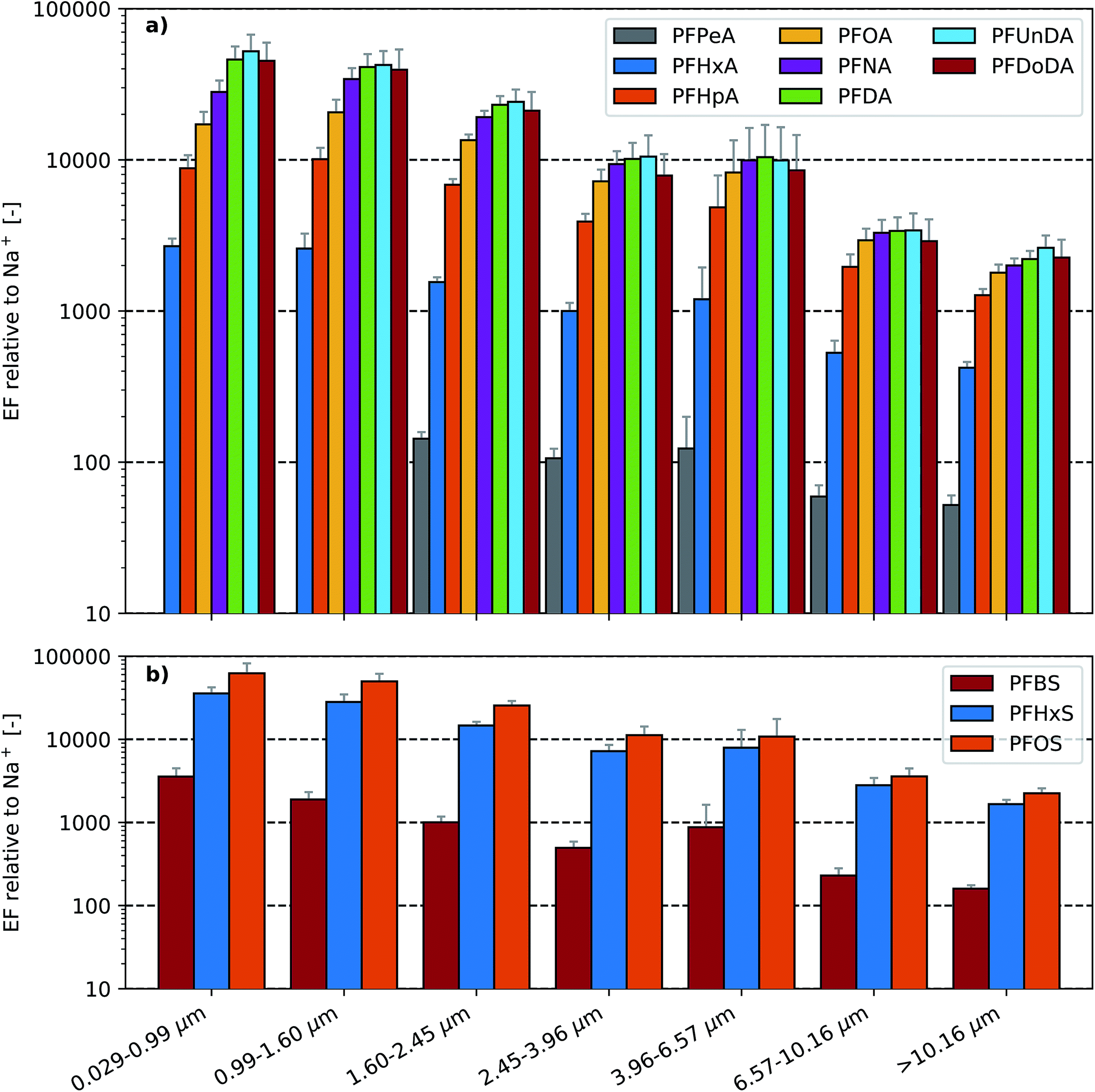 | ||
| Fig. 2 Enrichment factor, EF(X), relative to Na+ of (a) perfluoroalkyl carboxylic acids (PFCAs) and (b) perfluoroalkane sulfonic acids (PFSAs) as a function of particle aerodynamic diameter (experiment A). Note that PFPeA was not detectable in the two lowest stages of the impactor so no EF could be calculated. Here error bars represent 1 standard deviation following propagation of the standard deviation of the Na+ and PFAA concentrations measured in the seawater and aerosol samples. | ||
To investigate the enrichment behaviour of isomeric PFAA mixtures, we analysed the distribution between the linear and the sum of branched isomers in the different sample types (Fig. S9†), as well as the relationship between individual branched isomers (Table S5†). For all isomeric mixtures studied, a lower contribution of branched isomers was observed in the SML compared to the bulk water. PFOS displayed the largest difference in contribution of branched isomers between bulk water (28%) and SML (18%). The contribution of branched PFOS isomers was elevated in aerosols (24–28%) in relation to the SML, but no clear trend was observed in the comparison of different aerosol size ranges. Small aerosols (<1.60 μm) displayed a contribution of branched PFOA isomers closer to that of the SML (14%), while for larger aerosols (>1.60 μm) it was closer to that observed for bulk water (18%). A similar trend was observed for the individual branched PFOA isomers (Table S5†). For PFHxS, only slight differences were observed between bulk water, SML and aerosols. The only statistically significant differences were observed between bulk water and SML for PFOS (p = 0.046) and between the SML and 6.57–10.16 μm aerosols for PFOA (p = 0.029; see ESI† for the description of the statistical tests applied).
Branching of the chain of a fluorinated surfactant is expected to lead to a reduction in surface activity of the chemical due to less efficient molecular packing.99 However, measurements comparing the surface activity of branched and linear PFAA isomers are scarce. Shinoda et al.100 reported a critical micelle concentration of 8.5 mmol L−1 for 6-PFOA. The same author has also reported critical micelle concentrations for linear PFOA of 8.0 and 9.1 mmol L−1 in previous studies.101,102 McMurdo et al.45 hypothesised that the SML as well as SSA will be more enriched in linear PFOA than in branched PFOA isomers and therefore the processes of aerosol production would act as “a very effective filtering system for the branched isomers”. A number of previous studies have used the PFOA isomer pattern as a tool for source elucidation in environmental samples.6,31,103,104 If SSA act as a filter for branched PFOA isomers, as suggested by McMurdo et al.,45 use of this approach may be compromised, especially for study of atmospheric samples.31,103 Our data suggest that the surface activities of branched and linear PFAA isomers differ to some extent. However, SSA are not an efficient filter for branched PFOA isomers. As such, the distribution of branched PFAA isomers in SSA will likely be more influenced by spatial differences in isomer pattern occurrence in seawater104 than by fractionation in the formation of SSA.
