
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of bicarbonate and phosphate on arsenic release from mining-impacted sediments in the Cheyenne River watershed, South Dakota, USA†
Cherie L.
DeVore
a,
Lucia
Rodriguez-Freire
 b,
Abdul
Mehdi-Ali
c,
Carlyle
Ducheneaux
d,
Kateryna
Artyushkova
e,
Zhe
Zhou
f,
Drew E.
Latta
f,
Virgil W.
Lueth
g,
Melissa
Gonzales
h,
Johnnye
Lewis
i and
José M.
Cerrato
*a
b,
Abdul
Mehdi-Ali
c,
Carlyle
Ducheneaux
d,
Kateryna
Artyushkova
e,
Zhe
Zhou
f,
Drew E.
Latta
f,
Virgil W.
Lueth
g,
Melissa
Gonzales
h,
Johnnye
Lewis
i and
José M.
Cerrato
*a
aDepartment of Civil Engineering, University of New Mexico, MSC01 1070, Albuquerque, New Mexico 87131, USA. E-mail: jcerrato@unm.edu; Fax: +1 505-277-1918; Tel: +1 505-277-0870
bDepartment of Civil and Environmental Engineering, University Heights, New Jersey Institute of Technology, 266 Colton Hall, Newark, New Jersey 07102, USA
cDepartment of Earth and Planetary Sciences, University of New Mexico, Albuquerque, MSC03 2040, New Mexico 87131, USA
dDepartment of Environmental and Natural Resources, Cheyenne River Sioux Tribe, Eagle Butte, South Dakota 57625, USA
eDepartment of Chemical and Biological Engineering, Center for Microengineered Materials, University of New Mexico, Albuquerque, New Mexico 87131, USA
fDepartment of Civil and Environmental Engineering/IIHR, The University of Iowa, 4105 Seamans Center, Iowa City, Iowa 52242, USA
gNew Mexico Bureau of Geology & Mineral Resources, New Mexico Tech, 801 Leroy Place, Socorro, New Mexico 87801, USA
hSchool of Medicine, Department of Internal Medicine, University of New Mexico Health Sciences Center, MSC10 5550, Albuquerque, NM 87131, USA
iCommunity Environmental Health Program, College of Pharmacy, 1 University of New Mexico, MSC09 5360, Albuquerque, USA
First published on 4th February 2019
Abstract
The mobilization of arsenic (As) from riverbank sediments affected by the gold mining legacy in north-central South Dakota was examined using aqueous speciation chemistry, spectroscopy, and diffraction analyses. Gold mining resulted in the discharge of approximately 109 metric tons of mine waste into Whitewood Creek (WW) near the Homestake Mine and Cheyenne River at Deal Ranch (DR), 241 km downstream. The highest concentrations of acid-extractable As measured from solid samples was 2020 mg kg−1 at WW and 385 mg kg−1 at DR. Similar sediment mineralogy between WW and DR was identified using XRD, with the predominance of alumino-silicate and iron-bearing minerals. Alkalinity measured in surface water at both sites ranged from 1000 to 2450 mg L−1 as CaCO3 (10–20 mM HCO3− at pH 7). Batch laboratory experiments were conducted under oxidizing conditions to evaluate the effects of NaHCO3 (0.2 mM and 20 mM) and NaH2PO3 (0.1 and 10 mM) on the mobilization of As. These ions are relevant for the site due to the alkaline nature of the river and nutrient mobilization from the ranch. The range of As(V) release with the NaHCO3 treatment was 17–240 μg L−1. However, the highest release (6234 μg L−1) occurred with 10 mM NaH2PO3, suggesting that As release is favored by competitive ion displacement with PO43− compared to HCO3−. Although higher total As was detected in WW solids, the As(V) present in DR solids was labile when reacted with NaHCO3 and NaH2PO3, which is a relevant finding for communities living close to the river bank. The results from this study aid in a better understanding of As mobility in surface water sites affected by the mining legacy.
Environmental significanceThis study integrated laboratory experiments with aqueous chemistry and spectroscopy measurements to investigate arsenic (As) speciation and mobilization in sediments collected from a site in South Dakota affected by the mining legacy. The reaction of sediments with bicarbonate and phosphate can result in the release of considerable As to solution due to competitive ion displacement. This result has important implications given the alkaline nature of surface waters in the study site and the relevance of phosphate as a proxy to evaluate the effect of nutrient inputs into the watershed resulting from livestock and agricultural activities. The presence of labile As from sediments on tribal land should also be considered for the assessment of As exposures. |
1. Introduction
Arsenic (As) can naturally occur at a wide range of concentrations in waters world-wide.1 Additionally, anthropogenic activities such as mining can impact surface waters and associated sediments, causing the occurrence of elevated concentrations of As and other co-occurring metals.2 The western part of the United States has a legacy of over 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 abandoned mines and represents a particular area of exposure to mine wastes.3 A variety of adverse health effects ranging from skin and bladder cancers to cardiovascular and neurological diseases, have been associated with As exposure, primarily from ingestion of As-contaminated drinking water.4–6 The World Health Organization and the United States Environmental Protection Agency have set a standard of 10 μg As L−1 for drinking water.7,8
000 abandoned mines and represents a particular area of exposure to mine wastes.3 A variety of adverse health effects ranging from skin and bladder cancers to cardiovascular and neurological diseases, have been associated with As exposure, primarily from ingestion of As-contaminated drinking water.4–6 The World Health Organization and the United States Environmental Protection Agency have set a standard of 10 μg As L−1 for drinking water.7,8
The Cheyenne River watershed is a specific site that has experienced As release to surface water due to gold mining legacy.4 Mineral extraction and waste disposal near Lead, South Dakota directly discharged spent mine tailings into Whitewood Creek from 1876 until 1977. Approximately 109 metric tons of tailings containing As and other heavy metals have been deposited downstream of Whitewood Creek along the Belle Fourche and Cheyenne Rivers.9 Whitewood Creek drains the area of Homestake Mine and is a tributary of the Belle Fourche, which flows into the Cheyenne River.10 Physical and chemical processes have resulted in substantial transport and deposition of As-enriched materials within Whitewood Creek, the Belle Fourche, and Cheyenne River.11,12 Deposits from the mine wastes, which include sulfide and iron minerals, associated with gold ore and 9.5 metric tons of As in the form of arsenopyrite (FeAsS), were found along Whitewood Creek in the 1970s when production ceased at the gold mine.13 The Cheyenne River flows to the Missouri River, through the lands of the Cheyenne River Sioux Tribe (CRST), located 241 km downstream of the Homestake Mine. There are existing concerns about potential metal exposures in this community.3
Complex geochemical processes affect the mobilization of As in the environment. Arsenic release in source waters can result from the dissolution of iron oxyhydroxides and other metal oxides.14 Arsenic can occur as As(III) and As(V) under oxidation-reduction potential (Eh) and pH conditions that are relevant to most surface and groundwaters.1,15 Usually, these oxidation states of As are arsenite (As(III)) and arsenate (As(V)), and the As speciation is important due to the higher toxicity and enhanced the mobilization of As(III).1,16 Under reducing conditions, soluble As concentrations can be affected by desorption of As from dissolution of iron (Fe) and manganese (Mn) oxides and the sorption or co-precipitation of As with carbonates and sulfide minerals. These geochemical factors differ from those influencing soluble As concentrations under oxidizing conditions. For example, arsenopyrite is unstable under surface oxidizing conditions.17 The most frequently observed product of arsenopyrite weathering is scorodite (FeAsO4·2H2O). Other products of arsenopyrite oxidation are arseniosiderite [Ca2Fe3(AsO4)3O2·3H2O] and jarosite (KFe3(SO4)2(OH)6).12 Amorphous hydrous ferric arsenate-sulfate, pharmacosiderite [KFe4(AsO4)3(OH)4·6–7H2O], and arsenolite (As2O3) are less common products.11 Arsenic can be associated with jarosite, which is thermodynamically unstable at circumneutral pH. Jarosite dissolution can cause the release of metals in water at specific pH and redox conditions.18,19
