
Mineral identity, natural organic matter, and repeated contaminant exposures do not affect the carbon and nitrogen isotope fractionation of 2,4-dinitroanisole during abiotic reduction†
Matthew J.
Berens
 a,
Bridget A.
Ulrich
b,
Jennifer H.
Strehlau
a,
Thomas B.
Hofstetter
b and
William A.
Arnold
*a
a,
Bridget A.
Ulrich
b,
Jennifer H.
Strehlau
a,
Thomas B.
Hofstetter
b and
William A.
Arnold
*a
aDepartment of Civil, Environmental, and Geo- Engineering, University of Minnesota, 500 Pillsbury Drive SE, Minneapolis, MN 55455-0116, USA. E-mail: arnol032@umn.edu; Tel: +1 612-625-8582
bEawag, Swiss Federal Institute of Aquatic Science and Technology, Department of Environmental Chemistry, Überlandstrasse 133, CH-8600 Dübendorf, Switzerland
First published on 20th November 2018
Abstract
The recent development of insensitive munitions, such as 2,4-dinitroanisole (DNAN), as components of military explosives has generated concern for potential subsurface contamination and created a need to fully characterize their transformation processes. Compound specific isotope analysis (CSIA) has proven to be a useful means of assessing transformation pathways according to characteristic stable isotope fractionation patterns. The C and N isotope fractionation of DNAN associated with abiotic and enzymatic hydrolysis was recently assessed. The extent to which DNAN isotope fractionation will be affected by other potentially competing transformation pathways known for nitroaromatic compounds (e.g., reduction) and if previous knowledge can be extrapolated to other environmental matrices remains to be understood. Here, we investigated the C and N isotope fractionation and reaction rate constants of DNAN during abiotic reduction mediated by mineral-associated Fe(II) species as a function of mineral type, natural organic matter presence, and repeated exposures to DNAN. Though rate constants varied, N and C apparent kinetic isotope effects (AKIEs) remained consistent across all experiments (averaged values of 15N-AKIE = 1.0317 ± 0.0064 and 13C-AKIE = 1.0008 ± 0.0005) and revealed significant 15N- and minimal 13C-enrichment in agreement with previous work on nitroaromatic compounds. Moreover, the observed fractionation was clearly distinct from trends for abiotic and enzymatic hydrolysis. This study provides a strengthened basis for the use of CSIA as a robust tool for monitoring DNAN degradation in complex environmental matrices as a component of future remediation efforts.
Environmental significanceIron-bearing minerals can elicit the transformation of anthropogenic contaminants in subsurface environments where reliable quantification is often hampered by matrix conditions and non-degradative processes. While traditional concentration measurements fail to overcome these limitations, the use of stable isotope analysis has proven to be both a reliable and robust analytical tool for estimating the extent of contaminant degradation in complex environmental systems. Our results extend the scope of previous findings by applying stable isotope analysis to investigate novel insensitive munitions produced by the military. The results may be used to increase the understanding of the behavior and inherent risks of novel military-grade munitions in the environment. |
Introduction
Insensitive munitions have emerged as safer alternatives to traditional explosives such as 2,4,6-trinitrotoluene (TNT) and hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX). The U.S. military has recently begun development of two insensitive munitions formulations (IMX-101 and IMX-104) containing 2,4-dinitroanisole (DNAN).1 These formulations exhibit a decreased sensitivity to shock and high temperatures relative to traditional munitions and are designed to minimize the risk of accidental detonations during handling and storage.2,3 Previous contamination by traditional munitions at military installations has caused concern for future contamination by insensitive munitions, motivating considerable effort towards understanding their subsurface fate and transport properties. Limited data, however, is available concerning the fate and transport of these new energetic materials.4,5 Moreover, the toxicity and sediment-water partitioning behavior of DNAN and its transformation products have shown to be similar to that of TNT.1,6,7 A better understanding of the relevant natural attenuation processes for DNAN in subsurface environments is therefore needed.The degradation of DNAN in the environment occurs through several abiotic transformation pathways including alkaline hydrolysis,8,9 photolysis,10,11 and also reduction in systems containing zero valent iron or other iron-bearing mineral species.12,13 Previous work has shown that ferrous iron associated with iron oxides, iron (oxy)hydroxides, and sulfide-bearing minerals mediates the abiotic transformation of nitroaromatic compounds (NACs) under anoxic conditions, reducing NACs to the corresponding substituted anilines (e.g., Scheme 1).14–19 Such pathways have been observed for DNAN and are attributed to mineral-bound Fe(II) species as well as electron-donating functional groups of natural organic matter.15,20 Substituted anilines may also form during biotic processes facilitated by Fe(III)-reducing microorganisms and other concomitant suites of aerobic and anaerobic microbiota.4,20–24 Complete reduction of DNAN in subsurface environments is often achieved through coupled abiotic–biotic transformation mechanisms. For example, Niedźwiecka et al.25 observed rapid DNAN reduction in microcosms containing Fe(III) following reduction of Fe(III) to Fe(II) by Geobacter metallireducens, compared to microcosms without ferric iron.
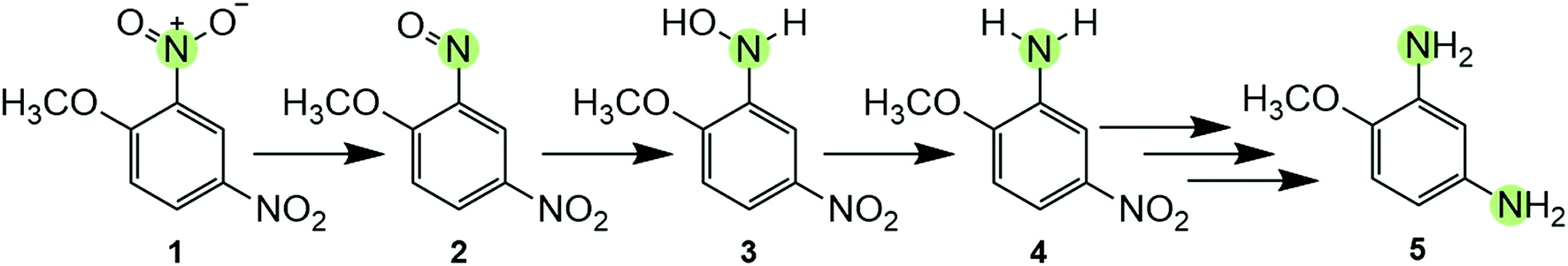 | ||
| Scheme 1 Reductive transformation pathway of DNAN, with one N atom at which the reduction reaction can occur highlighted. The pathway is initiated by the formation of a nitroso-moiety (2) from a NO2 group,13,20,26 followed by formation of hydroxylamine (3) and subsequently an amine derivative (e.g., 2-methoxy-5-nitroaniline (MENA) (4)). The mechanism of aromatic NO2 reduction is shown here arbitrarily for the ortho-substituent and does not imply preferential formation of MENA. However, most current studies are in agreement that reduction is initiated at the ortho substituent.20,26,27 In the presence of sufficient reductant, this process continues through other intermediates before reducing the second NO2-group to generate 2,4-diaminoanisole (DAAN) (5).15 | ||
Assessing the transformation of NACs in the environment is complicated by the possibility of several simultaneous (and potentially competing) transformation and transport processes (e.g., sorption, volatilization, (bio)degradation).4 Likewise, NACs in the subsurface may be present in different phases7,28 and exhibit high soil and sediment sorption, rendering the interpretation of concentration dynamics difficult.29,30 Compound specific isotope analysis (CSIA) has proven useful for assessing degradation processes of NACs in such complex environmental systems with potentially competing attenuation processes.31,32 CSIA of NAC (bio)transformations is based on characteristic changes in stable isotope ratios (e.g., 15N/14N, 13C/12C) that are indicative of a specific pathway.33,34 Thus, it is possible to use CSIA to determine the reaction process occurring without the need to detect reaction products, which may be not amenable to detection, further degraded, bound to organic matter, or sorbed to the matrix.35,36 These so-called isotope fractionation patterns arise from apparent kinetic isotope effects (AKIEs) controlled by changes in the bonding environment at the reactive position during the initial stages of NAC transformation.37 Indeed, AKIEs have been well-characterized for the abiotic reduction of several NACs including chloro- and methyl-substituted nitrobenzenes, dinitrobenzene isomers, and 2,4,6-trinitrotoluene (15N-AKIE ∼ 1.03–1.04, 13C-AKIE ∼ 1.001).38–41