3.2 Comparison to previous studies
Two previous studies have attempted to determine whether PFAAs are likely to be efficiently transferred from the ocean to the atmosphere.44,45 Both of these studies clearly highlighted the potential of SSA to act as an efficient vector for their transport. However, direct comparison to their results is impossible since (i) Reth et al.44 did not use seawater and (ii) McMurdo et al.45 used an aerosol generation mechanism very different from that which generates natural SSA and a highly unconventional aerosol sampling approach (as discussed in Mader et al.105). Further, neither of the studies presented aerosol EFs normalised to Na+.The SML EFs determined in our study are within a factor of two of those reported by Reth et al.44 for all tested substances except PFUnDA. For this substance Reth et al.44 reported an enrichment factor exceeding ours by a factor of 4. Furthermore, the SML EF for PFOS observed in experiment B exceeded that reported by Reth et al.44 by a factor of 3. Although they used a technique similar to that used in our study to obtain SML samples, a major difference in their experiment was the use of tap water rather than artificial seawater. Our measured SML enrichment factors also agree well with the SML enrichment factors measured in natural seawater samples by Ju et al.106 and Wang et al.107 These authors reported PFOA SML enrichment factors that were stable over periods of days across a variety of sampling sites (PFOA EFs: 1.2–1.8 and 1.3–4.7 respectively). However, their reported SML enrichment factors for PFOS across the same site and time period were more variable (2.0–109 and 3.3–13 respectively). While PFAAs are thought to have surface activities substantially higher than that of natural organic matter (NOM),4,108 competition with surface-active NOM may decrease the enrichment of PFAAs in the SML and in SSA. An alternative process, not tested in the current study, is SML and SSA enrichment of PFAAs sorbed to NOM. In other words, the presence of NOM could both enhance and reduce the enrichment of PFAAs in the SML and in SSA. Likely, the influence on PFAA EFs will depend on the type and concentration of NOM in the system.
The observation that PFAAs are predominantly enriched in submicron aerosols agrees with observations on the enrichment of organic carbon in SSAs65,68,70–72 and recent measurements of other carboxylic acids.109 However, Cochran et al.109 observed submicron aerosol EFs of up to about ∼1000 for non-fluorinated carboxylic acids (sum of three test substances), some two orders of magnitude lower than the highest EFs observed in the current study for the corresponding aerosol size range. This difference likely results from the much higher surface activity of PFAAs relative to non-fluorinated carboxylic acids. The increasing enrichment with chain length was also previously observed by Cochran et al.109 and suggests that the surface activity of PFAAs is a key driver of their enrichment in SSAs.
3.3 Modeling the transport of PFAAs via sea spray aerosols
The spatial distribution of the modeled PFOA emissions by SSA (Fig. 3a) and deposition to land areas by the same pathway (Fig. 3b) directly reflects the SSA production flux described previously by Salter et al.89 The emission of SSA, in turn, reflects the distribution of storms worldwide, since near surface wind speed is the dominant factor controlling SSA emissions. From Fig. 3b, it is clear that coastal regions are most impacted by transport and deposition of PFAAs following emission via SSA. However, large parts of inland Europe, Alaska and Central America are also affected. Fig. S6† presents similar maps for PFOS with the only difference being the magnitude of the emission and deposition in each grid cell which results from differences in the seawater concentration and the EF.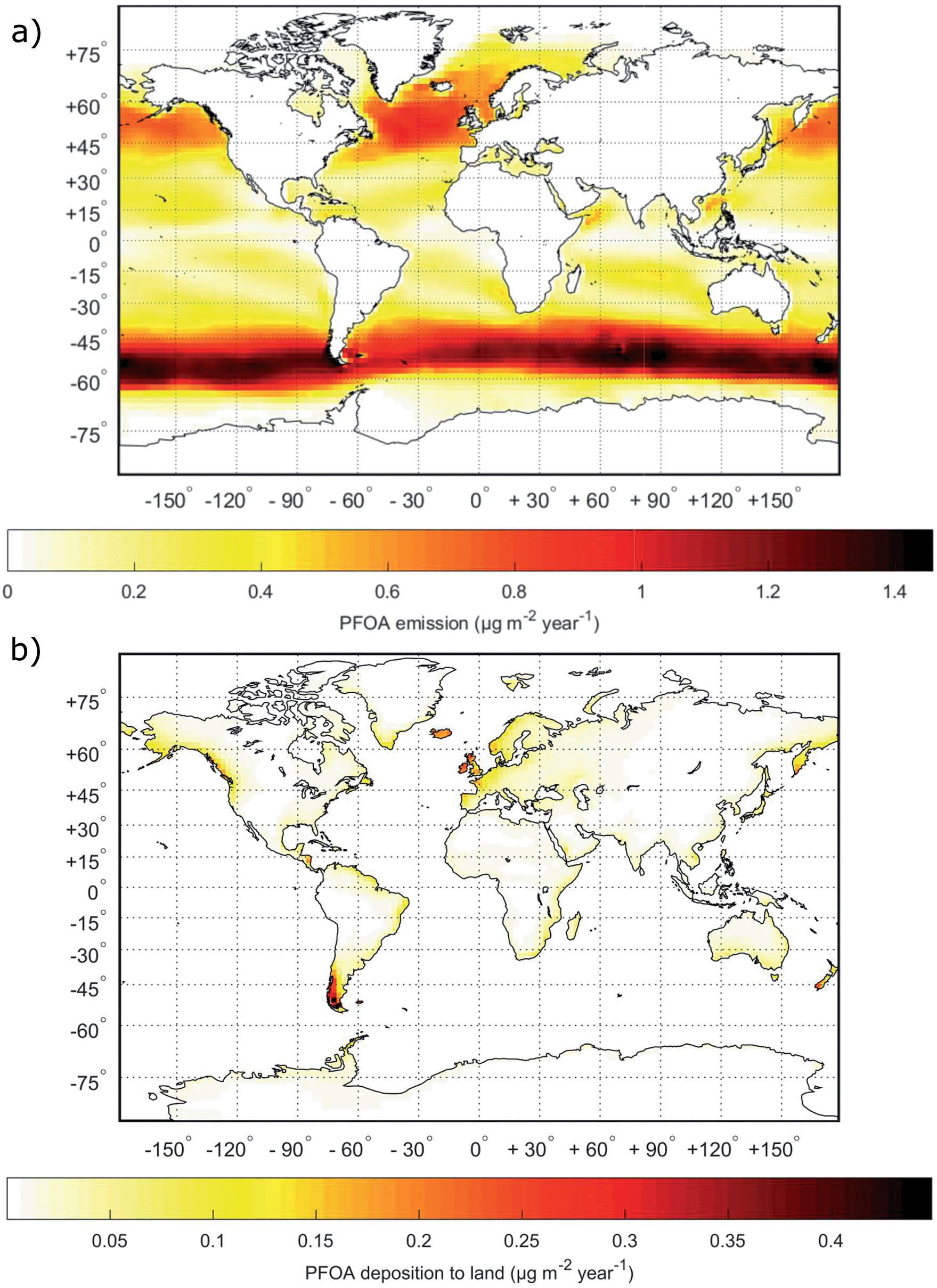 | ||
| Fig. 3 Global maps of (a) total yearly emissions of PFOA via SSAs and (b) total yearly deposition of PFOA transported to terrestrial environments by SSAs. | ||
We estimate that between 23 and 506 tonnes per year of PFOA and between 42 and 810 tonnes per year of PFOS are emitted to the atmosphere by SSAs with a best estimate of 122 tonnes per year and 183 tonnes per year of PFOA and PFOS, respectively (Table 1). Subsequently, between 1 and 13 tonnes per year of PFOA and between 1 and 20 tonnes per year of PFOS are deposited to the terrestrial environment with a best estimate of 3 tonnes per year and 5 tonnes per year of PFOA and PFOS, respectively. In other words, only about 3% of the PFOA and PFOS aerosolised as SSA is transported and deposited to land areas.