Interactions at the mineral-water interface play a fundamental role in controlling As transport. Arsenate can form inner-sphere surface complexes on both amorphous aluminum (Al) and Fe oxide, while arsenite forms both inner- and outer-sphere surface complexes on amorphous Fe-oxides.20 In circumneutral environmental conditions, the reduction from arsenate to arsenite can be important for As release, especially since arsenite adsorbs much less strongly to Fe and Mn oxides.21,22 Additionally, the presence of similar high-affinity anions for surface sites on Fe-oxides, such as phosphate, silicic acid, and bicarbonate can contribute to the mobility of As.16,23–25 Phosphate is particularly effective at competing with arsenate for sorption sites on Fe oxide minerals.26 Previous adsorption studies with carbonate and phosphate have focused on groundwater environments with the assumption that native phosphate in the subsurface is very low, so in these cases, equimolar phosphate with As(V) will not interfere with the adsorption of As(V) in groundwater systems.27 However, phosphate concentrations suppressed As(V) adsorption onto alluvial sediments when it was present at concentrations that were 10 times higher than that of As(V).27 Thus, competitive ion displacement and mobilization of As in the presence of phosphate represents a mechanism of release and transport into the aqueous phase.28 Areas with nutrient mobilization via agricultural runoff and phosphate-based fertilizers can be at risk for As mobilization.29 In addition, carbonate ions can play a role in contaminant retention and release via competitive adsorption. An investigation of bicarbonate (HCO3−) in groundwater found that concentrations of released As from subsurface core sediment strongly depended on concentrations of HCO3−, with enhanced mobility at higher concentrations.30 In addition, carbonate concentrations modeled after typical values found in Bangladesh (0.01 M) after receiving considerable attention in the past two decades,24 is only half the concentration of carbonate observed in South Dakota due to the alkaline nature of regional soils.11
Surface complexation models and adsorption studies have been used to predict As mobilization in groundwater environments.28,30,31 Some have combined these models with experimental data,32 but there is still a lack of field studies confirmed by experimental data. While there is extensive literature on As mobilization, many of the experimental and environmental conditions do not apply to the Cheyenne River. First, many studies have focused on the mobilization of As from enriched sediment and pore waters at the Eh range found in groundwaters.22,33–37 Arsenic release into groundwater can be due to anoxic conditions that may interfere with the stability of Fe-oxides.15,38–40 However, fewer studies have emphasized better understanding As mobilization in surface oxidizing environments. Second, leaching of As into groundwater by carbonation of As sulfide minerals was reported, but this process may not apply to both the surface water conditions and specific As minerals present in mining ore deposits along the Cheyenne River.25 This river is a relevant surface water source for the Cheyenne River Sioux Tribe and has high alkalinity values ranging from 1000 mg L−1 (10 mM) to 2400 mg L−1 (24 mM) as CaCO3 near the mine waste source. There are not many field or laboratory studies in the literature that have been conducted to evaluate the effect of well-buffered surface waters at such high alkalinity levels which justifies the need for the present study.
The objective of this study is to investigate the effect of competing environmentally relevant anions on the release of As from solids collected from the Cheyenne River watershed exposed to surface oxidizing conditions. We integrated laboratory experiments with aqueous chemistry and spectroscopy measurements to investigate As speciation and mobilization in sediment collected from Whitewood Creek (near the mine source) and Cheyenne River (241 km downstream on tribal land). A novel aspect of this study consists on the investigation of chemical mechanisms that affect As release under surface oxidizing conditions with high concentrations of HCO3− (20 mM), attempting to represent the alkaline nature (10–20 mM) of the Cheyenne River and Whitewood Creek. We also investigated the competing effect of phosphate (PO43−) on As mobilization in As-bearing soils collected from land that is used for agriculture and livestock. The results of this study have relevant implications for informing tribal and regulatory decision makers, as well as environmental risk assessments and mitigation efforts for communities located near abandoned mine wastes with similar characteristics to those observed in the Cheyenne River.
2. Study area
The Cheyenne and Moreau River watersheds lie within the Great Plains physiographic province. The Cheyenne River Indian Reservation lies on a gently northwest dipping flank of the Williston watershed in north-central South Dakota. The area of the Reservation is approximately 10956 km2 and encompasses Dewey and Ziebach Counties. The topography of the Reservation is largely rolling upland that has been eroded by the Missouri River and its tributaries.41 About 85 percent of the land area is native grass and is used primarily for grazing livestock. The Cheyenne and Moreau Rivers are the largest rivers traversing the Cheyenne River Sioux Tribe (CRST) Reservation.
The Cheyenne River forms the southern boundary of the Reservation and is the largest tributary to the Missouri River within South Dakota (drainage area of about 66045 km2). The Cheyenne River originates in eastern Wyoming, flows southeast around the southern Black Hills, and then flows northeast across South Dakota to its mouth at Lake Oahe. The Cheyenne River is joined by the Belle Fourche River approximately 24 km southwest of the Reservation boundary. The Belle Fourche River flows northeast in Wyoming and then flows southeast around the northern Black Hills to its confluence with the Cheyenne River in South Dakota. The Belle Fourche River is the largest tributary to the Cheyenne River and drains about one-third of the entire Cheyenne River Watershed.42 The Whitewood Creek is a major tributary of the Belle Fourche River. Throughout the study, we refer to the upstream source as WW (Whitewood Creek) and downstream sample on tribal land as DR (Deal Ranch) (Fig. 1).
 | ||
| Fig. 1 Map of the study area in north central South Dakota. | ||
3. Materials and methods
3.1 Materials
During 2016, we collected water and sediment samples from the study area in the locations indicated in the map in Fig. 1. In June 2016, two water samples from the Cheyenne River (impacted by the Homestake Mine), and two samples from the Moreau River (not impacted by the Homestake Mine) were analyzed for pH, alkalinity, anions, and total and dissolved elements (see section 3.2 for details on methods for anions and total and dissolved elements analyses). A second sampling trip was conducted in September 2016 to collect water samples upstream of the Cheyenne River, Belle Fourche River and Whitewood Creek close to the Homestake Mine. For each sample, one liter of surface water was collected using a clean polypropylene Nalgene bottle and preserved with 2% trace metal grade nitric acid (HNO3−). Water samples were filtered using a 0.45 μm Basix filter®, and a portion of the volume was filtered using a 0.22 μm Basix filter® in the laboratory. Bottles were rinsed three times with sample water in the field before filling and sealing with no headspace. Samples were cooled to 4 °C and shipped for analyses to the University of New Mexico (UNM). Field parameters were measured using a YSI 650 MDS pH/temp/conductivity probe and calibrated on the day of sample collection. The global positioning system (GPS) coordinates were collected at each site location (see Table S1 in the ESI†).Sixteen sediment samples were collected from the river bank profile at co-located sites in June 2016 from Fig. 1. Ten additional sediment samples were collected upstream of the Cheyenne River, Belle Fourche River and Whitewood Creek in close proximity to the Homestake Mine in September 2016. A hand trowel was rinsed with 10% nitric acid solution and used to collect half a gallon of sediment from the river bank and bed. Samples were placed in one-gallon plastic bags and cooled to 4 °C for shipment with water samples to UNM. Throughout the study, we refer to the upstream source as WW (Whitewood Creek) and downstream sample on tribal land as DR (Deal Ranch). The authors were escorted by the CRST Department of Environment and Natural Resources staff while conducting sampling. Travel and time constraints to South Dakota by UNM collaborators reduced the number of sampling events and possible sampling locations.