Abiotic reductions of NACs (Scheme 1) result in large primary 15N-AKIEs, for which cleavage of the first N–O bond to form a nitroso intermediate is the rate-limiting step and thus dictates shifts in the isotope ratios. In contrast, the corresponding 13C-AKIEs are typically small because C atoms are not directly involved in the rate-limiting step (i.e., secondary AKIEs).38–40 Whereas Ulrich et al.42 recently assessed 13C and 15N fractionation during the abiotic (i.e., alkaline) and enzymatic hydrolysis of DNAN, the isotope fractionation that arises from reductive transformation of DNAN remains to be assessed. Previous work on the abiotic reduction of NACs by Fe(II) associated with the surfaces of iron oxides and clay minerals suggests that DNAN will display similar isotopic fractionation behavior.38–41 Those studies, however, focused on isomers of chloro- and methyl-substituted compounds and on a limited set of minerals (i.e., Fe(II) associated with goethite and structural Fe(II) in magnetite and ferruginous smectite). Showing that fractionation is independent of environmental matrix conditions is imperative for accurately quantifying the extent of pollutant degradation at contaminated field sites where the subsurface composition may be complex, changing, or unknown. Potential matrix conditions could include the presence of different minerals with different reactivity towards NACs, interferences of natural organic matter (NOM) through association with the mineral surface, and the transformation of Fe minerals after repeated exposure to NACs. Previous work has revealed that transformation rates of NACs may span orders of magnitude depending on the type of mineral involved owing to modulations in the electrochemical potential (EH) of the oxide-associated Fe(II) system.14,16,43–45 The presence of NOM may also impact reaction rates through the formation of Fe(II)–NOM complexes.46 NOM may also alter particle dispersion or change the availability of reactive sites.47–50 Repeated contaminant exposure may induce alterations of the mineral structure, potentially interfering with Fe(II) uptake or developing a new surface oxide.50,51 Because the magnitude of isotope fractionation during the reduction of NACs was proposed to be determined by changes of bonding in the aromatic NO2 substituent(s) regardless of the rate of transformation (i.e., reduction of a nitro- to a nitroso-moiety), we hypothesize isotope fractionation associated with the reduction of new potential contaminants such as DNAN in the environment will be consistent with previously determined AKIE-values as well as independent of matrix conditions discussed above.31,38,40,41,52,53
The goal of this study was to investigate the effects of matrix conditions that could be encountered in environmental systems on the reaction kinetics and C and N isotope fractionation associated with abiotic DNAN reduction, thereby evaluating the robustness of CSIA for assessing reductive transformations of DNAN. To this end, we characterized the pseudo-first-order kinetics and the C and N isotope fractionation during abiotic DNAN reduction as a function of (1) the type of mineral (goethite, magnetite, or mackinawite), (2) the presence of natural organic matter (NOM), and (3) repeated exposures to DNAN. Measured C and N isotope signatures (δ13C and δ15N) as a function of reaction extent were used to derive the corresponding isotope enrichment factors (εC and εN) and apparent kinetic isotope effects (13C-AKIE and 15N-AKIE). These values were compared to previous evidence on isotope effects associated with NAC reduction as well as to those recently established for alkaline and enzymatic hydrolysis42 to confirm the efficacy of CSIA to distinguish between the most likely environmental DNAN degradation reactions. The findings of this work provide a strengthened basis for identifying and assessing these processes in subsurface environments by CSIA, a result that will be of particular significance to potential future efforts to monitor and remediate DNAN-contaminated subsurface environments.
Methods
Chemicals
Detailed chemical information is provided in the ESI (Section S1.1†). Fresh Fe(II) and DNAN stock solutions were prepared prior to each reduction experiment. Aqueous Fe(II) stock solutions were prepared by adding 0.44 g FeCl2·6H2O to 0.4 mL of 1 M HCl and enough deoxygenated ultrapure water to achieve a final concentration of 175 mM. DNAN (Alfa Aesar, 98%, CAS# 119-27-7) stock solutions were similarly prepared in deoxygenated methanol. It should be noted that DNAN was purchased in 2016 and is no longer available from Alfa Aesar in the United States. DNAN transformation products 2-methoxy-5-nitroaniline (MENA, 98%, CAS# 99-59-2) and 2,4-diaminoanisole (DAAN, ≥98%, CAS# 615-05-4) were purchased from Fisher Scientific. Elliot Soil humic acid (ESHA) was purchased from the International Humic Substances Society (IHSS, St. Paul, MN). With the exception of goethite synthesis and storage, all experiments were performed inside an anaerobic chamber (Coy, 95% N2/5% H2).Mineral synthesis
Detailed mineral synthesis and analysis procedures are provided in Section S2.† Mackinawite was precipitated using the method of Butler and Hayes54 and particles were stored in suspension in the anaerobic chamber at room temperature and pH 7. Synthetic goethite was prepared according to Anschutz and Penn55 and stored in suspension outside the anaerobic chamber at pH 4 and 4 °C. Magnetite synthesis was adapted from Schwertmann and Cornell.56 Synthetic magnetite was dried and stored as a powder inside the chamber. Magnetite stoichiometry (x = Fe(II)/Fe(III)) was determined by acid dissolution in 3 M HCl (Sigma, trace metals) as x = 0.50. Total Fe(II) and Fe(III) contents were quantified using the ferrozine colorimetric assay (Section S1.2†).57 Each mineral was characterized by X-ray diffraction (XRD) and compared to reference patterns to support the presence of each desired mineral phase (Fig. S1†). The XRD results for synthetic magnetite and goethite indicated the presence of only the desired phases. The pattern collected from mackinawite contained peaks consistent with mackinawite and greigite (Fe3S4, PDF no. 16-0713). Previous work has reported rapid phase transformation of mackinawite to greigite under oxic conditions at room temperature, which may suggest the presence of greigite as an artifact of sample preparation.58Mineral surface areas were determined by nitrogen adsorption analysis using the Brunauer–Emmett–Teller (BET) adsorption model59 as 103.8 m2 g−1 and 17.8 m2 g−1 for goethite and magnetite, respectively. Mackinawite decomposed under high vacuum and temperature during outgassing and an accurate surface area measurement could not be obtained. Mass loading experiments were performed, and the particle concentrations in goethite and mackinawite suspensions were approximately 10 g L−1 and 33 g L−1, respectively.
Batch reactions
Batch experiments to assess reaction kinetics were performed in triplicate in 50 mL borosilicate serum bottles capped with Teflon-lined butyl rubber septa. All reactors contained a mineral suspension in NaHCO3 buffer (10 mM, pH 7.0). Mineral loadings were 2.0 g L−1 for magnetite, 1.5 g L−1 for mackinawite, and 1.0 g L−1 for goethite. Aqueous Fe(II) was added to goethite and magnetite reactors at a concentration of 1 mM and equilibrated overnight by rotating on an end-over-end rotator (Glas-Col, 40 rpm). No Fe(II) was added to mackinawite reactors and only a 1 h equilibration was performed. For the subset of reactions containing NOM, ESHA was added at 5 mg C/L prior to equilibration. The pH was then measured and Fe(II) concentrations were determined using the ferrozine method.57 If necessary, the pH was adjusted back to 7.0 with 1 M HCl or 1 M NaOH (Fisher, ACS grade) and the Fe(II) was augmented to 1 mM in the magnetite and goethite reactors. Reactions were initiated by spiking DNAN from the methanolic stock solution to a concentration of 0.20 mM and returning reactors to the rotator. The DNAN concentration was selected to ensure that a sufficient concentration remained in solution to allow detection by CSIA after achieving our target of 95% reduction. Every hour, the pH and Fe(II) in the reactors were determined and adjusted to pH 7 and 1 mM, respectively. This was necessary because the initial 1 mM Fe(II) is not sufficient to completely reduce the 0.20 mM DNAN (a minimum of 2.4 mM e− is needed to reduce both –NO2 moieties to –NH2), and it allows for the assumption of pseudo-first-order kinetics by maintaining a high Fe(II) concentration. At appropriate time points, an aliquot from each reactor was removed and filtered with a 0.2 μm PTFE syringe filter. Aqueous DNAN, MENA, and DAAN concentrations were quantified at a wavelength of 230 nm using an Agilent 1200 series high pressure liquid chromatograph (HPLC, method in Section S1.3†). Pseudo-first-order rate constants were determined by regression using a log-linear relationship of ln(c/c0) versus time.To perform batch experiments for CSIA, eight replicate reactors were assembled as described above for each set of reaction conditions. At each sampling point, the entire contents of one reactor were removed, filtered, and stabilized by adjusting the pH to <4 with 1 M HCl (Sigma, trace metals). Approximately 14 mL of acidified filtrate were transferred to a clean 20 mL glass vial, fitted with a butyl rubber plug (Fisher, 20 mm) and crimp cap, and stored at 4 °C until isotope analysis (method described below).