| Lower estimate | Best estimate | Upper estimate | ||
|---|---|---|---|---|
| Inorganic sea spray | Global emission (pg per year) | 3.64 | 6.24 | 9.68 |
| Deposition to land (pg per year) | 0.09 | 0.16 | 0.24 | |
| PFOA | Seawater concentration (pg L−1) | 15 | 34 | 72 |
| Enrichment factor (—) | Mode 1 and 2: 13000 |
Mode 1 and 2: 17000 |
Mode 1 and 2: 22000 |
|
| Mode 3: 15000 |
Mode 3: 21000 |
Mode 3: 26000 |
||
| Global emission (tonnes per year) | 23 | 122 | 506 | |
| Deposition to land (tonnes per year) | 1 | 3 | 13 | |
| PFOS | Seawater concentration (pg L−1) | 11 | 20 | 44 |
| Enrichment factor (—) | Mode 1 and 2: 38000 |
Mode 1 and 2: 62000 |
Mode 1 and 2: 86000 |
|
| Mode 3: 36000 |
Mode 3: 50000 |
Mode 3: 64000 |
||
| Global emission (tonnes per year) | 42 | 183 | 810 | |
| Deposition to land (tonnes per year) | 1 | 5 | 20 | |
The total annual flux of PFOA to the atmosphere via SSA estimated in this study (Table 1) is comparable with PFOA emission estimates reported by Wang et al.1 These authors reported total emissions (including direct releases and precursor transformation) of 14–74 tonnes for PFOA in 2012.1 Current annual emissions of PFOS to air from industrial sources in China are estimated to be 1–1.4 tonnes.110 Current industrial emissions of PFOS outside of China are likely minor.111 Wang et al.111 estimated that between 2003 and 2015 less than 2.8 tonnes of PFOS was formed from precursors in the environment each year. Consequently, the lower estimate of SSA-borne releases of PFOS to the global atmosphere exceeds emission estimates for other potential sources of PFOS to air by one order of magnitude.
Comparison of our model results to existing inventories suggests that SSA may currently be an important source of PFAAs to the atmosphere. However, the large uncertainties in our modeling approach (revealed in Table 1) warrant discussion. The sea salt emissions that our estimates are based upon are uncertain by factors of between 2 and 10.95 Further, most of the PFAA mass will be associated with SSA particles with diameters larger than 1 μm which have concentrations that are even more uncertain. It is also important to note that our estimates of PFAA emissions via SSA do not include coastal wave-breaking (which are not directly wind-driven). Here, sea spray emissions are likely to be significantly greater and much closer to those of coastal regions where deposition may be important. It should also be noted that the lifetime of aerosols once airborne is highly uncertain, which contributes to the uncertainty in our estimates of deposition rate and extent.112
Further uncertainty is associated with the oceanic concentrations of PFAAs, which also vary over several orders of magnitude (Fig. S2†). As shown in our review of published seawater data (Fig. S2†), median concentrations of the studied PFAAs are all in the range 6–34 pg L−1. This homologue pattern is not in line with previous emission estimates, which state that the cumulative historical emissions of PFOA were between one and four orders of magnitude higher than those for other PFCA homologues.1 This suggests that the published data on occurrence of PFAAs in seawater may not accurately reflect the true environmental conditions. We therefore chose to perform model predictions for PFOA and PFOS only. These are the two most well-studied PFAAs and therefore analytical methods are often tailored to perform well for these substances.
Due to a lack of open ocean monitoring data in the published literature, it was not possible to account for the spatial distribution of PFAAs in the global oceans. The use of seawater concentrations of PFOA and PFOS that do not vary spatially adds further uncertainty to our estimates. For PFOA the median concentrations in the data used as a model input (Fig. S2†) were 55 pg L−1 (n = 307) and 13 pg L−1 (n = 139) in the Northern and the Southern hemispheres, respectively, while for PFOS the corresponding concentrations were 24 pg L−1 (n = 261) and 30 pg L−1 (n = 139), respectively. While there may be hemispheric differences in the emission of PFAAs, not captured by these initial estimates, the ranges of seawater concentrations used for the lower and upper emission scenarios account for this uncertainty. Although our best estimate emission scenario for PFOA may overestimate emissions in the Southern hemisphere it may also underestimate emissions in the Northern hemisphere, where transport to continents is likely to be more important. Furthermore, seawater concentrations of these compounds are often greater in coastal waters which are closer to anthropogenic PFAA sources.11,113 This, when combined with coastal wave-breaking, further increases the likelihood of transport to terrestrial coastal environments.
Another source of uncertainty stems from the aerosol EFs. We have used error propagation of the standard deviation of the Na+ and PFAA concentrations measured in the seawater and aerosol samples to obtain the error estimates included in Fig. 2 and these error estimates are included in our emission and deposition estimates (Table 1). The estimated standard deviations of the aerosol EFs all fall within a single order of magnitude. Despite the relatively low uncertainty in these values, when compared to SSA emissions and seawater PFAA concentrations, we do not include the effects of natural surface-active organic matter on the enrichment of these substances in our experiments, nor indeed potential interactions with other surface-active pollutants.
A number of studies have attempted to model the transport of PFAAs to the atmosphere via SSA.51,114,115 These studies assumed either that PFAAs were not enriched in the seawater droplets emitted as SSAs114 or that the enrichment of PFAAs in SSA was similar to the enrichment found in SML samples.51,115 Based upon the significant difference between the observed aerosol and SML EFs in the current study, this suggests that the approaches used by Qureshi et al.,114 Armitage et al.51 and Webster et al.115 underestimated the potential emission of PFAAs to the atmosphere via SSA dramatically.
The results of the current study highlight the potential of SSA to act as a vector for the transport of PFAAs from the oceans to the atmosphere. The approaches used allowed us to determine the potential magnitude of this transport pathway, so that it can be placed in the context of other sources.
The only way to rigorously test our modeling approach is through comparison to atmospheric aerosol samples collected within the marine boundary layer from which both the Na+ and PFAA concentrations have been determined. Unfortunately, such field data are currently not available in the scientific literature. Nevertheless, we have compared our model output to field measurements made by Jahnke et al.24 These authors reported concentrations of PFAAs in air samples collected during a cruise from Germany to South Africa. The air concentrations of PFOA and PFOS for the corresponding days and locations were calculated using model output for sodium concentrations in air and the SSA EFs measured for PFOS and PFOA in our experiment. The modelled and measured data are within one order of magnitude (Fig. S8†). However, the model overestimates the low measured concentrations and underestimates the high measured concentrations. This is not surprising, as the model outputs monthly averages for a 100 × 100 km grid cell and uses average weather (across five years) rather than the actual weather for a specific point in time, while each data point reported by Jahnke et al.24 represents a sample collected over three days. Day-to-day input of SSAs at a specific location may vary substantially and thus such short-term samples are not directly comparable to the model output.