3.2 Solution chemistry analyses
Aqueous elemental analyses for this study were performed using inductively coupled optical emission spectrometry (ICP-OES) (Perkin Elmer Optima 5300DV) and inductively coupled plasma-mass spectrometry (ICP-MS) (Perkin Elmer NexION 300D-Dynamic Reaction Cell) in aqueous solutions for trace element content (As, Cr, Fe, Mn, and U). Samples for ICP-MS were filtered using a 0.45 μm Basix filter and preserved with 2% HNO3−. A Thermo Fisher Scientific Ion Chromatogram (ICS-1100) was used to analyze 0.45 μm non-acidified water samples for major anion concentrations. Arsenic speciation was characterized using a Flexar 400 High-Performance Liquid Chromatography (HPLC) coupled with ICP-MS for As(III) and As(V) species. Samples were stabilized with a mobile phase solution (0.5 mM EDTA, 1 mM TBAoH and 1% MeOH) adjusted to pH 7 (±0.01). A CAPCELL PAK C18 column (250 mm × 4.6 mm, 5 μm particle size) was used for the separation of target As species. All reagents used in the experiments and analyses were analytical grade unless otherwise mentioned. Additional information about the HPLC-ICP-MS analyses can be found in ESI (Table S2†).3.3 Solid characterization analyses
Sediment samples were dried for 12 h at 60 °C in a controlled-temperature oven. Dried sediment samples were crushed and homogenized using a shatterbox. One gram of the dried sediment was weighted and added to a 50 mL digestion tube. All samples were acid digested (2.0 mL HNO3 (UHP) + 1 mL HCl (UHP)) in triplicate to determine extractable elemental concentrations. The mixture was digested for 60 min at 65 °C and then for an additional 60 min at 80 °C. Digested samples were diluted with deionized water to 25 mL. The digested and diluted samples were filtered through a 0.45 μm filter to remove any particulate matter for aqueous elemental analyses. Loss on Ignition (LOI) was used to estimate the organic matter content in sediment samples. Five grams of sample were weighed and dried at 104 °C for 12 hours. A calculation was made for mass loss difference after drying temperature increase from 104 °C to 550 °C for 5 hours (h).Dried solid samples from field and laboratory analyses were analyzed using X-ray photoelectron spectroscopy (XPS), X-ray powder diffraction (XRD), X-ray fluorescence (XRF) and Mössbauer spectroscopy. Measurements of X-ray fluorescence (XRF) were conducted to determine bulk elemental composition using a Rigaku ZSX with a rhodium X-ray rube that can be operated from 200 to 4000 watts (End window, Rh-anode, 4 kW, 60 kV). A Kratos AXIS-Ultra DLD X-ray Photoelectron Spectrometer was used to acquire elemental composition and oxidation states at the near surface (5–10 nm) of the solids. Pure (99%) Au powder was put onto samples for charge correction. Three areas per sample were analyzed using monochromatic Al Kα source and charge neutralization. CASAxps was used to process the spectra. The spectra were calibrated by adjusting the position of Au 4f spectra to 84 eV. Shirley background was used for Fe 3p and As 3d spectra processing. Fe 3p spectra were fitted with two symmetrical peaks due to Fe(II) and Fe(III). As 3d spectra were fitted with two symmetrical peaks due to As(III) and As(V) with each of them having a doublet separated by 0.77 eV. The positions of the peaks was determined previously from references materials analyzed and reported.43
A PanAnalytical XPert Pro Diffractometer was used for X-ray diffraction (XRD) analyses using CuK radiation for mineral identification of sediment samples. Samples were mounted in a rotating stage and were scanned from 5 to 70 degrees using 0.008 (°2θ) step size and 40.0 s scan step time. The XRD patterns were interpreted using PANALYTICAL X′PERT HIGHSCORE PLUS software. Mössbauer spectra were collected in transmission geometry with a constant acceleration drive system (SEE Co., Inc.) and a 57Co(Rh) source. The temperature was controlled in a Janis gas-exchange closed cycle cryostat down to a sample temperature of 15 K. Data were calibrated with an α-Fe foil at room temperature and fit with the Recoil software package using Voigt lineshapes. Quadrupole shift parameters are reported as 2ε, which is twice the parameter epsilon_0 in Recoil.
3.4. Laboratory experiments
Laboratory batch experiments were performed to evaluate the reaction of As in sediments from WW and DR with bicarbonate and phosphate concentrations relevant to the environment of the Cheyenne River Watershed. Batch reactors were operated in triplicate using deionized water, sodium bicarbonate (NaHCO3) stock solutions (0.2 mM and 20 mM) and sodium phosphate (NaH2PO3) stock solutions (0.1 mM and 10 mM), which were prepared based on aqueous chemistry results and literature review of the Cheyenne River.11 Five 100 mL centrifuge tubes were labeled for 20 mM NaHCO3; fifteen 100 mL for 0.2 mM NaHCO3 and 5 for DI water control. Five grams of dried WW and DR sediment was added per 100 mL centrifuge tube. A total of fifteen 100 mL centrifuge tubes was placed on a bench rotator for 96 h at 30 rpm. A volume of 3 mL of sample was collected at t = 0.5, 1, 2, 4, 6, 12, 24 and 96 h. Volume and pH were recorded at each time step. Samples were filtered using a 0.22 μm filter and cooled to 4 °C. 0.5 mL of sample was immediately placed in a 1.5 mL plastic HPLC vial and preserved in mobile phase solution to keep stable until aqueous elemental analyses were conducted.4. Results and discussion
4.1 Elemental composition of solid and water quality data from the site
Elevated As concentrations were measured in sediments in Whitewood Creek (WW) near the Homestake Mine, but also 241 km downstream in the Cheyenne River Sioux Tribe Reservation. Arsenic concentrations as high as 2020 mg kg−1 were measured in the sediments at WW. The WW flows into the Belle Fourche River and eventually into the Cheyenne River where high As concentrations were measured in the riverbank sediments. Arsenic concentrations in riverbank sediments more than 161 km downstream of the mine on tribal land at Deal Ranch (DR) and Cherry Creek were in the range of 90–394 mg kg−1 (Table 1). The As concentrations in these sediments exceed the regionally established background concentration of 10 mg kg−1, and it is in agreement with a 1989 USGS study which reported similar values in the Belle Fourche (>3500 mg kg−1) and the Cheyenne River (>530 mg kg−1) solids.4 However, low As concentrations were measured in the Moreau River, ranging from 7.4–14 mg kg−1, which was chosen as a reference location because it is not hydrologically connected to the Homestake Mine. Elemental bulk composition measured in sediments by XRF detected higher Fe and As content than acid extractable concentrations. Silicates are not readily dissolved by Aqua regia,44 which might explain the differences between XRF and acid digestion analyses for Fe in the samples (Table 2). The organic matter content in our samples was estimated by LOI and the mass loss observed in our sample was 2.23–2.99 (±0.005–0.006)%. The mass loss is likely attributed to the volatilization of organic matter within the sample. A previous study measured LOI at three sites along the Cheyenne River and were in the range of 0.5–1.0%,11 similar to the measurements in this study. The organic matter content found near the Cheyenne River is lower than what has been observed in other environmental systems. For instance, the LOI in agricultural soils is reported to be 65% and it ranges from 7% to 9% in soils that have been exposed to wildfires.45,46 These results suggest that, although mining operations have ceased for many decades, accumulation of As-impacted sediments has occurred on tribal land.| Site ID | Site | River | Distance from Mine (km) | As (mg kg−1) |