To assess the baseline reactivity of each mineral suspension for DNAN transformation, reactors were assembled containing only mineral, buffer, Fe(II) (for iron oxides), and DNAN. A control was performed with Fe(II) alone in NaHCO3 buffer, in which no reaction was observed over 21 days (Section S3†). To perform repeated contaminant exposure experiments, batch reactors were assembled in triplicate as described above. The first spike was initiated by injecting DNAN to a concentration of 0.20 mM. The Fe(II) content was determined in goethite and magnetite reactors after 1 h of reaction and adjusted accordingly to return each reactor to 1 mM Fe(II). A single aliquot (∼0.20 mL) was removed for analysis by HPLC after approximately 8 h to confirm complete DNAN transformation (<0.001 mM). Subsequently, to avoid the formation and accumulation of products over multiple reaction cycles, the aqueous solution from each reactor was removed and replaced with fresh buffer solution at the end of each reduction cycle. Suspensions were settled for at least 12 h or, in the case of magnetite, particles were isolated by using a neodymium magnet and discarding the supernatant. Fresh Fe(II) (1 mM) was added to goethite and magnetite reactors and the reactors were then equilibrated as described above. The pH was adjusted back to 7.0 in all reactors and a second DNAN spike was initiated and the reaction performed as described above. This procedure was carried out for five cycles, with concentrations measured as a function of time in triplicate during the desired cycle. For CSIA, a reactor was sacrificed at each time point during the fifth cycle and prepared for isotope analysis as described above.
Compound specific isotope analysis
Isotopic analysis of DNAN was conducted by gas chromatography isotope ratio mass spectrometry (GC/IRMS). DNAN was concentrated by solid phase micro extraction (SPME) prior to isotopic analysis. A method was modified from established SPME procedures42,60 using the PAL SPME Arrow (DVB/PDMS sorption phase, 120 μm phase thickness, 1.1 mm diameter) instead of a normal SPME fiber, such that improved sensitivity could be obtained with a larger sorption phase.61 Automated SPME was carried out using a PAL autosampler equipped with a PAL SPME Arrow Tool and a Heatex Stirrer. All samples were initially diluted with 10 mM phosphate buffer (pH 7, prepared with nanopure water) to obtain concentrations within a range of linear response, and sodium chloride (100 g L−1) was added to maximize extraction efficiency. Following equilibration at 50 °C for 10 min, the SPME Arrow was immersed in the sample for 70 minutes at 50 °C (600 RPM stir rate), and then DNAN was thermally desorbed from the arrow in an injector equipped with a deactivated liner (270 °C, 6 minutes).DNAN 15N/14N and 13C/12C ratios were determined using a Trace GC (Thermo Electron Corp.) coupled to an IRMS (DeltaPLUS XL, Thermo Electron Corp.) via a combustion interface (GC Combustion III, Thermo) equipped with a custom-made Ni/Pt reactor. The GC contained an Rtx-5MS capillary column (0.32 mm ID, 1 μm film thickness, 30 m length) and was operated in splitless mode (splitless time 6 minutes, purge flow 50 mL min−1) with the following temperature program: 50 °C for 1 minute, ramp 10 °C min−1 to 250 °C, hold for 5 minutes. During δ15N analysis a liquid nitrogen trap was used to trap CO2 produced from combustion. The oxidation reactor was operated as described previously.62 Method quantification limits (MQLs) for δ13C and δ15N according to the moving mean procedure were 0.2 μM and 1 μM, respectively.63
Isotope ratios were referenced against standard laboratory gases as well as DNAN of known isotopic composition (δ13C = 37.3 ± 0.1‰ and δ15N = −2.4 ± 0.1‰ as determined by an elemental analyzer IRMS) using standard bracketing procedures.64 Carbon and nitrogen isotope enrichment factors (εC and εN) were derived according to methods described by Pati et al.64 Linear regression analyses of the isotope signature data were carried out using the log-linearized form of eqn (1):
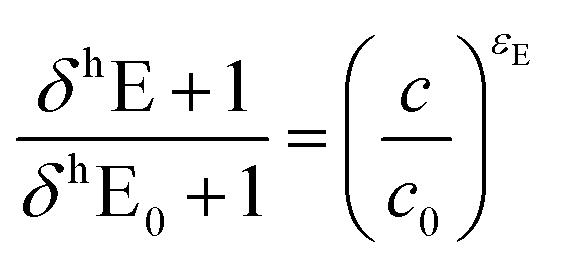 | (1) |
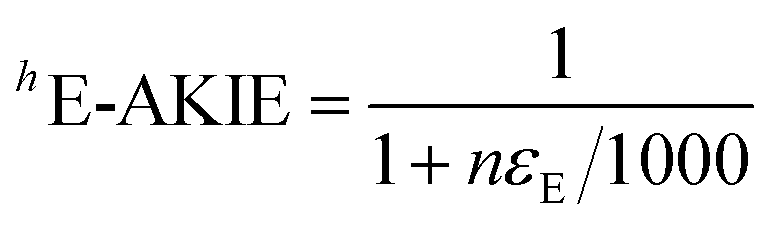 | (2) |
Results and discussion
Kinetic experiments