Deposition of PFAAs to terrestrial areas was modeled to generate an estimate of the proportion of SSA-borne PFAAs that deposit on land and to illustrate the regions predominantly influenced by this deposition. Published deposition fluxes span over orders of magnitude.28–30,32,35,36,116 This is likely related both to analytical issues and to large variations in factors that affect atmospheric deposition of aerosols. Most field studies only reported data for one or a few measurements from the same sampling site. Such data may not be suited for extrapolation to an average annual deposition flux, which is required in the comparison to our modeled data. Use of such field data to evaluate our model results is further complicated by the fact that deposition samples can be influenced by different sources of PFAAs, each of which may dominate under different sets of conditions.
4 Conclusions
Oceans are by far the largest environmental “reservoir” of historically released PFAAs. As PFAAs do not degrade in environmental waters and most PFAAs are not buried in sediments to a substantial degree, the substances are expected to persist in the global oceans indefinitely.117,118 Our results indicate that SSA have the capacity to circulate significant amounts of PFAAs between the oceans and the atmosphere. A portion of the mass emitted from the oceans will deposit on land, thus re-entering the terrestrial system. This suggests that human exposure to PFAAs will continue even if strict global emission controls are implemented. To determine whether the observations of water-to-air transport of PFAAs in our laboratory experiments are valid, field measurements of PFAAs in aerosols at remote locations affected by SSA using adequate aerosol sampling approaches are required. Ideally, these measurements should be conducted with high time resolution (days rather than weeks) so that statistical trajectory analysis techniques can be applied to determine the sources of PFAAs in the samples. Critically, the mass of a reference element present in bulk seawater, such as Na+, should be used to normalize PFAA measurements and calculate enrichment, as conducted in this study. Moreover, our results highlight the need for further study of the importance of SSA for the global transport of other persistent, water soluble and surface active substances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly thank Agneta Öhström for her assistance with the ion chromatography analysis, financially supported by the Swedish Research Council Vetenskapsrådet (project number 2016-03518). J. H. J. was funded by the Swedish Research Council FORMAS (project number 2011-1345). M. E. S. was financed by Vetenskapsrådet (project number 2016-05100). The sea spray simulation chamber was previously financed by Vetenskapsrådet projects MASC (2010-3821) and CESSA (2014-5354).Notes and references
- Z. Wang, I. T. Cousins, M. Scheringer, R. C. Buck and K. Hungerbühler, Global emission inventories for C4-C14 perfluoroalkyl carboxylic acid (PFCA) homologues from 1951 to 2030, part I: production and emissions from quantifiable sources, Environ. Int., 2014, 70, 62–75 CrossRef CAS PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. D. Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. J. van Leeuwen, Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- K. Prevedouros, I. T. Cousins, R. C. Buck and S. H. Korzeniowski, Sources, fate and transport of perfluorocarboxylates, 2006 Search PubMed.
- E. Kissa and E. Kissa, Fluorinated surfactants and repellents, Marcel Dekker, 2001, p. 615 Search PubMed.
- A. G. Paul, K. C. Jones and A. J. Sweetman, A first global production, emission, and environmental inventory for perfluorooctane sulfonate, Environ. Sci. Technol., 2009, 43, 386–392 CrossRef CAS PubMed.
- T. Wang, R. Vestergren, D. Herzke, J. Yu and I. T. Cousins, Levels, Isomer Profiles, and Estimated Riverine Mass Discharges of Perfluoroalkyl Acids and Fluorinated Alternatives at the Mouths of Chinese Rivers, Environ. Sci. Technol., 2016, 50, 11584–11592 CrossRef CAS PubMed.
- N. Yamashita, K. Kannan, S. Taniyasu, Y. Horii, G. Petrick and T. Gamo, A global survey of perfluorinated acids in oceans, Mar. Pollut. Bull., 2005, 51, 658–668 CrossRef CAS PubMed.
- S. Wei, L. Q. Chen, S. Taniyasu, M. K. So, M. B. Murphy, N. Yamashita, L. W. Y. Yeung and P. K. S. Lam, Distribution of perfluorinated compounds in surface seawaters between Asia and Antarctica, Mar. Pollut. Bull., 2007, 54, 1813–1818 CrossRef CAS PubMed.
- L. Ahrens, J. L. Barber, Z. Xie and R. Ebinghaus, Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean, Environ. Sci. Technol., 2009, 43, 3122–3127 CrossRef CAS PubMed.
- L. Ahrens, Z. Xie and R. Ebinghaus, Distribution of perfluoroalkyl compounds in seawater from Northern Europe, Atlantic Ocean, and Southern Ocean, Chemosphere, 2010, 78, 1011–1016 CrossRef CAS PubMed.
- J. P. Benskin, D. C. G. Muir, B. F. Scott, C. Spencer, A. O. De Silva, H. Kylin, J. W. Martin, A. Morris, R. Lohmann, G. Tomy, B. Rosenberg, S. Taniyasu and N. Yamashita, Perfluoroalkyl acids in the Atlantic and Canadian arctic oceans, Environ. Sci. Technol., 2012, 46, 5815–5823 CrossRef CAS PubMed.
- M. Cai, Z. Zhao, Z. Yin, L. Ahrens, P. Huang, M. Cai, H. Yang, J. He, R. Sturm, R. Ebinghaus and Z. Xie, Occurrence of perfluoroalkyl compounds in surface waters from the North Pacific to the Arctic Ocean, Environ. Sci. Technol., 2012, 46, 661–668 CrossRef CAS PubMed.
- K. Y. Kwok, X. H. Wang, M. Ya, Y. Li, X. H. Zhang, N. Yamashita, J. C. W. Lam and P. K. S. Lam, Occurrence and distribution of conventional and new classes of per- and polyfluoroalkyl substances (PFASs) in the South China Sea, J. Hazard. Mater., 2015, 285, 389–397 CrossRef CAS PubMed.
- L. S. Haug, C. Thomsen and G. Becher, Time trends and the influence of age and gender on serum concentrations of perfluorinated compounds in archived human samples, Environ. Sci. Technol., 2009, 43, 2131–2136 CrossRef CAS PubMed.
- K. Kato, L. Y. Wong, L. T. Jia, Z. Kuklenyik and A. M. Calafat, Trends in exposure to polyfluoroalkyl chemicals in the U.S. population: 1999-2008, Environ. Sci. Technol., 2011, 45, 8037–8045 CrossRef CAS PubMed.
- T. Zhang, Q. Wu, H. W. Sun, X. Z. Zhang, S. H. Yun and K. Kannan, Perfluorinated compounds in whole blood samples from infants, children, and adults in China, Environ. Sci. Technol., 2010, 44, 4341–4347 CrossRef CAS PubMed.
- M. Houde, A. O. De Silva, D. C. G. Muir and R. J. Letcher, Monitoring of perfluorinated compounds in aquatic biota, Environ. Sci. Technol., 2011, 45(19), 7962–7973 CrossRef CAS PubMed.