|---|---|---|---|---|
| a NC = not hydrologically connected to Cheyenne River. | ||||
| WW | Whitewood creek | Whitewood creek | 0.49 | 2040 (±118) |
| BF/WW | Belle/Whitewood | Belle Fourche | 5.70 | 18.2 (±1.52) |
| DR | Deal Ranch | Cheyenne River | 230 | 235 (±119) |
| CC | Cherry Creek | Cheyenne River | 237 | 394 (±54.7) |
| RL | Ross Lawrence | Moreau River-reference | NC | 14.1 (±1.85) |
| TB | Thunder Butte | Moreau River- reference | NC | 7.40 (±0.79) |
| WW XRF (mg kg−1) | WW acid-extractable (mg kg−1) | DR XRF (mg kg-−1) | DR acid-extractable (mg kg−1) | |
|---|---|---|---|---|
| Fe | 199143 (±2578) |
34541 (±12186) |
95156 (±256) |
244 (±9.10) |
| As | 4584 (±20) | 2040 (±118) | 1008 (±8.5) | 235 (±119) |
While the sediment concentration of As in the sediments was elevated, relatively low As concentrations were detected in surface waters at different locations along the Whitewood Creek, Belle Fourche River, Cheyenne River and Moreau River (Table 2). For instance, As measurements in surface water ranged from 2.25–62.2 μg L−1 in Whitewood Creek (WW) near the mine waste, and ranged from 3.74–5.84 μg L−1 (DR, CC) downstream along the Cheyenne River. The As concentrations in waters from the Moreau River ranged from 1.38–2.06 μg L−1. Additional samples were collected between WW and DR along the Belle Fourche River and were comparable to values measured in the Cheyenne River.11,47 The results from this study at Whitewood Creek are consistent with As measurements in a previous study, where concentrations vary between about 20 and 80 μg L−1 during the year in response to variations in groundwater inflow and dilution.4 As a reference for freshwater sources, aquatic life chronic level criterion continuous concentration is 150 μg As L−1.48
Additional water quality data were collected in these surface waters that are relevant to describing conditions that may mobilize metals in the environment. The pH in Whitewood and Belle Fourche Rivers closer to the mine waste ranged from 8.21–8.90 and alkalinity values ranged from 1342–2650 mg L−1 as CaCO3. The pH in the Cheyenne River 241 km downstream of Homestake Mine ranged from 7.69–8.32, with alkalinity 580–1000 mg L−1 as CaCO3 (Table 3). These alkalinity values are relatively high considering that this is a surface water source; the range of alkalinity values measured in this study are characteristic of waters associated with alkaline soils from this region of South Dakota.11 The major-ion composition of the water results from the cumulative effects of the interaction with alluvial aquifers, underlying shale units and transported mine waste. Sodium and calcium are the predominant cations, and sulfate is the predominant anion. Sulfate concentrations exceed the secondary drinking-water standard of 250 mg L−1 at all sampling sites,42 and range from about 1100 to more than 2500 mg L−1. The sources of sulfate in the alluvium can be attributed to both the shale and mine tailings.49
| Site ID | Site Name | River | Total As (μg L−1) | Alkalinity mg L−1 (mM) as CaCO3 | pH |
|---|---|---|---|---|---|
| WW | Whitewood Creek | Whitewood Creek | 62.16 | 2650 (2.7) | 8.50 |
| BF | Belle Fourche River | Belle Fourche | 19.0 | 2867 (28.7) | 8.21 |
| BF/WW | Belle Fourche/Whitewood | Belle Fourche | 2.25 | 1342 (13.4) | 8.90 |
| WA | Wasta | Cheyenne River | 6.68 | 1647 (16.5) | 7.39 |
| BR | Bridger | Cheyenne River | 6.10 | 1464 (14.6) | 8.30 |
| DR | Deal Ranch | Cheyenne River | 5.14 | 720 (7.2) | 7.82 |
| CC | Cherry Creek | Cheyenne River | 5.84 | 1000 (10) | 7.69 |
| RL | Ross Lawrence | Moreau River | 1.47 | 580 (5.8) | 8.32 |
| TB | Thunder Butte | Moreau River | 2.05 | 164 (1.6) | 8.23 |
4.2 Laboratory batch experiment: reactivity of As with bicarbonate
The release of As was investigated in batch experiments reacting sediments (WW, DR) using HCO3− (0.2 mM and 20 mM) and PO43− (0.1 mM and 10 mM) under surface oxidizing conditions. Note that concentrations used in previous studies for bicarbonate found in typical groundwaters are too low to be representative of Cheyenne River's surface water environment.24,27 Thus, the bicarbonate concentrations and pH selected for this study are characteristic of WW and DR surface waters. Also, phosphate was added to the experiment as a proxy to evaluate the effect of nutrient inputs into the Cheyenne River watershed resulting from livestock and agricultural activities in the area. The goal of these experiments is to use controlled laboratory settings to better understand processes that could be relevant to the mobilization of As in the study site.Increasing bicarbonate concentrations promote As mobilization from the sediments (Fig. 2). Low bicarbonate concentration (0.2 mM) representing typical values found in drinking waters (0.2–2.0 mM as CaCO3) released a maximum of 2.4 μg L−1 As(V) in WW near the mine (Fig. 2A) and a maximum of 48 μg L−1 as As(V) downstream of the mine at DR (Fig. 2B) over a period of 96 h. No As(III) was released with either treatment and fluctuations within 1 pH unit (7.34–8.23) were observed (Fig. S1†). The highest bicarbonate concentration (20 mM) emulating the high alkalinity measured in the Cheyenne River (1000 to 2100 mg L−1 as CaCO3, about 10–20 mM) mobilized a maximum of 14 μg L−1 As(V) from WW sediments (Fig. 2A) and a maximum 204 μg L−1 As(V) was released from DR sediments (Fig. 2B). Additionally, a control without bicarbonate (only DI water) was conducted in parallel and As(V) concentrations were below 100 μg L−1. Hence, the results from this experiment suggest that the presence of bicarbonate in surface water could cause As mobilization from the sediments.
 | ||
| Fig. 2 Release of As(V) after reaction of sediments reacted with de-ionized water (DI control), 0.2 mM, and 20 mM sodium bicarbonate solutions for: (A) Whitewood Creek (WW) and (B) Deal Ranch (DR). | ||
The presence of carbonates in groundwater systems with adsorbed As has been examined in previous studies that integrated controlled laboratory experiments with surface complexation modeling.21,26,50,51 A slight decrease in arsenate adsorption was observed at pH 7.75–8.00 in early reaction times in one study. This decrease in As adsorption could be due to the increased negative charge at the surface of ferric oxides such as hematite caused by adsorbed carbonate.52 In addition, the importance of carbonate-complexes on up to 70% of ferrihydrite surface sites in the subsurface was illustrated in surface chemistry models.30 There are relevant implications for As mobility in groundwaters where these complexes can either enhance or suppress adsorption.31,53 The competitive effect of carbonates has been mostly studied in batch experiments reacting synthetic solid samples with solutions having chemical characteristics similar to those in groundwater.25,30,54 These observations are important to consider, given the abundant carbonate minerals interacting with the alluvium and overlying surface water.49 Future studies are necessary to investigate surface complexation reactions in natural surface water systems. Additional experiments were conducted to evaluate the release of As after reaction with phosphate, a known chemical analog of arsenate (note the similarities in pKa values indicated in Table S3†) and a proxy for nutrient inputs into the surface waters evaluated in this study.