Pseudo-first-order rate constants (kobs) from triplicate reactors were calculated for all conditions tested (Table 1). The only reaction products observed were MENA and DAAN. Aqueous concentrations of DNAN and its transformation products are highlighted in Fig. 1. Mass balances were ∼80%, and given the high initial concentration, the incomplete mass balance could be due to formation of coupling products between hydroxylamine and nitroso intermediates.68,69 The limited accumulation of DAAN is likely due to lower reactivity of MENA compared to DNAN, because Fe(II) was maintained at 1 mM throughout the experiments with goethite and magnetite. The greater accumulation of MENA with mackinawite may be due to changing mineralogy (see below). Because the focus of CSIA analysis was on DNAN, monitoring the reaction to complete conversion to DAAN was not necessary. The highest transformation rates were observed for the first contaminant exposure (i.e., Spike 1) in Fe(II)/goethite suspensions followed by mackinawite and Fe(II)/magnetite, respectively. Aqueous Fe(II) was supplemented (and added throughout the experiment to maintain a 1 mM concentration) into goethite and magnetite suspensions because these minerals contain little to no inherent surface associated Fe(II) reactive sites. In contrast, mackinawite is an Fe(II)-bearing mineral known to directly participate in reduction reactions.54,70,71 Mineral concentrations in batch reactors decreased in the order of magnetite (2.0 g L−1), mackinawite (1.5 g L−1), and goethite (1.0 g L−1), resulting in the same order of increasing kobs when normalized to mineral loadings. Mineral concentrations were selected such that DNAN reduction experiments occurred within a similar experimental time frame. Organic matter (5 mg C/L from ESHA) had no effect on kobs in all samples tested. Previous work has shown that organic matter decreases reduction rates of NACs, likely via competitive adsorption with Fe(II) or blocking of reactive sites at the mineral surface.50 One possible explanation for these differences is that our work uses higher mineral loadings and a constant supply of Fe(II), thus increasing the abundance of Fe(II) and mineral reactive sites while decreasing the competition for each. Alternatively, higher mineral loadings and Fe(II) content may overcome any increases in EH imposed by NOM functional groups which have been linked to decreased rates of contaminant reduction.45| System | Mackinawite | Goethite kobs (h−1) | Magnetite |
|---|---|---|---|
| a Rate constants obtained from linear regression of natural log of concentration versus time data. b Uncertainties represent standard deviations of triplicate reactors. | |||
| Spike 1 | 1.15 ± 0.12 | 2.55 ± 0.14 | 0.79 ± 0.05 |
| ESHA | 1.16 ± 0.09 | 2.49 ± 0.18 | 0.82 ± 0.04 |
| Spike 5 | 0.19 ± 0.04 | 2.52 ± 0.11 | 0.81 ± 0.07 |
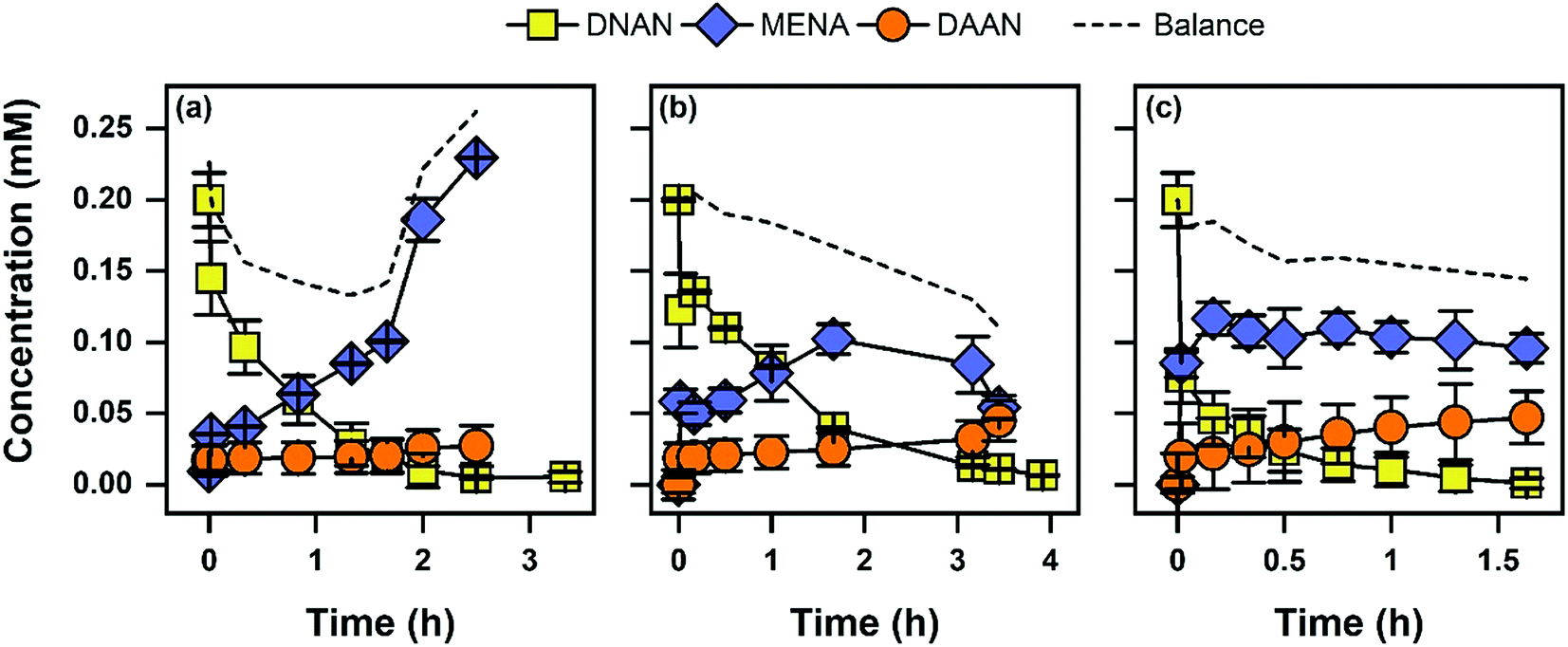 | ||
| Fig. 1 Aqueous concentrations of DNAN, MENA, and DAAN during DNAN kinetics experiments in suspensions of (a) mackinawite (1.5 g L−1), (b) magnetite (2 g L−1), and (c) goethite (1.0 g L−1). Reactions were performed at pH 7 with 1 mM Fe(II) and are for the first spike of DNAN. Error bars represent standard deviations of triplicate reactors. Dotted lines represent mass balances (moles) of the three compounds. | ||
Rate constants calculated during repeated contaminant exposure experiments revealed that reaction kinetics were independent of previous contaminant exposure in reactors containing goethite and magnetite. This is consistent with previous work with iron oxides, confirming that minimal variation in the rate constant can be expected if pH is carefully maintained over multiple contaminant exposures.50 Moreover, these results suggest that any mineral transformations that may have occurred during repeated exposures did not significantly affect the reduction potential of the oxide-bound Fe(II) in each system.45
In contrast to goethite and magnetite, the observed pseudo-first order reaction rate constant for mackinawite decreased with each exposure to DNAN (Fig. 2a). This observation could be attributed to the development of lepidocrocite (γ-FeOOH, PDF no. 44-1415) over the course of five contaminant spikes (Fig. 2b). While it is likely the rate constant was changing over the course of each contaminant exposure, a pseudo-first order model adequately fits the data for each spike and allows quantification of the loss of reactivity over time (Fig. 2a). Lepidocrocite is a polymorph of goethite and could be expected to exhibit similar reactive properties to goethite if sufficient Fe(II) was available,43 which it likely is not in this system given that none was added. Therefore, the observation that kobs decreases during lepidocrocite formation is supported by the observation that lepidocrocite has a lower kobs for NAC transformation and the requirement of Fe(II) supplementation for reaction to occur on Fe(III)-oxides.14 These results highlight the importance of understanding the effects of phase evolution on reaction kinetics at highly contaminated sites.
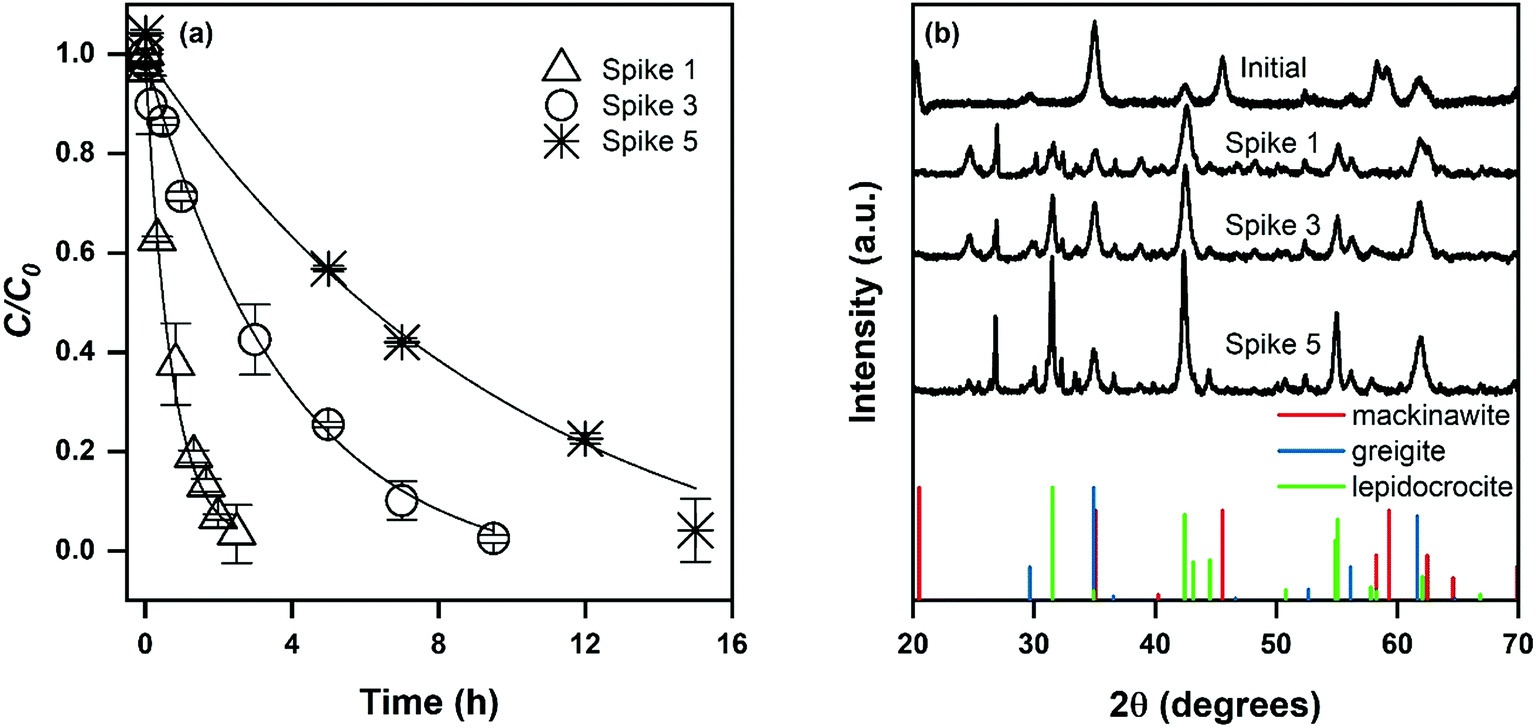 | ||
| Fig. 2 (a) Kinetic profiles and (b) XRD patterns of mackinawite reactions during repeated exposures to DNAN. Error bars represent standard deviations of triplicate reactors, and lines are pseudo-first order fits to the data. | ||
CSIA of DNAN reduction