- T. Frömel and T. P. Knepper, Biodegradation of fluorinated alkyl substances, Rev. Environ. Contam. Toxicol., 2010, 208, 161–177 CrossRef PubMed.
- J. M. Conder, R. A. Hoke, W. D. Wolf, M. H. Russell and R. C. Buck, Are PFCAs bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds, Environ. Sci. Technol., 2008, 42(4), 995–1003 CrossRef CAS PubMed.
- C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens and J. Seed, Perfluoroalkyl acids: a review of monitoring and toxicological findings, 2007 Search PubMed.
- European Union, Commission Regulation (EU) No. 757/2010, 2010.
- UNEP, Stockholm Convention on Persistent Organic Pollutants, Annex B, 2001 Search PubMed.
- S. K. Kim and K. Kannan, Perfluorinated acids in air, rain, snow, surface runoff, and lakes: relative importance of pathways to contamination of urban lakes, Environ. Sci. Technol., 2007, 41, 8328–8334 CrossRef CAS PubMed.
- A. Jahnke, U. Berger, R. Ebinghaus and C. Temme, Latitudinal gradient of airborne polyfluorinated alkyl substances in the marine atmosphere between Germany and South Africa (53° N-33° S), Environ. Sci. Technol., 2007, 41, 3055–3061 CrossRef CAS PubMed.
- A. Dreyer, I. Weinberg, C. Temme and R. Ebinghaus, Polyfluorinated compounds in the atmosphere of the Atlantic and Southern Oceans: evidence for a global distribution, Environ. Sci. Technol., 2009, 43, 6507–6514 CrossRef CAS PubMed.
- A. Dreyer, V. Matthias, C. Temme and R. Ebinghaus, Annual time series of air concentrations of polyfluorinated compounds, Environ. Sci. Technol., 2009, 43, 4029–4036 CrossRef CAS PubMed.
- C. E. Müller, A. C. Gerecke, C. Bogdal, Z. Wang, M. Scheringer and K. Hungerbühler, Atmospheric fate of poly- and per fluorinated alkyl substances (PFASs): I. Day–night patterns of air concentrations in summer in Zurich, Switzerland, Environ. Pollut., 2012, 169, 196–203 CrossRef PubMed.
- K. Y. Kwok, S. Taniyasu, L. W. Y. Yeung, M. B. Murphy, P. K. S. Lam, Y. Horii, K. Kannan, G. Petrick, R. K. Sinha and N. Yamashita, Flux of perfluorinated chemicals through wet deposition in Japan, the United States, and several other countries, Environ. Sci. Technol., 2010, 44, 7043–7049 CrossRef CAS PubMed.
- B. F. Scott, C. Spencer, S. A. Mabury and D. C. G. Muir, Poly and perfluorinated carboxylates in north American precipitation, Environ. Sci. Technol., 2006, 40, 7167–7174 CrossRef CAS PubMed.
- S. Taniyasu, N. Yamashita, H. B. Moon, K. Y. Kwok, P. K. S. Lam, Y. Horii, G. Petrick and K. Kannan, Does wet precipitation represent local and regional atmospheric transportation by perfluorinated alkyl substances?, Environ. Int., 2013, 55, 25–32 CrossRef CAS PubMed.
- G. Shan, X. Chen and L. Zhu, Occurrence, fluxes and sources of perfluoroalkyl substances with isomer analysis in the snow of northern China, J. Hazard. Mater., 2015, 299, 639–646 CrossRef CAS PubMed.
- A. Dreyer, V. Matthias, I. Weinberg and R. Ebinghaus, Wet deposition of poly- and perfluorinated compounds in Northern Germany, Environ. Pollut., 2010, 158, 1221–1227 CrossRef CAS PubMed.
- M. Loewen, F. Wania, F. Wang and G. Tomy, Altitudinal transect of atmospheric and aqueous fluorinated organic compounds in Western Canada, Environ. Sci. Technol., 2008, 42, 2374–2379 CrossRef CAS PubMed.
- J. P. Benskin, V. Phillips, V. L. St. Louis and J. W. Martin, Source elucidation of perfluorinated carboxylic acids in remote alpine lake sediment cores, Environ. Sci. Technol., 2011, 45, 7188–7194 CrossRef CAS PubMed.
- T. Kirchgeorg, A. Dreyer, J. Gabrieli, N. Kehrwald, M. Sigl, M. Schwikowski, C. Boutron, A. Gambaro, C. Barbante and R. Ebinghaus, Temporal variations of perfluoroalkyl substances and polybrominated diphenyl ethers in alpine snow, Environ. Pollut., 2013, 178, 367–374 CrossRef CAS PubMed.
- X. Wang, C. Halsall, G. Codling, Z. Xie, B. Xu, Z. Zhao, Y. Xue, R. Ebinghaus and K. C. Jones, Accumulation of perfluoroalkyl compounds in Tibetan mountain snow: temporal patterns from 1980 to 2010, Environ. Sci. Technol., 2014, 48, 173–181 CrossRef CAS PubMed.
- C. A. Barton, L. E. Butler, C. J. Zarzecki, J. Flaherty and M. Kaiser, Characterizing perfluorooctanoate in ambient air near the fence line of a manufacturing facility: comparing modeled and monitored values, J. Air Waste Manage. Assoc., 2006, 56, 48–55 CrossRef CAS.
- G. Shan, M. Wei, L. Zhu, Z. Liu and Y. Zhang, Concentration profiles and spatial distribution of perfluoroalkyl substances in an industrial center with condensed fluorochemical facilities, Sci. Total Environ., 2014, 490, 351–359 CrossRef CAS PubMed.
- J. W. Martin, D. A. Ellis, S. A. Mabury, M. D. Hurley and T. J. Wallington, Atmospheric chemistry of perfluoroalkanesulfonamides: kinetic and product studies of the OH radical and Cl atom initiated oxidation of N-ethyl perfluorobutanesulfonamide, Environ. Sci. Technol., 2006, 40, 864–872 CrossRef CAS PubMed.
- G. Yarwood, S. Kemball-Cook, M. Keinath, R. L. Waterland, S. H. Korzeniowski, R. C. Buck, M. H. Russell and S. T. Washburn, High-resolution atmospheric modeling of fluorotelomer alcohols and perfluorocarboxylic acids in the North American troposphere, Environ. Sci. Technol., 2007, 41, 5756–5762 CrossRef CAS PubMed.
- C. J. Young and S. A. Mabury, Atmospheric perfluorinated acid precursors: chemistry, occurrence, and impacts, Rev. Environ. Contam. Toxicol., 2010, 208, 1–109 CrossRef CAS PubMed.
- U. Schenker, M. Scheringer, M. Macleod, J. W. Martin, I. T. Cousins and K. Hungerbühler, Contribution of volatile precursor substances to the flux of perfluorooctanoate to the arctic, Environ. Sci. Technol., 2008, 42, 3710–3716 CrossRef CAS PubMed.
- M. H. Plumlee, K. Mcneill and M. Reinhard, Indirect photolysis of perfluorochemicals: hydroxyl radical-initiated oxidation of N-ethyl perfluorooctane sulfonamido acetate (N-EtFOSAA) and other perfluoroalkanesulfonamides, Environ. Sci. Technol., 2009, 43, 3662–3668 CrossRef CAS PubMed.