4.3 Batch experiment: reactivity of As with phosphate
The release of As(V) was at least 10-times higher after reacting mine waste solids with phosphate, compared to the experiments with bicarbonate. Our experiments were conducted with a low (0.1 mM) and high (10 mM) phosphate concentration. The low concentration was selected because phosphate measured in the surface water was not detectable during our field sampling at sites shown in Fig. 1. However, previous monitoring activities conducted by the CRST Environment Department have reported total phosphorus concentrations in the range of 0.17–3.4 mg L−1 (0.01–0.24 mM).41 Most of the land base in the Cheyenne River watershed is dedicated to livestock purposes and several irrigation areas exist within the basin. Previous studies have used a range of 0.57–16.67 mM PO43− in microcosm studies with P amended contaminated soil to understand the influence of phosphate on As leaching.29,55 The 0.1 and 10 mM concentrations selected for our study are similar to the treatments in the batch and column experiments with P and As-amended soils from these previous studies29,55 A maximum release of 1.6 μg L−1 As was observed in experiments reacting 0.1 mM phosphate with WW solids (Fig. 3B) and the range of pH changes during this experiment were 6.90–8.25 (±0.01–0.16) (Fig. S2†). The release of As in this experiment is a relatively low concentration as it only represents 0.03% of the total As in the solids. However, a maximum concentration of 244 μg L−1 As was observed in experiments reacting 0.1 mM phosphate with DR solids (Fig. 3D), which is more than two orders of magnitude higher than the As released from WW solids.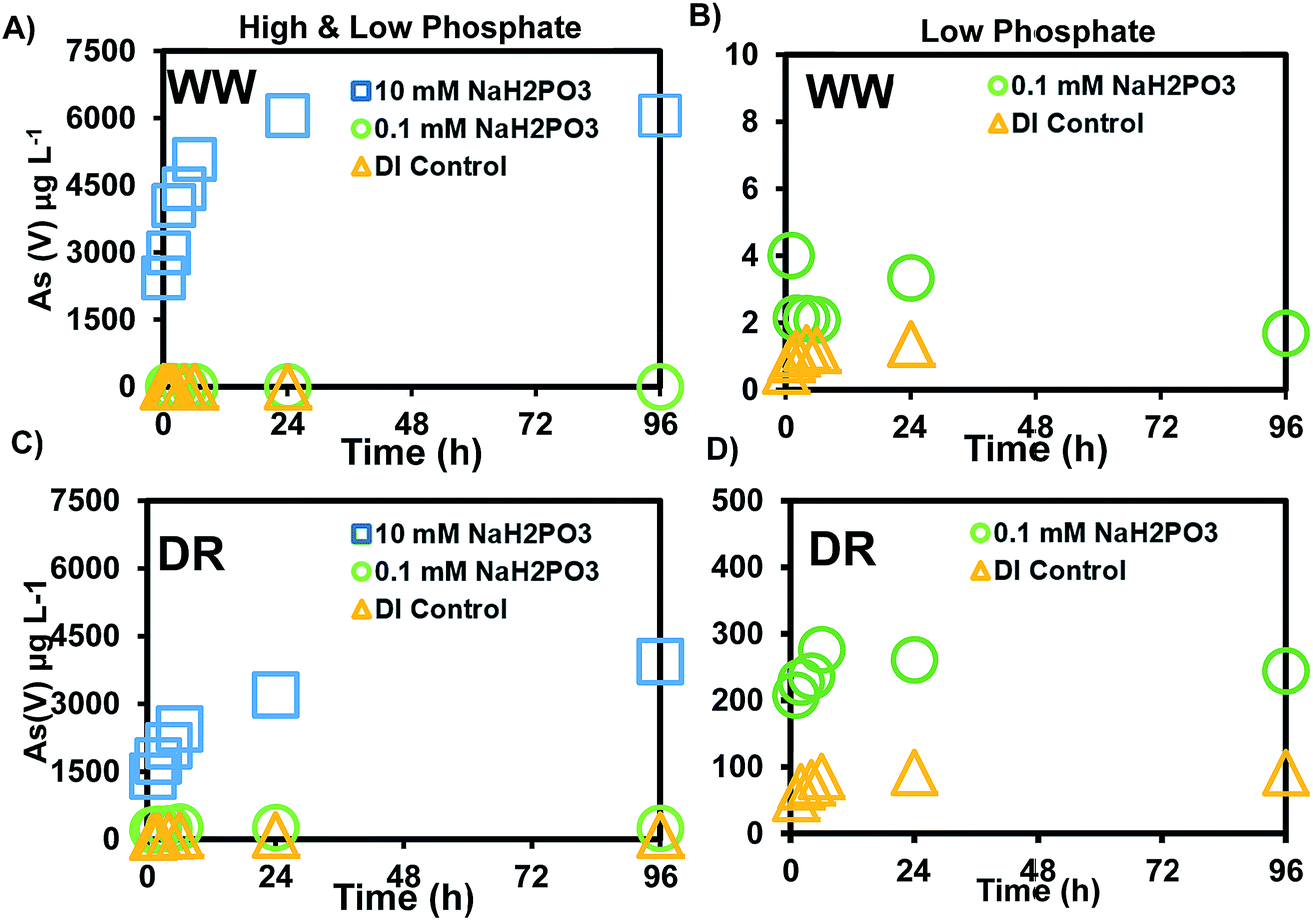 | ||
| Fig. 3 Release of As(V) after reaction of sediments with de-ionized water (DI Control), 0.1 mM, and 10 mM sodium phosphate solutions for: (A) Whitewood Creek (WW) high & low phosphate (B) Whitewood Creek (WW) zoomed into low phosphate (C) Deal Ranch (DR) high & low phosphate and (D) Deal Ranch (DR) zoomed into low phosphate. | ||
Experiments reacting sediments with a higher phosphate concentration (10 mM) released a maximum of 6056 μg L−1 from WW (Fig. 3A) and 3939 μg L−1 from DR (Fig. 3C). More than 95% of the total As was in the form As(V). A minor fraction was released as As(III) with the phosphate treatment (10 mM) and the range of pH during this experiment was 5.58–6.59 (Fig. S1†). Compared to the bicarbonate treatment where no As(III) was detected, phosphate released As(III) in the range of 1–5 μg L−1. A maximum of 4.89% As(V) of the solid concentration was released with the PO43− treatment from WW solids compared to 0.025% with bicarbonate. A maximum of 33% As(V) was released with the PO43− treatment from DR solids, compared to 1.33% with the bicarbonate treatment (Table 4). Although more total As concentration (2040 mg kg−1) was measured in WW sediments, solids from DR (285 mg kg−1) mobilized more As(V) on a mass basis. Our results using field solids suggest that As(V) is more easily released from DR solids compared to WW solids after reaction with DI water, bicarbonate and phosphate solutions. Additional analyses of the solids are discussed in later sections to help us understand As release under these experimental conditions.
| Total As (mg kg−1) | % mobilized with HCO3 | % mobilized with PO4−3 | % mobilized DI Control | |
|---|---|---|---|---|
| WW | 2040 | 0% | 4.9% | 0% |
| DR | 235 | 1.3% | 33% | 0.7% |
It is known that the adsorption of As(V) and As(III) onto Fe-oxides can be influenced by the presence of competing anions in the system and their relative binding affinities. Competition for surface sites between similar anions influence adsorption processes by changing electrostatic charges at the solid surface.28 Our results indicate that As release is favored by competitive ion displacement with 10 mM PO43− compared to 20 mM HCO3−. This is likely due to the displacement of HAsO42− related to the higher affinity of the solid surface for the dominant species HPO42−, which has a more negative charge than HCO3−. Since less than 5% As(III) was released into solution, the As likely occurs as weakly bound As(V) at the surface of the sediments. It is possible that inorganic phosphorus associated with livestock runoff could be an important factor for mobilizing surface-bound As in Cheyenne River sediments, mainly after storm events. The potential for nutrient runoff to affect As release is an important factor to consider based on the results from these experiments with phosphate.
Previous studies have shown the effects of phosphate on the decrease of arsenate adsorption by reacting synthesized iron oxide solids with As solution concentrations synthesized in the laboratory.28,53 The dominance of As(III) released using similar concentrations of competitive anions used in this study (22.7 mM CO3 and 0.25 mM PO43−) was measured. Solids from one study were synthetic iron coated sands operated in controlled column experiments. The distribution of As species released from this study can be used to show the effects of different surface sites from field derived solids compared to synthesized materials. Experimental data using a synthesized iron oxide-based sorbent showed the individual and competitive adsorption of As(V) and HPO4−2.26 The addition of 129 μM phosphate to a suspension with 6.67 μM As(V) (926 μg L−1) induced desorption of As(V). In addition, a 10-fold molar mass of phosphate to arsenate suppressed arsenate adsorption in experiments conducted with unsaturated alluvial sediments from a recharge pond in Antelope Valley, CA.27
Chemical equilibrium modeling (MINEQL) used to simulate aqueous species (solids not considered) in this system under relevant laboratory and field conditions indicated that HPO42−, HAsO42−, H2AsO4−, and HCO3− were the predominant species. Considering the complex speciation of As(V) in sediments and its reactivity with naturally occurring anions, modeling results suggest that HCO3− and HPO42− should be considered to understand As mobilization in the Cheyenne River and other environments with similar characteristics at pH 7. Additional analyses were conducted to further investigate chemical characteristics of unreacted and reacted solids from these experiments.