The transformation of DNAN in suspensions of mackinawite, Fe(II)/goethite, and Fe(II)/magnetite was associated with strong enrichment of 15N of up to 60‰ (Fig. 3) in the remaining substrate whereas enrichment of 13C was minimal (below 2.4‰) in agreement with previous work on the reductive transformation of other NACs.33,34,37,38,41 The extent of DNAN degradation was related to isotope fractionation by eqn (1). Average εN-values for the reduction of DNAN derived after the first vs. fifth spike to the mineral suspension decreased by 1‰ to 5‰ as shown in Table 2 (e.g., −19 ± 1‰ in mackinawite experiment Spike 1 vs. −16 ± 2‰ for Spike 5). All changes, however, were within the uncertainty (95% confidence intervals). Variations of εC were much smaller (with errors of the same magnitude as the measurements) and did not show a discernable trend (Fig. 3b). The minimum and maximum 15N-AKIE-values derived from εN (eqn (2)) ranged from 1.018 ± 0.002 (magnetite, Spike 5) to 1.039 ± 0.001 (mackinawite, Spike 1) and fall within the range of data reported for the abiotic reduction of substituted mono-, di-, and trinitroaromatic compounds by mineral-bound Fe(II) reported previously.33,34,37,38,41 Whereas large 15N-AKIEs are due to the rate-limiting cleavage of the first N–O bond of the aromatic NO2 groups, 13C-AKIEs are small (i.e., secondary) and vary between 1.000 and 1.002 (Table 2) because no changes in bonds to C occur. Identical observations were made with experiments where DNAN was reduced in mineral suspension in the presence of ESHA in that 15N- and 13C-AKIEs were identical within experimental error to experiments without humic acids.| System | ε N (‰) | ε C (‰) | 15N-AKIE (−) | 13C-AKIE (−) |
|---|---|---|---|---|
| a Values derived from log-linear regression analysis of eqn (1). b Uncertainties represent 95% confidence intervals c Averaged values according to the methods of Scott et al.67 using the Pitman estimator. d Data from Ulrich et al.34,38–40,42,60 | ||||
| Mackinawite | ||||
| Spike 1 | −19 ± 1 | −0.8 ± 0.6 | 1.039 ± 0.001 | 1.0008 ± 0.0008 |
| ESHA | −16 ± 1 | −0.7 ± 1.4 | 1.034 ± 0.001 | 1.0007 ± 0.0014 |
| Spike 5 | −16 ± 2 | −1.5 ± 1.2 | 1.034 ± 0.002 | 1.0015 ± 0.0012 |
| Average | −19 ± 2 | −0.3 ± 0.6 | 1.039 ± 0.004 | 1.0003 ± 0.0006 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Goethite | ||||
| Spike 1 | −17 ± 3 | −0.1 ± 0.3 | 1.035 ± 0.003 | 1.0000 ± 0.0003 |
| ESHA | −16 ± 5 | −0.6 ± 0.4 | 1.034 ± 0.005 | 1.0006 ± 0.0004 |
| Spike 5 | −11 ± 1 | −1.2 ± 0.9 | 1.022 ± 0.001 | 1.0012 ± 0.0009 |
| Average | −16 ± 3 | −0.5 ± 0.3 | 1.033 ± 0.007 | 1.0005 ± 0.0003 |
|
||||
| Magnetite | ||||
| Spike 1 | −15 ± 3 | −0.7 ± 0.6 | 1.031 ± 0.003 | 1.0007 ± 0.0006 |
| ESHA | −17 ± 3 | −1.3 ± 0.2 | 1.036 ± 0.003 | 1.0013 ± 0.0005 |
| Spike 5 | −9 ± 2 | −0.1 ± 0.2 | 1.018 ± 0.002 | 1.0000 ± 0.0002 |
| Average | −17 ± 3 | −0.3 ± 0.5 | 1.035 ± 0.006 | 1.0000 ± 0.0005 |
| Alkaline hydrolysis | −2.7 ± 0.4 | −6.0 ± 0.5 | 1.0027 ± 0.0004 | 1.0445 ± 0.0028 |
| Enzymatic hydrolysis by O-demethylase | −3.2 ± 0.1 | −3.7 ± 0.1 | 1.0032 ± 0.0003 | 1.0269 ± 0.0053 |
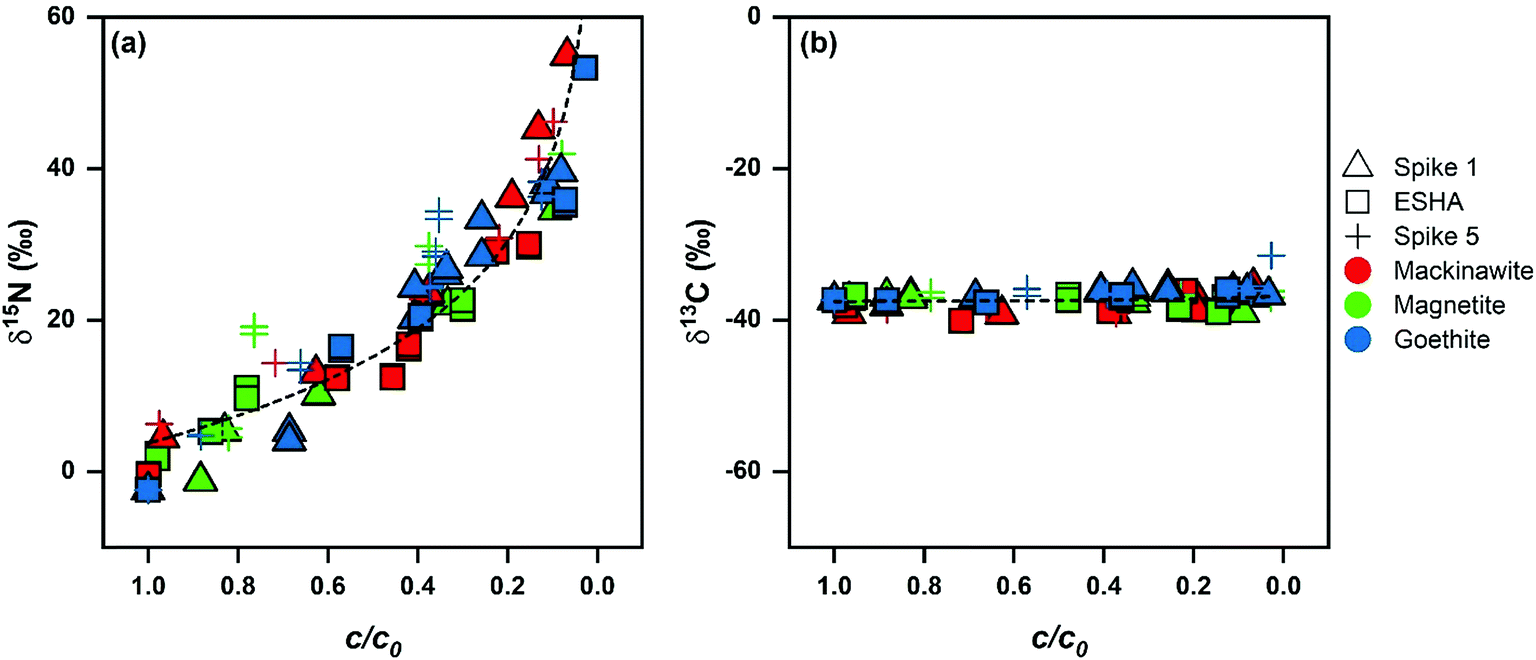 | ||
| Fig. 3 Nitrogen and carbon isotope fractionation of DNAN during abiotic reduction. (a) δ15N and (b) δ13C vs. fraction of remaining substrate (c/c0) with dotted lines provided to guide the eye along the data. Detailed N and C enrichment values are provided in Table 2 (see Fig. S3† for δ15N and δ13C vs. c/c0 data separated by reaction condition). | ||
Slight decreases in fractionation were observed during repeated contaminant exposure in Fe(II)/goethite and Fe(II)/magnetite systems (Table 2). This suggests that morphological changes (e.g., phase evolution and growth) occurring on the mineral structure during repeated surface oxidation and Fe(II) exposure may have limited the accessibility of reactive Fe(II) and thus slightly masked the isotope fractionation (Section S4.3, Fig. S4†). These changes were within experimental error of the initial spike experiment (εN = −17 ± 3‰ to −11 ± 1‰ and −15 ± 3‰ to −9 ± 2‰ between the single and multiple spike experiments for goethite and magnetite, respectively).
The collective C and N isotope fractionation data from all DNAN reduction experiments is plotted in Fig. 3. Plots separated by mineral are in Fig. S3.† The values of δ15N follow the general trend of εN values between −19 ± 1‰ to −9 ± 2‰ from individual DNAN reduction experiments. The scatter of δ15N values illustrates that experimental and analytical uncertainties are larger in experiments with ESHA and after repeated spikes of DNAN. We conclude that those uncertainties are primarily responsible for the observed variations of εN values from the individual experiments as well as for the large confidence intervals (typically <0.5‰).64 Based on this interpretation, we derived average 15N-AKIE for DNAN reduction by each mineral, that is 1.039 ± 0.004, 1.033 ± 0.007, and 1.035 ± 0.005 for mackinawite, goethite, and magnetite, respectively (Table 2) which are again identical within uncertainty.