- M. Reth, U. Berger, D. Broman, I. T. Cousins, E. D. Nilsson and M. S. McLachlan, Water-to-air transfer of perfluorinated carboxylates and sulfonates in a sea spray simulator, Environ. Chem., 2011, 8, 381–388 CrossRef CAS.
- C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen and L. K. Reid, Aerosol enrichment of the surfactant PFO and mediation of the water-air transport of gaseous PFOA, Environ. Sci. Technol., 2008, 42, 3969–3974 CrossRef CAS PubMed.
- D. A. Ellis and E. Webster, Response to Comment on “Aerosol Enrichment of the Surfactant PFO and Mediation of the Water-Air Transport of Gaseous PFOA”, Environ. Sci. Technol., 2009, 43, 1234–1235 CrossRef CAS.
- K. U. Goss, The pKa values of PFOA and other highly fluorinated carboxylic acids, Environ. Sci. Technol., 2008, 42, 456–458 CrossRef CAS PubMed.
- J. Cheng, E. Psillakis, M. R. Hoffmann and A. J. Colussi, Acid dissociation versus molecular association of perfluoroalkyl oxoacids: environmental implications, J. Phys. Chem. A, 2009, 113, 8152–8156 CrossRef CAS PubMed.
- L. Vierke, U. Berger and I. T. Cousins, Estimation of the acid dissociation constant of perfluoroalkyl carboxylic acids through an experimental investigation of their water-to-air transport, Environ. Sci. Technol., 2013, 47, 11032–11039 CrossRef CAS PubMed.
- J. H. Johansson, H. Yan, U. Berger and I. T. Cousins, Water-to-air transfer of branched and linear PFOA: influence of pH, concentration and water type, Emerging Contam., 2017, 3, 46–53 CrossRef.
- J. M. Armitage, M. Macleod and I. T. Cousins, Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources, Environ. Sci. Technol., 2009, 43, 5830–5836 CrossRef CAS PubMed.
- J. M. Armitage, M. Macleod and I. T. Cousins, Modeling the global fate and transport of perfluorooctanoic acid (PFOA) and perfluorooctanoate (PFO) emitted from direct sources using a multispecies mass balance model, Environ. Sci. Technol., 2009, 43, 1134–1140 CrossRef CAS PubMed.
- J. M. Armitage, U. Schenker, M. Scheringer, J. W. Martin, M. Macleod and I. T. Cousins, Modeling the global fate and transport of perfluorooctane sulfonate (PFOS) and precursor compounds in relation to temporal trends in wildlife exposure, Environ. Sci. Technol., 2009, 43, 9274–9280 CrossRef CAS PubMed.
- R. J. Cipriano and D. C. Blanchard, Bubble and aerosol spectra produced by a laboratory “breaking wave”, J. Geophys. Res., 1981, 86, 8085 CrossRef.
- X. Wang, G. B. Deane, K. A. Moore, O. S. Ryder, M. D. Stokes, C. M. Beall, D. B. Collins, M. V. Santander, S. M. Burrows, C. M. Sultana and K. A. Prather, The role of jet and film drops in controlling the mixing state of submicron sea spray aerosol particles, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 201702420 Search PubMed.
- D. E. Spiel, The number and size of jet drops produced by air bubbles bursting on a fresh water surface, J. Geophys. Res., 1994, 99296, 289–310 Search PubMed.
- D. E. Spiel, On the births of film drops from bubbles bursting on seawater surfaces, J. Geophys. Res.: Oceans, 1998, 103, 24907–24918 CrossRef.
- A. H. Woodcock, Salt Nuclei in Marine Air as a Function of Altitude and Wind Force, J. Meteorol., 1953, 10, 362–371 CrossRef.
- E. D. Nilsson, Ã. Rannik, E. Swietlicki, C. Leck, P. P. Aalto, J. Zhou and M. Norman, Turbulent aerosol fluxes over the Arctic Ocean: 2. Wind-driven sources from the sea, J. Geophys. Res.: Atmos., 2001, 106, 32139–32154 CrossRef.
- E. R. Lewis and S. E. Schwartz, Sea salt aerosol production: mechanisms, methods, measurements and models – a critical review, American Geophysical Union, Washington, D. C., 2004, vol. 152, pp. 1–408 Search PubMed.
- R. L. Modini, L. M. Russell, G. B. Deane and M. D. Stokes, Effect of soluble surfactant on bubble persistence and bubble-produced aerosol particles, J. Geophys. Res.: Atmos., 2013, 118, 1388–1400 Search PubMed.
- F. P. Parungo, C. T. Nagamoto, J. Rosinski and P. L. Haagenson, A study of marine aerosols over the Pacific Ocean, J. Atmos. Chem., 1986, 4, 199–226 CrossRef CAS.
- D. C. Blanchard, The ejection of drops from the sea and their enrichment with bacteria and other materials: a review, Estuaries, 1989, 12, 127–137 CrossRef CAS.
- C. Leck and E. K. Bigg, Biogenic particles in the surface microlayer and overlaying atmosphere in the central Arctic Ocean during summer, Tellus, Ser. B: Chem. Phys. Meteorol., 2005, 57, 305–316 CrossRef.
- M. C. Facchini, M. Rinaldi, S. Decesari, C. Carbone, E. Finessi, M. Mircea, S. Fuzzi, D. Ceburnis, R. Flanagan, E. D. Nilsson, G. de Leeuw, M. Martino, J. Woeltjen and C. D. O'Dowd, Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates, Geophys. Res. Lett., 2008, 35, L17814 CrossRef.
- Applied Chemistry at Protein Interfaces, ed. R. E. Baier, 1975, vol. 145, ch. 18, pp 360–387, DOI:10.1021/ba-1975-0145.ch018.
- F. MacIntyre, The Sea, John Wiley and Sons, New York, 1974, vol. 5, pp. 245–299 Search PubMed.
- C. Oppo, S. Bellandi, N. Degli Innocenti, A. M. Stortini, G. Loglio, E. Schiavuta and R. Cini, Surfactant components of marine organic matter as agents for biogeochemical fractionation and pollutant transport via marine aerosols, Mar. Chem., 1999, 63, 235–253 CrossRef CAS.
- A. Cincinelli, A. M. Stortini, M. Perugini, L. Checchini and L. Lepri, Organic pollutants in sea-surface microlayer and aerosol in the coastal environment of Leghorn – (Tyrrhenian Sea), Mar. Chem., 2001, 76, 77–98 CrossRef CAS.
- C. D. O'Dowd, M. C. Facchini, F. Cavalli, D. Ceburnis, M. Mircea, S. Decesari, S. Fuzzi, Y. J. Yoon and J.-P. Putaud, Biogenically driven organic contribution to marine aerosol, Nature, 2004, 431, 676–680 CrossRef PubMed.
- W. C. Keene, H. Maring, J. R. Maben, D. J. Kieber, A. A. P. Pszenny, E. E. Dahl, M. A. Izaguirre, A. J. Davis, M. S. Long, X. Zhou, L. Smoydzin and R. Sander, Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface, J. Geophys. Res.: Atmos., 2007, 112, D21202 CrossRef.