4.4 Solid analyses
 | ||
| Fig. 4 XRD patterns and atomic weight% of Whitewood Creek (WW) and Deal Ranch (DR) sediments. | ||
 | ||
| Fig. 5 High-resolution XPS spectra for oxidation state percent content in sediment samples from Whitewood Creek (WW) and Deal Ranch (DR) before and after reaction with phosphate for: (A) Fe (based on fitting of Fe 3p high-resolution spectra); and (B) As (based on fitting of As 3d high-resolution spectra). | ||
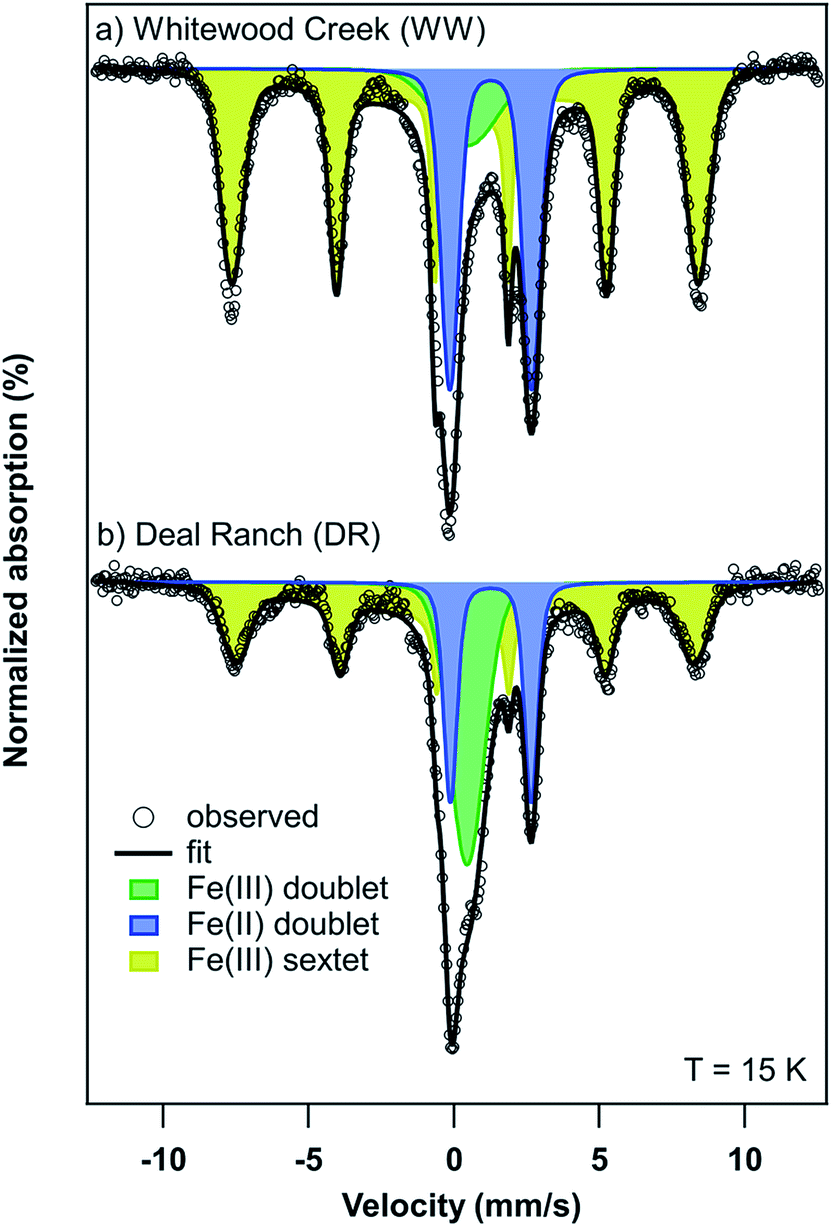 | ||
| Fig. 6 Mössbauer spectra of (a) Whitewood Creek (WW) and (b) Deal Ranch (DR) sediments collected at a temperature of 15 K. | ||
| Sample | Component | Relative area (%) | Center shift, CS (mm s−1) | Quadrupole splitting, QS or 2εb (mm s−1) | σ(Δ)c (mm s−1) | Hyperfine field, H (Tesla) | σ(H)d (Tesla) | χ ν 2 |
|---|---|---|---|---|---|---|---|---|
| a value in parenthesis reflects the error (1σ) in determination of the relative area for each component. b 2ε = quadrupole shift parameter in sextet. c σ(Δ) = standard deviation of quadrupole splitting component. d σ(H) = standard deviation of hyperfine field component, * = parameter held constant during fitting. | ||||||||
| Whitewood Creek (WW) | Fe(II) doublet | 27 (0.2) | 1.26 | 2.82 | 0.51 | N/A | N/A | 3.72 |
| Fe(III) doublet | 10 (0.4) | 0.45 | 1.63 | 1.23 | N/A | N/A | ||
| Fe(III) sextet | 63 (0.4) | 0.50 | −0.22 | 43.4 | 13.66 | |||
| Deal Ranch (DR) | Fe(II) doublet | 18 (0.1) | 1.27 | 2.78 | 0.35 | N/A | N/A | 1.22 |
| Fe(III) doublet | 27 (0.3) | 0.45 | 0.88 | 0.67 | N/A | N/A | ||
| Fe(III) sextet | 56 (0.4) | 0.51 | −0.26 | 42.3 | 20.91 | |||
The presence of goethite identified by Mössbauer spectroscopy in sediments from WW and DR is relevant as this could explain the association of As in these sediments. For example, the association of As and goethite has been widely reported in the literature.16,59–62 Given the similar bulk mineralogy of the two sediments, as observed by Mössbauer spectroscopy, the following explanations for As release are plausible and include: (i) different As-binding minerals present in the two sediments (e.g. a different surface or Fe-As mineral), (ii) different surface Fe-to-PO4/CO3 ratios in the two sediments during extraction, and (iii) greater potential for As(V) occlusion within Fe minerals or Fe mineral aggregates in the WW sample making As poorly extractable.63 Additionally, the potential for Fe and As influenced by interactions with natural organic matter (NOM) is possible. However, detailed mechanistic studies of NOM are challenging in environmental systems due to the heterogenous nature of NOM and the estimated organic content in our samples is only 2–3%.64 Based on our present data, we cannot definitely conclude which of these or other potential mechanisms affect anion-induced As release in the WW and DR sediments.
5. Summary and conclusions
This study evaluated the concentration and reactivity of As in sediments impacted by an abandoned gold mine (Homestake Mine), in the Whitewood Creek (near the mine) and in the Cheyenne River (241 km downstream and inside the Cheyenne River Sioux Tribe reservation). This study suggests that the presence of environmentally relevant ions, bicarbonate and phosphate, can favor the release of As, through competitive ion displacement. More specifically, the highest As release was observed in batch experiments after reaction of mine waste solids with 10 mM sodium phosphate, which released 30 times as much as 20 mM sodium bicarbonate. However, a considerable release of As was observed after reaction with 20 mM bicarbonate, which has important implications for water quality conditions of the Cheyenne River.These results are relevant given that As release to water sources continues to be a concern in CRST and in many other communities located near mine waste sites. For instance, the high alkalinity (ranging from 1000 to 2650 mg L−1 and 10–26 mM as CaCO3) in the Cheyenne River and Whitewood Creek could affect As reactivity. In the current study, the reaction of mine waste sediments with 20 mM bicarbonate in batch experiments resulted in a maximum release of 240 μg L−1 of As(V). In addition, the role of inorganic and organic phosphorus on As release into surface water sources is an important consideration. For example, in the current study, the reaction of 10 mM phosphate with mine waste solids resulted in the release of up to 6900 μg L−1As(V). Land use within CRST boundaries includes 85% livestock use and agricultural activities, so the potential for the effect of organic and inorganic phosphorus on the release of As from sediments after runoff events should be considered. More research is necessary to better understand the effect of agricultural inputs on As reactivity in sediments from CRST.
Though As can be released by changes in associated Fe mineralogy and reductive dissolution, this study also suggests that the mobilization of As can be affected by phosphate and to some extent bicarbonate by competitive ion displacement of weakly bound As(V) at the surface of the solids. This is an interesting finding given that, although the DR solids have lower total As, the concentrations of As(V) mobilized after reaction with bicarbonate and phosphate are considerable. The presence of labile As(V) from sediments is relevant to consider in the assessment of As exposure to the local community. Existing literature has indicated that As has the affinity to associate with goethite in environmental systems. Given that Mössbauer analyses indicate that goethite is present in samples from WW and DR, it is likely that goethite plays an important role in As binding in these sediments. However, additional research is necessary to better understand the specific binding mechanisms for As in sediments from our system.