An analysis of eqn (1) and (2) illustrates that a variation of εN for DNAN by ± 2.8‰, that is variations of 15N-AKIE of ±0.006 as observed in the average uncertainty of 15N-AKIEs in this study, will introduce uncertainty in estimating the extent of transformation (i.e., 100 × (1 − c/c0)). This uncertainty will be higher when the observed N isotope fractionation is small (e.g., 15% for Δ15N of 5‰) and level off as the N isotope fractionation increases (e.g., <7.5% at Δ15N of 25‰ (ref. 31) see Fig. 4). Given the large magnitude of 15N-AKIEs observed here, the extent of DNAN reduction has to exceed only approximately 12% to generate N isotope fractionation beyond the typical total uncertainties of N isotope ratio measurements of ±1‰ (ref. 33) based on an average εN value of −16‰.
 | ||
| Fig. 4 Changes in N isotope ratios (Δ15N = δ15N − δ15N0) vs. the extent of DNAN reaction (100 × (1 − c/c0)). Eqn (1) and (2) were used to illustrate variations in the extent of reaction given uncertainties in calculated AKIEs. Isotope fractionation profiles were generated using εN values of −16.0‰ (red) and −18.8‰ (blue), values typical for abiotic reductions of NACs but also within the range of isotope effects derived for the reduction of other NACs by other minerals. These εN values represent the extrema observed in this work. Dotted lines represent uncertainties in calculating the extent of reaction from uncertainties in 15N-AKIEs. | ||
The N isotope fractionation data for DNAN reduction reveal two important findings. First, abiotic reduction of DNAN will give rise to similar isotope enrichment factors across a variety of reaction conditions (i.e., regardless of the presence of NOM and previous contaminant exposures). Moreover, it is notable that 15N-AKIE and εN values calculated in this study closely agree with those observed for the reduction of other model NACs by Fe-bearing mineral phases.38–41 This comparison confirms our hypothesis that previous N isotope fractionation data for the abiotic reduction of NACs by Fe(II) bearing minerals can be extrapolated to new contaminants such as DNAN. This observation also indicates that N isotope fractionation is a robust indicator for monitoring the extent of reduction of DNAN and other NACs in complex environmental matrices, even in the absence of knowledge relating to reaction kinetics and product formation. Indeed, this technique has previously been employed to provide quantitative estimates of the extent of organic contaminant transformations at contaminated field sites.72 Our work, therefore, serves to qualify the prospective use of CSIA to assess the abiotic reduction of novel insensitive munitions in the environment.31,73
The combined C and N isotope analysis for DNAN reduction derived here is compared to the data for alkaline hydrolysis and aerobic biodegradation from Ulrich et al.42 in Fig. 5 and confirms that the different transformation pathways can also be discerned by CSIA. These data support the claim that N fractionation of DNAN during abiotic reduction is distinct from the isotopic fractionation observed in alkaline hydrolysis (nucleophilic aromatic substitution) and enzymatic hydrolysis (nucleophilic aliphatic substitution). Due to the vertical nature of the ΛN/C for the abiotic reduction data, a standard linear regression failed. Thus, the slope was found by plotting all of the δ13C vs. δ15N data, finding the slope, and then taking the inverse of this value. The ΛN/C calculated via this method for abiotic reduction was 50.5 ± 23.2 (shown as the dotted line in Fig. 5) in contrast to 0.46 ± 0.04 and 0.87 ± 0.18 for alkaline hydrolysis and biodegradation, respectively.42 These results therefore suggest that an identification of those DNAN reaction pathways and their respective contributions in the environment would be possible.74 Abiotic and biotic transformations of NACs typically elicit variable degree of 15N, 13C, and 2H enrichment,34,39,75 thus the observable isotope fractionation of processes occurring simultaneously can, in principle, be described by linear combinations of the enrichment factors pertinent to the individual, contributing processes.52,74 Such dual or triple isotope analysis31 can also circumvent masking interferences, which so far, have been reported primarily for oxidative NAC degradation processes.32,75–79
 | ||
| Fig. 5 Two-dimensional isotope analysis for reductive and hydrolytic DNAN transformation pathways. Changes in δ13C and δ15N values were monitored during abiotic reduction (circles), alkaline hydrolysis (squares), and biodegradation (diamonds). Alkaline hydrolysis and biodegradation data were reproduced with permission from Ulrich et al.42 Reduction data separated by mineral type are provided in Fig. S5.† The dotted line along the reduction data is provided simply to guide the eye. Moreover, any apparent inverse fractionation is an artifact of uncertainties in 13C-AKIEs that are close to 1. | ||
Conclusion
Knowledge of the stable isotope fractionation associated with DNAN reduction will allow for stakeholders to make quantitative estimates of the extent of DNAN degradation from measured δ15N and δ13C values of DNAN at contaminated sites. Our findings revealed that N and C isotope fractionation during the mineral-mediated reduction of DNAN is consistent despite the presence of different minerals, NOM, and repeated contaminant exposures while remaining independent of reaction kinetics. The potential changes of N isotope enrichment factors with repetitive exposure merit further investigation, especially considering that surface modification on natural minerals may be different than those for synthetic materials while those effects do not affect the interpretation of dual isotope analysis. Moreover, we showed that isotope fractionation during DNAN reduction will elicit ΛN/C values that are distinct from transformation pathways initiated by oxidation and substitution reactions.38 By showing that our data reflects previous results for reductions of other NACs,30,33,34,36,38 this work illustrates how predictive comparisons can be made to assess the extent of pollutant transformation from existing data. Such practices provide a strengthened basis for CSIA to serve as a reliable tool for validating future remediation efforts.Funding sources
This work was supported by the Strategic Environmental Research and Development Program (SERDP).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no competing interests.Acknowledgements
This work was supported by the Strategic Environmental Research and Development Program (SERDP, Project No. ER2618). We thank Lee Penn (University of Minnesota) for providing access to XRD instrumentation. In addition, acknowledgments to the laboratories of Andreas Stein and Marc Hillmyer (University of Minnesota) for allowing the use of instrumentation for nitrogen adsorption analysis.References
- S. G. Dodard, M. Sarrazin, J. Hawari, L. Paquet, G. Ampleman, S. Thiboutot and G. I. Sunahara, Ecotoxicological assessment of a high energetic and insensitive munitions compound: 2,4-Dinitroanisole (DNAN), J. Hazard. Mater., 2013, 262, 143–150 CrossRef CAS PubMed.
- W. Trzcinski, S. Cudzilo, S. Dyjak and M. Nita, A comparison of the sensitivity and performance characteristics of melt-pour explosives with TNT and DNAN binder, Cent. Eur. J. Energ. Mater., 2014, 11, 443–455 Search PubMed.
- P. J. Davies and A. Provatas, Characterization of 2,4-dinitroanisole: an ingredient for use in low sensitivity melt cast formulations, DSTO-TR-1904, Edinburgh, South Australia, 2006, vol. DSTO-TR-19 Search PubMed.
- J. C. Spain, J. B. Hughes and H. Knackmuss, Biodegradation of nitroaromatic compounds, Lewis Publishers, Inc., Boca Raton, 2000 Search PubMed.
- R. D. Albright, Cleanup of chemical and explosive munitions – locating, identifying contaminants, and planning for environmental remediation of land and sea military ranges and ordnance dumpsites, William Andrew, Norwich, NY, USA, 2nd edn, 2012 Search PubMed.
- M. S. Johnson, W. S. Eck and E. M. Lent, Toxicity of Insensitive Munition (IMX) Formulations and Components, Propellants, Explos., Pyrotech., 2017, 42, 9–16 CrossRef.
- V. M. Boddu, K. Abburi, S. W. Maloney and R. Damavarapu, Thermophysical properties of an insensitive munitions compound, 2,4-dinitroanisole, J. Chem. Eng. Data, 2008, 53, 1120–1125 CrossRef CAS.
- L. Sviatenko, C. Kinney, L. Gorb, F. C. Hill, A. J. Bednar, S. Okovytyy and J. Leszczynski, Comprehensive investigations of kinetics of alkaline hydrolysis of TNT (2,4,6-trinitrotoluene), DNT (2,4-dinitrotoluene), and DNAN (2,4-dinitroanisole), Environ. Sci. Technol., 2014, 48, 10465–10474 CrossRef CAS.
- A. J. Salter-Blanc, E. J. Bylaska, J. J. Ritchie and P. G. Tratnyek, Mechanisms and kinetics of alkaline hydrolysis of the energetic nitroaromatic compounds 2,4,6-trinitrotoluene (TNT) and 2,4-dinitroanisole (DNAN), Environ. Sci. Technol., 2013, 47, 6790–6798 CrossRef CAS.
- H. W. Schroer, X. Li, H. J. Lehmler and C. L. Just, Metabolism and photolysis of 2,4-dinitroanisole in Arabidopsis, Environ. Sci. Technol., 2017, 51, 13714–13722 CrossRef CAS PubMed.
- A. Halasz, J. Hawari and N. N. Perreault, New insights into the photochemical degradation of the insensitive munition formulation IMX-101 in water, Environ. Sci. Technol., 2018, 52, 589–596 CrossRef CAS PubMed.
- S. C. Ahn, D. K. Cha, B. J. Kim and S.-Y. Oh, Detoxification of PAX-21 ammunitions wastewater by zero-valent iron for microbial reduction of perchlorate, J. Hazard. Mater., 2011, 192, 909–914 CrossRef CAS PubMed.
- J. Hawari, F. Monteil-Rivera, N. N. Perreault, A. Halasz, L. Paquet, Z. Radovic-Hrapovic, S. Deschamps, S. Thiboutot and G. Ampleman, Environmental fate of 2,4-dinitroanisole (DNAN) and its reduced products, Chemosphere, 2015, 119, 16–23 CrossRef CAS.
- J. Klausen, S. P. Troeber, S. B. Haderlein and R. P. Schwarzenbach, Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral suspensions, Environ. Sci. Technol., 1995, 29, 2396–2404 CrossRef CAS PubMed.