- E. Fuentes, H. Coe, D. Green and G. McFiggans, Laboratory-generated primary marine aerosol via bubble-bursting and atomization, Atmospheric Measurement Techniques Discussions, 2010, 3, 141–162 CrossRef CAS.
- C. Leck and E. Svensson, Importance of aerosol composition and mixing state for cloud droplet activation over the Arctic pack ice in summer, Atmos. Chem. Phys., 2015, 15, 2545–2568 CrossRef CAS.
- M. D. Stokes, G. B. Deane, K. Prather, T. H. Bertram, M. J. Ruppel, O. S. Ryder, J. M. Brady and D. Zhao, A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols, Atmos. Meas. Tech., 2013, 6, 1085–1094 CrossRef.
- I. Ebersbach, S. M. Ludwig, M. Constapel and H. W. Kling, An alternative treatment method for fluorosurfactant-containing wastewater by aerosol-mediated separation, Water Res., 2016, 101, 333–340 CrossRef CAS PubMed.
- T. Kudo, K. Sekiguchi, K. Sankoda, N. Namiki and S. Nii, Effect of ultrasonic frequency on size distributions of nanosized mist generated by ultrasonic atomization, Ultrason. Sonochem., 2017, 37, 16–22 CrossRef CAS PubMed.
- D. B. Collins, D. F. Zhao, M. J. Ruppel, O. Laskina, J. R. Grandquist, R. L. Modini, M. D. Stokes, L. M. Russell, T. H. Bertram, V. H. Grassian, G. B. Deane and K. A. Prather, Direct aerosol chemical composition measurements to evaluate the physicochemical differences between controlled sea spray aerosol generation schemes, Atmos. Meas. Tech., 2014, 7, 3667–3683 CrossRef.
- E. M. Mårtensson, E. D. Nilsson, G. de Leeuw, L. H. Cohen and H.-C. Hansson, Laboratory simulations and parameterization of the primary marine aerosol production, J. Geophys. Res., 2003, 108, 1–12 CrossRef.
- C. A. Tyree, V. M. Hellion, O. A. Alexandrova and J. O. Allen, Foam droplets generated from natural and artificial seawaters, J. Geophys. Res.: Atmos., 2007, 112, D12204 CrossRef.
- J. Y. Park, S. Lim and K. Park, Mixing state of submicrometer sea spray particles enriched by insoluble species in bubble-bursting experiments, J. Atmospheric Ocean. Technol., 2014, 31, 93–104 CrossRef.
- N. W. May, J. L. Axson, A. Watson, K. A. Pratt and A. P. Ault, Lake spray aerosol generation: a method for producing representative particles from freshwater wave breaking, Atmos. Meas. Tech., 2016, 9, 4311–4325 CrossRef CAS.
- M. E. Salter, E. D. Nilsson, A. Butcher and M. Bilde, On the seawater temperature dependence of the sea spray aerosol generated by a continuous plunging jet, J. Geophys. Res.: Atmos., 2014, 119, 9052–9072 Search PubMed.
- G. W. Harvey, Microlayer collection from the sea surface: a new method and initial results, Limnol. Oceanogr., 1966, 11, 608–613 CrossRef.
- J. Löfstedt Gilljam, J. Leonel, I. T. Cousins and J. P. Benskin, Is Ongoing Sulfluramid Use in South America a Significant Source of Perfluorooctanesulfonate (PFOS)? Production Inventories, Environmental Fate, and Local Occurrence, Environ. Sci. Technol., 2016, 50, 653–659 CrossRef PubMed.
- J. P. Benskin, M. G. Ikonomou, M. B. Woudneh and J. R. Cosgrove, Rapid characterization of perfluoralkyl carboxylate, sulfonate, and sulfonamide isomers by high-performance liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 2012, 1247, 165–170 CrossRef CAS PubMed.
- R. A. Duce, W. Stumm and J. M. Prospero, Working symposium on sea-air chemistry: summary and recommendations, J. Geophys. Res., 1972, 77, 5059–5061 CrossRef.
- T. Iversen, M. Bentsen, I. Bethke, J. B. Debernard, A. Kirkevåg, O. Seland, H. Drange, J. E. Kristjánsson, I. Medhaug, M. Sand and I. A. Seierstad, The Norwegian Earth System Model, NorESM1-M – Part 2: Climate Response and Scenario Projections, Geoscientific Model Development Discussions, 2012, 5, 2933–2998 CrossRef.
- M. Bentsen, I. Bethke, J. B. Debernard, T. Iversen, A. Kirkevåg, O. Seland, H. Drange, C. Roelandt, I. A. Seierstad, C. Hoose and J. E. Kristjánsson, The Norwegian Earth System Model, NorESM1-M – Part 1: Description and Basic Evaluation, Geoscientific Model Development Discussions, 2012, 5, 2843–2931 CrossRef.
- M. E. Salter, P. Zieger, J. C. Acosta Navarro, H. Grythe, A. Kirkeväg, B. Rosati, I. Riipinen and E. D. Nilsson, An empirically derived inorganic sea spray source function incorporating sea surface temperature, Atmos. Chem. Phys., 2015, 15, 11047–11066 CrossRef CAS.
- L. Ahrens, W. Gerwinski, N. Theobald and R. Ebinghaus, Sources of polyfluoroalkyl compounds in the North Sea, Baltic Sea and Norwegian Sea: evidence from their spatial distribution in surface water, Mar. Pollut. Bull., 2010, 60, 255–260 CrossRef CAS PubMed.
- Z. Zhao, Z. Xie, A. Möller, R. Sturm, J. Tang, G. Zhang and R. Ebinghaus, Distribution and long-range transport of polyfluoroalkyl substances in the Arctic, Atlantic Ocean and Antarctic coast, Environ. Pollut., 2012, 170, 71–77 CrossRef CAS PubMed.
- B. González-Gaya, J. Dachs, J. L. Roscales, G. Caballero and B. Jiménez, Perfluoroalkylated Substances in the Global Tropical and Subtropical Surface Oceans, Environ. Sci. Technol., 2014, 48, 13076–13084 CrossRef PubMed.
- L. W. Yeung, C. Dassuncao, S. Mabury, E. M. Sunderland, X. Zhang and R. Lohmann, Vertical Profiles, Sources, and Transport of PFASs in the Arctic Ocean, Environ. Sci. Technol., 2017, 51, 6735–6744 CrossRef CAS PubMed.
- C. Textor, M. Schulz, S. Guibert, S. Kinne, Y. Balkanski, S. Bauer, T. Berntsen, T. Berglen, O. Boucher, M. Chin, F. Dentener, T. Diehl, R. Easter, H. Feichter, D. Fillmore, S. Ghan, P. Ginoux, S. Gong, A. Grini, J. Hendricks, L. Horowitz, P. Huang, I. Isaksen, I. Iversen, S. Kloster, D. Koch, A. Kirkevåg, J. E. Kristjansson, M. Krol, A. Lauer, J. F. Lamarque, X. Liu, V. Montanaro, G. Myhre, J. Penner, G. Pitari, S. Reddy, Ã. Seland, P. Stier, T. Takemura and X. Tie, Analysis and quantification of the diversities of aerosol life cycles within AeroCom, Atmos. Chem. Phys., 2006, 6, 1777–1813 CrossRef CAS.