The Cheyenne River watershed continues to receive sediment loads from upstream, and the alterations of As as a function of sediment burial and redox transformations can be an important component of As release. Therefore, future investigations of redox processes can aid in our understanding of As mobility in the riverbank profile along the Cheyenne River, where the community of CRST harvests fruiting trees and medicinal plants. Additional studies are necessary to better understand the effect of microbial and plant processes on As mobility. The results of this investigation on As reactivity have important implications for environmental exposure and risk assessments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Cheyenne River Sioux Tribe Environment Department and community members that were instrumental in accessing field sites for this work. Funding for this work has been provided by the National Institute for Environmental Health Sciences and the UNM Center for Native Environmental Health Equity Research- A Center of Excellence In Environmental Health Disparities Research- funded jointly by grants from NIEHS & NIMHD (1P50ES026102) & USEPA (#83615701) and, the Center for Water and the Environment, funded by the National Science Foundation (CREST Grant Number 1345169). This material was developed in part under Assistance Agreement No. 83615701 awarded by the U.S. Environmental Protection Agency to the University of New Mexico Health Sciences Center. It has not been formally reviewed by EPA. The views expressed are solely those of the speakers and do not necessarily reflect those of the agencies. EPA does not endorse any products or commercial services mentioned in this publication.References
- P. Smedley and D. Kinniburgh, A review of the source, behaviour and distribution of arsenic in natural waters, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- S. Murcott, Arsenic Contamination in the World, IWA Publishing, 2012 Search PubMed.
- J. Lewis, J. Hoover and D. MacKenzie, Mining and Environmental Health Disparities in Native American Communities, Curr. Environ. Health Rep., 2017, 4, 130–141 CrossRef PubMed.
- K. E. Goddard, Composition, distribution, and hydrologic effects of contaminated sediments resulting from 13 the discharge of gold milling wastes to Whitewood Creek at Lead and Deadwood, South Dakota, Rapid 14 City, S. D., Dept. of the Interior, U.S. Geological Survey, Denver, CO, Open-File Reports, 1989.
- M. Gonzales, E. Erdei, J. Hoover and J. Nash, A Review of Environmental Epidemiology Studies in Southwestern and Mountain West Rural Minority Populations, Current Epidemiology Reports, 2018, 2, 101–113 CrossRef.
- W. Yu, C. Harvey and C. Harvey, Arsenic in groundwater in Bangladesh: A geostatistical and epidemiological framework for evaluating health effects and potential remedies, Water Resour. Res., 2003, 39(6), 1146 CrossRef.
- WHO, Arsenic in drinking water, Geneva, Switzerland, 2011, vol. 75, Rev. 1 Search PubMed.
- USEPA, https://www.epa.gov/sites/production/files/2015-22-09/documents/<?pdb_no 2005?>2005<?pdb END?>_11_10_arsenic_ars_final_app_c.pdf, Accessed January 2018.
- C. M. Donna, Floodplain storage of mine tailings in the belle fourche river system: A sediment budget approach, Earth Surf. Process. Landforms, 1992, 17, 675–685 CrossRef.
- J. F. Stamm and G. K. Hoogestraat, Concentrations of Selected Metals in Quaternary-Age Fluvial Deposits along the Lower Cheyenne and Middle Belle Fourche Rivers, 2009-10, Western South Dakota, USGS Data 27 Series 695, 2012 Search PubMed.
- B. Pfeifle, J. Stamm and J. Stone, Arsenic Geochemistry of Alluvial Sediments and Pore Waters Affected by Mine Tailings along the Belle Fourche and Cheyenne River Floodplains, Water Air Soil Pollut., 2018, vol. 229 Search PubMed.
- A. J. Horowitz, K. A. Elrick and E. Callender, The effect of mining on the sediment trace-element geochemistry of cores from the Cheyenne River arm of Lake Oahe, South Dakota, USA, Chem. Geol., 1988, 67, 17–33 CrossRef CAS.
- J. Noble, Ore Mineralization in the Homestake Gold Mine, Lead, South Dakota, Geol. Soc. Am. Bull., 1950, 34 61, 221 CrossRef.
- E. Smith, R. Naidu and A. Alston, Arsenic in the soil environment: A review, Adv. Agron., 1998, 64, 149–195 CAS.
- M. Herbel and S. Fendorf, Biogeochemical processes controlling the speciation and transport of arsenic within iron coated sands, Chem. Geol., 2006, 228, 16–32 CrossRef CAS.
- S. Dixit and J. Hering, Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: Implications for arsenic mobility, Environ. Sci. Technol., 2003, 37, 4182–4189 CrossRef CAS PubMed.
- M. Fillipi, P. Drahota, V. Machovic, B. Bohmova and M. Mihaljevic, Arsenic mineralogy and mobility in the arsenic-rich historical mine waste dump, Sci. Total Environ., 2015, 536, 713–728 CrossRef PubMed.
- C. Saup, K. Williams, L. Rodriguez-Freire, J. Cerrato, M. Johnston and M. Wilkins, Anoxia stimulates microbially catalyzed metal release from Animas River sediments, Environ. Sci.: Processes Impacts, 2017, 19, 578–585 RSC.
- L. Rodriguez-Freire, S. Avasarala, A. Ali, D. Agnew, J. Hoover, K. Artyushkova, D. Latta, E. Peterson, J. Lewis, L. Crossey, A. Brearley and J. Cerrato, Post Gold King Mine Spill Investigation of Metal Stability in Water and Sediments of the Animas River Watershed, Environ. Sci. Technol., 2016, 50, 11539–11548 CrossRef CAS PubMed.
- S. Goldberg and C. T. Johnston, Mechanisms of Arsenic Adsorption on Amorphous Oxides Evaluated Using Macroscopic Measurements, Vibrational Spectroscopy, and Surface Complexation Modeling, J. Colloid Interface Sci., 2001, 234, 204–216 CrossRef CAS PubMed.
- J. Rawson, H. Prommer, A. Siade, J. Carr, M. Berg, J. Davis and S. Fendorf, Numerical Modeling of Arsenic Mobility during Reductive Iron-Mineral Transformations, Environ. Sci. Technol., 2016, 50, 2459–2467 CrossRef CAS PubMed.
- T. Borch, R. Kretzschmar, A. Kappler, P. Van Cappellen, M. Ginder-Vogel, A. Voegelin and K. Campbell, Biogeochemical Redox Processes and their Impact on Contaminant Dynamics, Environ. Sci. Technol., 2010, 44, 15–23 CrossRef CAS PubMed.
- S. N. Fendorf, P. S. Kocar, B. D. Masue and Y. Tufano KJ, in Developments in Soil Science, 2010, vol. 34, ch. 12, pp. 357–378 Search PubMed.
- M. Stachowicz, T. Hiemstra and W. Van Riemsdijk, Arsenic-bicarbonate interaction on goethite particles, Environ. Sci. Technol., 2007, 41, 5620–5625 CrossRef CAS PubMed.
- M. Kim, J. Nriagu and S. Haack, Carbonate ions and arsenic dissolution by groundwater, Environ. Sci. Technol., 2000, 34, 3094–3100 CrossRef CAS.
- H. Zeng, B. Fisher and D. Giammar, Individual and competitive adsorption of arsenate and phosphate to a high-surface-area iron oxide-based sorbent, Environ. Sci. Technol., 2008, 42, 147–152 CrossRef CAS PubMed.
- S. Goldberg and D. Suarez, Arsenate Adsorption by Unsaturated Alluvial Sediments, Soil Sci. Soc. Am. J., 2013, 77, 782–791 CrossRef CAS.
- A. Jain and R. Loeppert, Effect of competing anions on the adsorption of arsenate and arsenite by ferrihydrite, J. Environ. Qual., 2000, 29, 1422–1430 CrossRef CAS.
- F. Peryea and R. Kammereck, Phosphate-enhanced movement of arsenic out of lead arsenate-contaminated topsoil and through uncontaminated subsoil, Water, Air, Soil Pollut., 1997, 93, 243–254 CAS.
- H. Anawar, J. Akai and H. Sakugawa, Mobilization of arsenic from subsurface sediments by effect of bicarbonate ions in groundwater, Chemosphere, 2004, 54, 753–762 CrossRef CAS PubMed.
- C. Appelo, M. Van der Weiden, C. Tournassat and L. Charlet, Surface complexation of ferrous iron and carbonate on ferrihydrite and the mobilization of arsenic, Environ. Sci. Technol., 2002, 36, 3096–3103 CrossRef CAS PubMed.
- B. Manning and S. Goldberg, Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals, Soil Sci. Soc. Am. J., 1996, 60, 121–131 CrossRef CAS.
- R. Islam, R. Salminen and P. Lahermo, Arsenic and other toxic elemental contamination of groundwater, surface: Water and soil in Bangladesh and its possible effects on human health, Environ. Geochem. Health, 2000, 22, 33–53 CrossRef.
- S. Beauchemin and Y. Kwong, Impact of redox conditions on arsenic mobilization from tailings in a wetland with neutral drainage, Environ. Sci. Technol., 2006, 40, 6297–6303 CrossRef CAS PubMed.
- A. Lizama, T. Fletcher and G. Sun, Removal processes for arsenic in constructed wetlands, Chemosphere, 2011, 84, 1032–1043 CrossRef PubMed.
- K. Tufano, C. Reyes, C. Saltikov and S. Fendorf, Reductive Processes Controlling Arsenic Retention: Revealing the Relative Importance of Iron and Arsenic Reduction, Environ. Sci. Technol., 2008, 42, 8283–8289 CrossRef CAS PubMed.
- J. Gorny, G. Billon, L. Lesven, D. Dumoulin, B. Made and C. Noiriel, Arsenic behavior in river sediments under redox gradient: A review, Sci. Total Environ., 2015, 505, 423–434 CrossRef CAS PubMed.
- J. Kumpiene, D. Ragnvaldsson, L. Lovgren, S. Tesfalidet, B. Gustavsson, A. Lattstrom, P. Leffler and C. Maurice, Impact of water saturation level on arsenic and metal mobility in the Fe-amended soil, Chemosphere, 2009, 74, 206–215 CrossRef PubMed.
- P. O'Day, D. Vlassopoulos, R. Root and N. Rivera, The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 13703–13708 CrossRef PubMed.
- H. Pedersen, D. Postma and R. Jakobsen, Release of arsenic associated with the reduction and transformation of iron oxides, Geochim. Cosmochim. Acta, 2006, 70, 4116–4129 CrossRef CAS.
- A. J. Heakin, Water quality trends for the Cheyenne and Moreau Rivers, Cheyenne River Indian 49 Reservation, South Dakota, 1972-94, Report 98-4092, USGS Water-Resources Investigations, 1998 Search PubMed.
- USGS, Mineral and water resources of South Dakota, South Dakota Geological Survey, South Dakota 2 School of Mines, US Bureau of Reclamation, US Bureau of Mines, 1975 Search PubMed.
- J. Blake, C. De Vore, S. Avasarala, A. Ali, C. Roldan, F. Bowers, M. Spilde, K. Artyushkova, M. Kirk, E. Peterson, L. Rodriguez-Freire and J. Cerrato, Uranium mobility and accumulation along the Rio Paguate, Jackpile Mine in Laguna Pueblo, NM, Environ. Sci.: Processes Impacts, 2017, 19, 605–621 RSC.
- S. Gaudino, C. Galas, M. Belli, S. Barbizzi, P. de Zorzi, R. Jacimovic, Z. Jeran, A. Pati and U. Sansone, The role of different soil sample digestion methods on trace elements analysis: a comparison of ICP-MS and INAA measurement results, Accred Qual. Assur., 2007, 12, 84–93 CrossRef CAS.
- K. Cawley, A. Hohner, D. Podgorski, W. Cooper, J. Korak and F. Rosario-Ortiz, Molecular and Spectroscopic Characterization of Water Extractable Organic Matter from Thermally Altered Soils Reveal Insight into Disinfection Byproduct Precursors, Environ. Sci. Technol., 2017, 51, 771–779 CrossRef CAS PubMed.
- S. Paramananthan, P. Lee, M. Wong, E. Van Ranst, R. Wust and J. Vijiandran, A Comparative Study of the Use of Organic Carbon and Loss on Ignition in Defining Tropical Organic Soil Materials, Commun. Soil Sci. Plant Anal., 2018, 49, 626–634 CrossRef CAS.
- R. Stach, R. Helberson, R. Bretz, M. Tipton, D. R. Beissel and J. C. Harksen, Arsenic Levels in the Surface and 16 ground waters along Whitewood Creek, Belle Fourche River, and a portion of the Cheyenne River South Dakota, South Dakota Geological Survey, Vermillion, South Dakota, 1978 Search PubMed.
- USEPA, https://www.epa.gov/wqc/national-recommended-water-quality-criteria-aquatic-life-criteria19-table, Accessed January 2018.
- J. A. Cherry, F. M. Morel, J. V. Rouse, J. L. Schnoor and M. G. Wolman, Hydrochemistry of sulfide and arsenic-rich tailings and alluvium along Whitewood Creek, South Dakota, Colorado School of Mines, Mineral and Energy Resources, 1986, vol. 29 Search PubMed.
- G. Waychunas, B. Rea, C. Fuller and J. Davis, Surface Chemistry of Ferrihydrite. 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate, Geochim. Cosmochim. Acta, 1993, 57, 2251–2269 CrossRef CAS.
- H. Zhao and R. Stanforth, Competitive adsorption of phosphate and arsenate on goethite, Environ. Sci. Technol., 2001, 35, 4753–4757 CrossRef CAS.
- Y. Arai, D. Sparks and J. Davis, Effects of dissolved carbonate on arsenate adsorption and surface speciation at the hematite-water interface, Environ. Sci. Technol., 2004, 38, 817–824 CrossRef CAS PubMed.
- T. Radu, J. Subacz, J. Phillippi and M. Barnett, Effects of dissolved carbonate on arsenic adsorption and mobility, Environ. Sci. Technol., 2005, 39, 7875–7882 CrossRef CAS PubMed.
- X. Meng, S. Bang and G. Korfiatis, Effects of silicate, sulfate, and carbonate on arsenic removal by ferric chloride, Water Res., 2000, 34, 1255–1261 CrossRef CAS.
- T. Lin, C. Wei, C. Huang, C. Chang, F. Hsu and V. Liao, Both Phosphorus Fertilizers and Indigenous Bacteria Enhance Arsenic Release into Groundwater in Arsenic-Contaminated Aquifers, J. Agric. Food Chem., 2016, 64, 2214–2222 CrossRef CAS PubMed.
- S. Ying, B. Kocar and S. Fendorf, Oxidation and competitive retention of arsenic between iron- and manganese oxides, Geochim. Cosmochim. Acta, 2012, 96, 294–303 CrossRef CAS.
- M. Dyar, D. Agresti, M. Schaefer, C. Grant and E. Sklute, Mössbauer spectroscopy of earth and planetary materials, Annu. Rev. Earth Planet Sci., 2006, 34, 83–125 CrossRef CAS.
- Y. Yoshida and G. Langouche, Mössbauer spectroscopy, Springer-Verlag Berlin Heidelberg, 2013, vol. 1, p. 549 Search PubMed.
- B. Manning, S. Fendorf and S. Goldberg, Surface structures and stability of arsenic(III) on goethite: Spectroscopic evidence for inner-sphere complexes, Environ. Sci. Technol., 1998, 32, 2383–2388 CrossRef CAS.
- S. Fendorf, M. Eick, P. Grossl and D. Sparks, Arsenate and chromate retention mechanisms on goethite.1. Surface structure, Environ. Sci. Technol., 1997, 31, 315–320 CrossRef CAS.
- X. Sun and H. Doner, Adsorption and oxidation of arsenite on goethite, Soil Sci., 1998, 163, 278–287 CrossRef CAS.
- R. Bowell, Sorption of arsenic by iron oxides and oxyhydroxides in soils, Appl. Geochem., 1994, 9, 279–286 CrossRef CAS.
- R. Ford, Rates of hydrous ferric oxide crystallization and the influence on coprecipitated arsenate, Environ. Sci. Technol., 2002, 36, 2459–2463 CrossRef CAS PubMed.
- D. P. Martin, J. M. Seiter, B. J. Lafferty and A. J. Bednar, Exploring the ability of cations to facilitate binding between inorganic oxyanions and humic acid, Chemosphere, 2017, 166, 192–196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8em00461g |
| This journal is © The Royal Society of Chemistry 2019 |