- T. B. Hofstetter, C. G. Heijman, S. B. Haderlein, C. Holliger and R. P. Schwarzenbach, Complete reduction of TNT and other (poly)nitroaromatic compounds under iron-reducing subsurface conditions, Environ. Sci. Technol., 1999, 33, 1479–1487 CrossRef CAS.
- M. Elsner, R. P. Schwarzenbach and S. B. Haderlein, Reactivity of Fe(II)-bearing minerals toward reductive transformation of organic contaminants, Environ. Sci. Technol., 2004, 38, 799–807 CrossRef CAS.
- D. Colón, E. J. Weber and J. L. Anderson, QSAR study of the reduction of nitroaromatics by Fe(II) species, Environ. Sci. Technol., 2006, 40, 4976–4982 CrossRef.
- F. H. Crocker, K. J. Indest and H. L. Fredrickson, Biodegradation of the cyclic nitramine explosives RDX, HMX, and CL-20, Appl. Microbiol. Biotechnol., 2006, 73, 274–290 CrossRef CAS PubMed.
- P. Larese-Casanova and M. M. Scherer, Abiotic transformation of hexahydro-1,3,5-triazine (RDX) by Green Rust, Environ. Sci. Technol., 2008, 42, 3795–3981 CrossRef.
- C. Olivares, J. Liang, L. Abrell, R. Sierra-Alvarez and J. A. Field, Pathways of reductive 2,4-dinitroanisole (DNAN) biotransformation in sludge, Biotechnol. Bioeng., 2013, 110, 1595–1604 CrossRef CAS.
- J. B. Niedźwiecka and K. T. Finneran, Combined biological and abiotic reactions with iron and Fe(III)-reducing microorganisms for remediation of explosives and insensitive munitions (IM), Environ. Sci.: Water Res. Technol., 2015, 1, 34–39 RSC.
- N. N. Perreault, D. Manno, A. Halasz, S. Thiboutot, G. Ampleman and J. Hawari, Aerobic biotransformation of 2,4-dinitroanisole in soil and soil Bacillus sp., Biodegradation, 2012, 23, 287–295 CrossRef CAS.
- M. J. Kwon, E. J. O'Loughlin, D. A. Antonopoulos and K. T. Finneran, Geochemical and microbiological processes contributing to the transformation of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) in contaminated aquifer material, Chemosphere, 2011, 84, 1223–1230 CrossRef CAS.
- T. T. Fida, S. Palamuru, G. Pandey and J. C. Spain, Aerobic biodegradation of 2,4-dinitroanisole by Nocardioides sp. strain JS1661, Appl. Environ. Microbiol., 2014, 80, 7725–7731 CrossRef.
- J. B. Niedźwiecka, S. R. Drew, M. A. Schlautman, K. A. Millerick, E. Grubbs, N. Tharayil and K. T. Finneran, Iron and electron shuttle mediated (bio)degradation of 2,4-dinitroanisole (DNAN), Environ. Sci. Technol., 2017, 51, 10729–10735 CrossRef.
- C. Olivares, R. Sierra-Alvarez, L. Abrell, J. Chorover, J. Field and R. Khatiwada, (Bio)transformation of 2,4-dinitroanisole (DNAN) in soils, J. Hazard. Mater., 2016, 304, 214–221 CrossRef CAS.
- S. E. Barrows, C. J. Cramer, D. G. Truhlar, M. S. Elovitz and E. J. Weber, Factors controlling regioselectivity in the reduction of polynitroaromatics in aqueous solution, Environ. Sci. Technol., 1996, 30, 3028–3038 CrossRef CAS.
- S. L. Larson, W. A. Martin, B. L. Escalon and M. Thompson, Dissolution, sorption, and kinetics involved in systems containing explosives, water, and soil, Environ. Sci. Technol., 2008, 42, 786–792 CrossRef CAS PubMed.
- B. R. Linker, R. Khatiwada, N. Pedrial, L. Abrell, R. Sierra, J. A. Field and J. Chorover, Adsorption of novel insensitive munitions compounds at clay mineral and metal oxide surfaces, Environ. Chem., 2015, 12, 74–84 CrossRef CAS.
- K. W. Weissmahr, M. Hildenbrand, R. P. Schwarzenbach and S. B. Haderlein, Laboratory and field scale evaluation of geochemical controls on groundwater transport of nitroaromatic ammunition residues, Environ. Sci. Technol., 1999, 33, 2593–2600 CrossRef CAS.
- R. S. Wijker, J. Bolotin, S. F. Nishino, J. C. Spain and T. B. Hofstetter, Using compound-specific isotope analysis to assess biodegradation of nitroaromatic explosives in the subsurface, Environ. Sci. Technol., 2013, 47, 6872–6883 CrossRef CAS.
- M. Elsner, Stable isotope fractionation to investigate natural transformation mechanisms of organic contaminants: principles, prospects and limitations, J. Environ. Monit., 2010, 12, 2005–2031 RSC.
- M. Elsner, M. A. Jochmann, T. B. Hofstetter, D. Hunkeler, A. Bernstein, T. C. Schmidt and A. Schimmelmann, Current challenges in compound-specific stable isotope analysis of environmental organic contaminants, Anal. Bioanal. Chem., 2012, 403, 2471–2491 CrossRef CAS PubMed.
- T. B. Hofstetter, P. Schwarzenbach and S. M. Bernasconi, Assessing transformation processes of organic compounds using stable isotope fractionation, Environ. Sci. Technol., 2008, 42, 7737–7743 CrossRef CAS.
- E. J. Weber, D. L. Spidle and K. A. Thorn, Covalent binding of aniline to humic substances. 1. Kinetic studies, Environ. Sci. Technol., 1996, 30, 2755–2763 CrossRef CAS.
- D. Colón, E. J. Weber and J. L. Anderson, Effect of natural organic matter on the reduction of nitroaromatics by Fe(II) species, Environ. Sci. Technol., 2008, 42, 6538–6543 CrossRef.
- T. B. Hofstetter, J. Bolotin, S. G. Pati, M. Skarpeli-Liati, S. Spahr and R. S. Wijker, Isotope effects as new proxies for organic pollutant transformation, Chimia, 2014, 68, 788–792 CrossRef CAS PubMed.
- A. E. Hartenbach, T. B. Hofstetter, M. Aeschbacher, M. Sander, D. Kim, T. J. Strathmann, W. A. Arnold, C. J. Cramer and R. P. Schwarzenbach, Variability of nitrogen isotope fractionation during the reduction of nitroaromatic compounds with dissolved reductants, Environ. Sci. Technol., 2008, 42, 8352–8359 CrossRef CAS PubMed.
- A. Hartenbach, T. B. Hofstetter, M. Berg, J. Bolotin and R. P. Schwarzenbach, Using nitrogen isotope fractionation to assess abiotic reduction of nitroaromatic compounds, Environ. Sci. Technol., 2006, 40, 7710–7716 CrossRef CAS PubMed.
- T. B. Hofstetter, A. Neumann, W. A. Arnold, A. E. Hartenbach, J. Bolotin, C. J. Cramer and R. P. Schwarzenbach, Substituent effects on nitrogen isotope fractionation during abiotic reduction of nitroaromatic compounds, Environ. Sci. Technol., 2008, 42, 1997–2003 CrossRef CAS.
- C. A. Gorski, J. T. Nurmi, P. G. Tratnyek, T. B. Hofstetter and M. M. Scherer, Redox behavior of magnetite: implications for contaminant reduction, Environ. Sci. Technol., 2010, 44, 55–60 CrossRef CAS PubMed.
- B. A. Ulrich, M. Palatucci, J. Bolotin, J. C. Spain and T. B. Hofstetter, Different mechanisms of alkaline and enzymatic hydrolysis of the insensitive munition component 2,4-dinitroanisole lead to identical products, Environ. Sci. Technol. Lett., 2018, 5, 456–461 CrossRef CAS.
- N. B. Tobler, T. B. Hofstetter, K. L. Straub, D. Fontana and R. P. Schwarzenbach, Iron-mediated microbial oxidation and abiotic reduction of organic contaminants under anoxic conditions, Environ. Sci. Technol., 2007, 41, 7765–7772 CrossRef CAS PubMed.
- C. A. Gorski, R. Edwards, M. Sander, T. B. Hofstetter and S. M. Stewart, Thermodynamic characterization of iron oxide-aqueous Fe(II) redox couples, Environ. Sci. Technol., 2016, 50, 8538–8547 CrossRef CAS PubMed.
- S. M. Stewart, T. B. Hofstetter, P. Joshi and C. A. Gorski, Linking thermodynamics to pollutant reduction kinetics by Fe2+ bound toiron oxides, Environ. Sci. Technol., 2018, 52, 5600–5609 CrossRef CAS PubMed.
- E. E. Daugherty, B. Gilbert, P. S. Nico and T. Borch, Complexation and redox buffering of iron(II) by dissolved organic matter, Environ. Sci. Technol., 2017, 51, 11096–11104 CrossRef CAS PubMed.
- A. M. Vindedahl, J. H. Strehlau, W. A. Arnold and R. L. Penn, Organic matter and iron oxide nanoparticles: aggregation, interactions, and reactivity, Environ. Sci.: Nano, 2016, 3, 494–505 RSC.
- A. M. Vindedahl, W. A. Arnold and R. L. Penn, Impact of Pahokee Peat humic acid and buffer identity on goethite aggregation and reactivity, Environ. Sci.: Nano, 2015, 2, 509–517 RSC.
- A. M. Vindedahl, M. S. Stemig, W. A. Arnold and R. L. Penn, Character of humic substances as a predictor for goethite nanoparticle reactivity and aggregation, Environ. Sci. Technol., 2016, 50, 1200–1208 CrossRef CAS PubMed.
- J. H. Strehlau, M. S. Stemig, R. L. Penn and W. A. Arnold, Facet-dependent oxidative goethite growth as a function of aqueous solution conditions, Environ. Sci. Technol., 2016, 50, 10406–10412 CrossRef CAS PubMed.
- C. L. Chun, R. L. Penn and W. A. Arnold, Kinetic and microscopic studies of reductive transformations of organic contaminants on goethite, Environ. Sci. Technol., 2006, 40, 3299–3304 CrossRef CAS PubMed.
- T. B. Hofstetter, J. C. Spain, S. F. Nishino, J. Bolotin and R. P. Schwarzenbach, Identifying competing aerobic nitrobenzene biodegradation pathways by compound-specific isotope analysis, Environ. Sci. Technol., 2008, 42, 4764–4770 CrossRef CAS PubMed.
- R. S. Wijker, J. Zeyer and T. B. Hofstetter, Isotope fractionation associated with the simultaneous biodegradation of multiple nitrophenol isomers by Pseudomonas putida B2, Environ. Sci.: Processes Impacts, 2017, 19, 775–784 RSC.
- E. C. Butler and K. F. Hayes, Effects of solution composition and pH on the reductive dechlorination of hexachloroethane by iron sulfide, Environ. Sci. Technol., 1998, 32, 1276–1284 CrossRef CAS.
- A. J. Anschutz and R. L. Penn, Reduction of crystalline iron(III) oxyhydroxides using hydroquinone: Influence of phase and particle size, Geochem. Trans., 2005, 6, 60–66 CrossRef CAS.
- U. Schwertmann and R. M. Cornell, in Iron Oxides in the Laboratory: Preparation and Characterization, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2nd edn, 2000, pp. 135–140 Search PubMed.
- E. Viollier, P. W. Inglett, K. Hunter, A. N. Roychoudhury and P. Van Cappellen, The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters, Appl. Geochem., 2000, 15, 785–790 CrossRef CAS.
- J. A. Bourdoiseau, M. Jeannin, C. Rémazeilles, R. Sabot and P. Refait, The transformation of mackinawite into greigite studied by Raman spectroscopy, J. Raman Spectrosc., 2011, 42, 496–504 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, Gases in multimolecular layers, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- M. Berg, J. Bolotin and T. B. Hofstetter, Compound-specific nitrogen and carbon isotope analysis of nitroaromatic compounds in aqueous samples using solid-phase microextraction coupled to GC/IRMS, Anal. Chem., 2007, 79, 2386–2393 CrossRef CAS PubMed.
- A. Kremser, M. A. Jochmann and T. C. Schmidt, PAL SPME Arrow – evaluation of a novel solid-phase microextraction device for freely dissolved PAHs in water, Anal. Bioanal. Chem., 2016, 408, 943–952 CrossRef CAS PubMed.
- S. Spahr, S. Huntscha, J. Bolotin, M. P. Maier, M. Elsner, J. Hollender and T. B. Hofstetter, Compound-specific isotope analysis of benzotriazole and its derivatives, Anal. Bioanal. Chem., 2013, 405, 2843–2856 CrossRef CAS PubMed.
- S. Spahr, J. Bolotin, J. Schleucher, I. Ehlers, U. von Gunten and T. B. Hofstetter, Compound-specific carbon, nitrogen, and hydrogen isotope analysis of N-nitrosodimethylamine in aqueous solutions, Anal. Chem., 2015, 87, 2916–2924 CrossRef CAS PubMed.
- S. G. Pati, H.-P. E. Kohler and T. B. Hofstetter, Characterization of Substrate, Cosubstrate, and Product Isotope Effects Associated With Enzymatic Oxygenations of Organic Compounds Based on Compound-Specific Isotope Analysis, in Methods in Enzymology, Elsevier Inc., 1st edn, 2017, vol. 596, pp. 291–329 Search PubMed.
- R. U. Meckenstock, B. Morasch, C. Griebler and H. H. Richnow, Stable isotope fractionation analysis as a tool to monitor biodegradation in contaminated acquifers, J. Contam. Hydrol., 2004, 75, 215–255 CrossRef CAS PubMed.
- M. Elsner, L. Zwank, D. Hunkeler and R. P. Schwarzenbach, A new concept linking observable stable isotope fractionation to transformation pathways of organic pollutants, Environ. Sci. Technol., 2005, 39, 6896–6916 CrossRef CAS PubMed.
- K. M. Scott, X. Lu, C. M. Cavanaugh and J. S. Liu, Optimal methods for estimating kinetic isotope effects from different forms of the Rayleigh distillation equation, Geochim. Cosmochim. Acta, 2004, 68, 433–442 CrossRef CAS.
- P. Zuman and B. Shah, Addition, reduction, and oxidation reactions of nitrosobenzene, Chem. Rev., 1994, 94, 1621–1641 CrossRef CAS.
- G. C. Wallace, M. Sander, Y.-P. Chin and W. A. Arnold, Quantifying the electron donating capacities of sulfide and dissolved organic matter in sediment pore waters of wetlands, Environ. Sci.: Processes Impacts, 2017, 19, 758–767 RSC.
- Y. Lan, A. S. Elwood Madden and E. C. Butler, Transformation of mackinawite to greigite by trichloroethylene and tetrachloroethylene, Environ. Sci.: Processes Impacts, 2016, 1–8 CAS.
- M. Tobiszewski and J. Namieśnik, Abiotic degradation of chlorinated ethanes and ethenes in water, Environ. Sci. Pollut. Res., 2012, 19, 1994–2006 CrossRef CAS PubMed.
- D. Hunkeler, R. U. Meckenstock, B. S. Lollar, T. C. Schmidt and J. T. Wilson, A Guide for Assessing Biodegradation and Source Identification of Organic Ground Water Contaminants using Compound Specific Isotope Analysis (CSIA), Oklahoma, USA, 2008 Search PubMed.
- A. Bernstein, E. Adar, Z. Ronen, H. Lowag, W. Stichler and R. U. Meckenstock, Quantifying RDX biodegradation in groundwater using δ15N isotope analysis, J. Contam. Hydrol., 2010, 111, 25–35 CrossRef CAS PubMed.
- B. M. Van Breukelen, Extending the Rayleigh equation to allow competing isotope fractionating pathways to improve quantification of biodegradation, Environ. Sci. Technol., 2007, 41, 4004–4010 CrossRef CAS PubMed.
- N. B. Tobler and T. B. Hofstetter, Carbon and hydrogenisotope toluene oxidation by Geobacter metallireducens with different Fe(III) phases as terminal electron acceptors, Environ. Sci. Technol., 2008, 42, 7786–7792 CrossRef CAS PubMed.
- S. G. Pati, H. P. E. Kohler, J. Bolotin, R. E. Parales and T. B. Hofstetter, Isotope effects of enzymatic dioxygenation of nitrobenzene and 2-nitrotoluene by nitrobenzene dioxygenase, Environ. Sci. Technol., 2014, 48, 10750–10759 CrossRef CAS PubMed.
- R. S. Wijker, S. G. Pati, J. Zeyer and T. B. Hofstetter, Enzyme kinetics of different types of flavin-dependent monooxygenases determine the observable contaminant stable isotope fractionation, Environ. Sci. Technol. Lett., 2015, 2, 329–334 CrossRef CAS.
- S. G. Pati, H.-P. E. Kohler, A. Pabis, P. Paneth, R. E. Parales and T. B. Hofstetter, Substrate and enzyme specificity of the kinetic isotope effects associated with the dioxygenation of nitroaromatic contaminants, Environ. Sci. Technol., 2016, 50, 6708–6716 CrossRef CAS PubMed.
- S. A. Mancini, S. K. Hirschorn, M. Elsner, G. Lacrampe-Couloume, B. E. Sleep, E. A. Edwards and B. Sherwood Lollar, Effects of trace element concentration on enzyme controlled stable isotope fractionation during aerobic biodegradation of toluene, Environ. Sci. Technol., 2006, 40, 7675–7681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed report of additional analytical methods, materials synthesis and characterization techniques, batch reaction procedures, CSIA calculations, and further kinetics and isotope results. See DOI: 10.1039/c8em00381e |
| This journal is © The Royal Society of Chemistry 2019 |