- G. de Leeuw, E. Andreas, M. Anguelova, C. Fairall, R. Ernie, C. O'Dowd, M. Schulz and S. Schwartz, Production Flux of Sea-Spray Aerosol, Rev. Geophys., 2011, 49, 1–39 CrossRef.
- H. M. Scholberg, R. A. Guenthner and R. I. Coon, Surface Chemistry of Fluorocarbons and their Derivatives, J. Phys. Chem., 1953, 57, 923–925 CrossRef CAS.
- H. B. Klevens, Association in fluoroacids. Spectral charakteristics of added dyes, Kolloid-Z., 1958, 158, 53–58 CrossRef CAS.
- M. Bernett and W. Zisman, Wetting of Low-Energy Solids by Aqueous Solutions of Highly Fluorinated Acids and Salts, J. Phys. Chem., 1959, 63, 1911–1916 CrossRef CAS.
- M. K. Bernett and W. A. Zisman, Surface properties of perfluoro acids as affected by terminal branching and chlorine substitution, Phys. Chem., 1967, 71, 2075–2082 CrossRef CAS.
- K. Shinoda, M. Hato and T. Hayashi, Physicochemical properties of aqueous solutions of fluorinated surfactants, J. Phys. Chem., 1972, 76, 909–914 CrossRef CAS.
- K. Shinoda and H. Nakayama, Separate determinations of the surface excesses of surface-active ions and of gegenions at the air-solution interface, J. Colloid Sci., 1963, 18, 705–712 CrossRef CAS.
- H. Nakayama and K. Shinoda, The Effect of Added Sodium Dodecyl Salts on the Solubilities and Krafft Points Sulfate and Potassium, Bull. Chem. Soc. Jpn., 1967, 40, 1797–1799 CrossRef CAS.
- J. H. Johansson, Y. Shi, M. Salter and I. T. Cousins, Spatial variation in the atmospheric deposition of perfluoroalkyl acids: source elucidation through analysis of isomer patterns, Environ. Sci.: Processes Impacts, 2018, 20, 997–1006 RSC.
- J. P. Benskin, L. Ahrens, D. C. G. Muir, B. F. Scott, C. Spencer, B. Rosenberg, G. Tomy, H. Kylin, R. Lohmann and J. W. Martin, Manufacturing origin of perfluorooctanoate (PFOA) in Atlantic and Canadian Arctic seawater, Environ. Sci. Technol., 2012, 46, 677–685 CrossRef CAS PubMed.
- B. T. Mader, Comment on “Aerosol Enrichment of the Surfactant PFO and Mediation of the Water-Air Transport of Gaseous PFOA”, Environ. Sci. Technol., 2009, 43, 1232–1233 CrossRef CAS PubMed.
- X. Ju, Y. Jin, K. Sasaki and N. Saito, Perfluorinated surfactants in surface, subsurface water and microlayer from Dalian coastal waters in China, Environ. Sci. Technol., 2008, 42, 3538–3542 CrossRef CAS PubMed.
- S. Wang, H. Wang, W. Zhao, Y. Cao and Y. Wan, Investigation on the distribution and fate of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in a sewage-impacted bay, Environ. Pollut., 2015, 205, 186–198 CrossRef CAS PubMed.
- K. A. Hunter and P. S. Liss, The input of organic material to the oceans: air-sea interactions and the organic chemical composition of the sea surface, Mar. Chem., 1977, 5, 361–379 CrossRef CAS.
- R. E. Cochran, T. Jayarathne, E. A. Stone and V. H. Grassian, Selectivity Across the Interface: A Test of Surface Activity in the Composition of Organic-Enriched Aerosols from Bubble Bursting, J. Phys. Chem. Lett., 2016, 7, 1692–1696 CrossRef CAS PubMed.
- S. Xie, Y. Lu, T. Wang, S. Liu, K. Jones and A. Sweetman, Estimation of PFOS emission from domestic sources in the eastern coastal region of China, Environ. Int., 2013, 59, 336–343 CrossRef CAS PubMed.
- Z. Wang, J. M. Boucher, M. Scheringer, I. T. Cousins and K. Hungerbuehler, Toward a comprehensive global emission inventory of C4–C10 perfluoroalkane sulfonic acids (PFSAs) and related precursors: focus on the life cycle of C8-based products and ongoing industrial transition, Environ. Sci. Technol., 2017, 51(8), 4482–4493 CrossRef CAS PubMed.
- N. I. Kristiansen, A. Stohl, D. J. Olivié, B. Croft, O. A. Søvde, H. Klein, T. Christoudias, D. Kunkel, S. J. Leadbetter, Y. H. Lee, K. Zhang, K. Tsigaridis, T. Bergman, N. Evangeliou, H. Wang, P. L. Ma, R. C. Easter, P. J. Rasch, X. Liu, G. Pitari, G. Di Genova, S. Y. Zhao, Y. Balkanski, S. E. Bauer, G. S. Faluvegi, H. Kokkola, R. V. Martin, J. R. Pierce, M. Schulz, D. Shindell, H. Tost and H. Zhang, Evaluation of observed and modelled aerosol lifetimes using radioactive tracers of opportunity and an ensemble of 19 global models, Atmos. Chem. Phys., 2016, 16, 3525–3561 CrossRef CAS.
- Z. Zhao, Z. Xie, J. Tang, R. Sturm, Y. Chen, G. Zhang and R. Ebinghaus, Seasonal variations and spatial distributions of perfluoroalkyl substances in the rivers Elbe and lower Weser and the North Sea, Chemosphere, 2015, 129, 118–125 CrossRef CAS PubMed.
- A. Qureshi, M. MacLeod and K. Hungerbühler, Modeling aerosol suspension from soils and oceans as sources of micropollutants to air, Chemosphere, 2009, 77, 495–500 CrossRef CAS PubMed.
- E. Webster and D. A. Ellis, Potential role of sea spray generation in the atmospheric transport of perfluorocarboxylic acids, Environ. Toxicol. Chem., 2010, 29, 1703–1708 CrossRef CAS PubMed.
- C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir and S. A. Mabury, Perfluorinated acids in arctic snow: new evidence for atmospheric formation, Environ. Sci. Technol., 2007, 41, 3455–3461 CrossRef CAS PubMed.
- N. Yamashita, S. Taniyasu, G. Petrick, S. Wei, T. Gamo, P. K. S. Lam and K. Kannan, Perfluorinated acids as novel chemical tracers of global circulation of ocean waters, Chemosphere, 2008, 70, 1247–1255 CrossRef CAS PubMed.
- R. Lohmann, E. Jurado, H. A. Dijkstra and J. Dachs, Vertical eddy diffusion as a key mechanism for removing perfluorooctanoic acid (PFOA) from the global surface oceans, Environ. Pollut., 2013, 179, 88–94 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8em00525g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |