
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformal monolayer contacts with lossless interfaces for perovskite single junction and monolithic tandem solar cells†
Amran
Al-Ashouri
 *a,
Artiom
Magomedov
*b,
Marcel
Roß
a,
Marko
Jošt
a,
Martynas
Talaikis
c,
Ganna
Chistiakova
d,
Tobias
Bertram
e,
José A.
Márquez
f,
Eike
Köhnen
a,
Ernestas
Kasparavičius
b,
Sergiu
Levcenco
f,
Lidón
Gil-Escrig
a,
Charles J.
Hages
f,
Rutger
Schlatmann
e,
Bernd
Rech
dg,
Tadas
Malinauskas
b,
Thomas
Unold
f,
Christian A.
Kaufmann
e,
Lars
Korte
d,
Gediminas
Niaura
c,
Vytautas
Getautis
b and
Steve
Albrecht
*ag
*a,
Artiom
Magomedov
*b,
Marcel
Roß
a,
Marko
Jošt
a,
Martynas
Talaikis
c,
Ganna
Chistiakova
d,
Tobias
Bertram
e,
José A.
Márquez
f,
Eike
Köhnen
a,
Ernestas
Kasparavičius
b,
Sergiu
Levcenco
f,
Lidón
Gil-Escrig
a,
Charles J.
Hages
f,
Rutger
Schlatmann
e,
Bernd
Rech
dg,
Tadas
Malinauskas
b,
Thomas
Unold
f,
Christian A.
Kaufmann
e,
Lars
Korte
d,
Gediminas
Niaura
c,
Vytautas
Getautis
b and
Steve
Albrecht
*ag
aYoung Investigator Group Perovskite Tandem Solar Cells, Helmholtz-Zentrum Berlin, Kekuléstraße 5, 12489 Berlin, Germany. E-mail: amran.al-ashouri@helmholtz-berlin.de; steve.albrecht@helmholtz-berlin.de
bDepartment of Organic Chemistry, Kaunas University of Technology, Radvilenu pl. 19, Kaunas LT-50254, Lithuania. E-mail: artiom.magomedov@ktu.lt
cDepartment of Organic Chemistry, Center for Physical Sciences and Technology, Sauletekio Ave. 3, Vilnius LT-10257, Lithuania
dInstitute for Silicon Photovoltaics, Helmholtz-Zentrum Berlin, Kekuléstraße 5, 12489 Berlin, Germany
ePVcomB, Helmholtz Zentrum Berlin, Schwarzschildstr. 3, 12489 Berlin, Germany
fDepartment of Structure and Dynamics of Energy Materials, Helmholtz-Zentrum-Berlin für Materialien und Energie GmbH, Hahn-Meitner Platz 1, 14109 Berlin, Germany
gFaculty of Electrical Engineering and Computer Science, Technical University Berlin, Marchstraße 23, 10587 Berlin, Germany
First published on 2nd October 2019
Abstract
The rapid rise of perovskite solar cells (PSCs) is increasingly limited by the available charge-selective contacts. This work introduces two new hole-selective contacts for p–i–n PSCs that outperform all typical p-contacts in versatility, scalability and PSC power-conversion efficiency (PCE). The molecules are based on carbazole bodies with phosphonic acid anchoring groups and can form self-assembled monolayers (SAMs) on various oxides. Besides minimal material consumption and parasitic absorption, the self-assembly process enables conformal coverage of arbitrarily formed oxide surfaces with simple process control. The SAMs are designed to create an energetically aligned interface to the perovskite absorber without non-radiative losses. For three different perovskite compositions, one of which is prepared by co-evaporation, we show dopant-, additive- and interlayer-free PSCs with stabilized PCEs of up to 21.1%. Further, the conformal coverage allows to realize a monolithic CIGSe/perovskite tandem solar cell with as-deposited, rough CIGSe surface and certified efficiency of 23.26% on an active area of 1 cm2. The simplicity and diverse substrate compatibility of the SAMs might help to further progress perovskite photovoltaics towards a low-cost, widely adopted solar technology.
Broader contextPerovskite-based photovoltaics promises three main benefits: low cost, high efficiency and large versatility. However, combining all three factors into one solar cell design is still a difficult endeavor. In particular, one of the main bottlenecks towards large-scale production is the available choice of hole-selective contacts. The best standards in both polarities, n–i–p (Spiro-OMeTAD) and p–i–n (PTAA), are highly unsuitable for commercial production due to their very high prices and limited processing versatility. Thus, with this work, we present a new generation of self-assembled monolayers (SAMs) as hole-selective contacts that are intrinsically scalable, simple to process, dopant-free and cheap. In addition, they enable highly efficient p–i–n perovskite solar cells and a record-efficiency monolithic perovskite/CIGSe tandem device. While self-assembly offers the crucial advantage of conformally covering rough surfaces within a self-limiting process, one of the herein used SAMs creates an energetically well-aligned interface to the perovskite absorber with minimal non-radiative recombination. Our model system further provides insights into the influence of molecular design on surface passivation and open-circuit voltage, beneficially adding to future prospects of rationally engineering perfect charge-selective contacts. |
Introduction
Metal-halide perovskites triggered intensive research activities throughout the last 5 years with over 3000 published papers on perovskite solar cells (PSCs) only in 2018.1 Typical metal-halide perovskite absorbers are composed of a mixture of different cations (methylammonium MA, formamidinium FA, Cs, Rb etc.) and anions (I−, Br−, Cl−). These compositions have attracted attention due to their outstanding optoelectronic properties including a steep absorption onset together with strong solar absorption2 and high defect tolerance.3,4 Furthermore, the low non-radiative recombination rates enabled voltage deficits that are only ∼65 mV below the radiative limit,5 which is striking for a material processed at low temperatures of around 100 °C. The perovskite layers can be fabricated through a variety of techniques, including vacuum deposition by co-evaporation6 and versatile solution processing methods like spin coating or printing.7 The relatively high band gap of 1.6–1.7 eV, together with the ability of band gap tuning by compositional engineering,8 also renders these materials suitable for integration into tandem solar cell architectures to overcome the efficiency limit of single solar cells.9 In efficient tandem devices, the PSC is used as the top cell absorber with either crystalline silicon,10,11 Cu(In,Ga)Se2 (CIGSe)12,13 or a Sn-based perovskite forming the lower band gap bottom cell.14Although highest reported power conversion efficiencies (PCEs) of over 23% are demonstrated for PSCs with the “regular” n–i–p device architecture,15 the p–i–n (so called “inverted”) architecture is gaining increasing popularity due to its ease of processing and superior suitability for perovskite-based tandem solar cells.16–19 Moreover, p–i–n PSCs carry the promise of low-temperature fabrication, high stability20 without the use of dopants that cause degradation,21–23 low current–voltage hysteresis24 and compatibility to flexible substrates.25,26 However, compared to their n–i–p single junction counterparts, p–i–n PSCs still lack behind in maximum power-conversion efficiency. This is predominantly due to a higher loss in potential, i.e., energetic difference between open-circuit potential (eVOC, with elementary charge e) and band gap. This loss was identified to be dominated by the interfaces to charge-selective contacts.27 Thus, recent efforts were dedicated to reduce these losses through surface passivation, mostly by processing nanometer-thick interfacial layers between absorber and charge-selective contacts.28–34 Recently, changes of the perovskite precursor (e.g., addition of Sr35 or an organic molecule with passivating functional groups,36 or a substitution5 of PbI2) led to open-circuit voltages of well over 1.20 V with comparable loss-in-potential values as obtained in best n–i–p PSCs. However, the mentioned strategies often require finely tuned processing that might be complicated to implement on a large scale. Additionally, for most high-VOC approaches, acceptable stability of the PSCs has yet to be shown.
In the scope of future high-throughput commercialization, it is crucial to keep the simplicity and robustness that p–i–n PSCs exhibit even at high PCEs > 20%. Additionally, it is desirable to minimize parasitic absorption and to use low-cost materials that are suitable for a variety of substrates with arbitrary surfaces and large areas, in order to expand the fields of PSC applications. These ambitions could be realized by using self-assembled monolayers (SAMs) as charge-selective contacts: the required material quantities are minimal; the substrate compatibility is manifold and process control is simple, with the molecules autonomously forming a functional layer in a self-limiting process by design. Functionalization of surfaces with SAMs already has a rich history in surface chemistry.37–39 With the rise of miniaturized electronics, e.g., SAM-based field-effect transistors were built.40,41 After first occurrences in PSCs as electrode modifications,42,43 the first hole-selective SAMs were introduced in 2018.44,45 These molecules covalently bind to the transparent conductive oxide (TCO), e.g., indium tin oxide (ITO), on which the perovskite absorber crystallizes. Due to their hole-selectivity, the SAMs can replace the classical hole-transporting layer. To date, however, the SAMs in PSCs did not enable high PCEs of over 20% that would surpass those reached with the typically used polymeric hole contact material PTAA (poly[bis(4-phenyl)(2,4,6-trime-thylphenyl)amine]). Here, we reach this important objective by using a new generation of SAMs in which the molecules are based on carbazole bodies with phosphonic acid anchoring groups.
We show that the SAMs act as simple hole-selective contacts that can be prepared by classical dip-coating or spin-coating within wide processing windows. By replacing PTAA with a SAM, we demonstrate a maximum PCE of over 21%, which is comparable to current record-efficiencies in the p–i–n architecture.27,34,36 Notably, this PCE is achieved without any perovskite post-treatments, additives, dopants or interlayers that are usually used for high PCEs after delicate fine-tuning. Ultraviolet photoelectron spectroscopy reveals that both new SAMs show a stronger hole-selectivity than PTAA, and photoluminescence (PL) studies show that the SAM/perovskite interface does not introduce non-radiative losses. This enables a VOC of up to 1.19 V and a PL decay time of ∼2 μs. The investigated SAMs work efficiently for three different perovskite compositions, including a 19.6%-efficient PSC which is fabricated by co-evaporation, assuring that hole-extraction by SAMs is a universal approach. We further demonstrate that self-assembly leads to conformal coverage of rough surfaces like as-deposited CIGSe. By integrating a SAM into a tandem architecture, we realize a 23.26%-efficient monolithic CIGSe/perovskite tandem solar cell (certified) with an active area of 1.03 cm2, embodying a low-cost, facile way of realizing all-thin-film tandem solar cells, which has proven to be a hard endeavor in the past.46,47
Results
A schematic representation of the used p–i–n device structure is displayed in Fig. 1a. The glass/ITO serves as a substrate for covalent bonding of the molecules to the ITO, forming a SAM. Afterwards, the perovskite is deposited on top of the SAM. As the electron-selective contact, C60 is thermally evaporated on top of the perovskite absorber. The device is completed by thermal evaporation of a bathocuproine (BCP)/Cu electrode. More details on sample fabrication and methods are provided in the supporting information. Fig. 1c–e displays the molecular structures of the molecules that form the SAMs. Fig. 1f shows the molecule structure of PTAA, which is currently used in the highest performing p–i–n PSCs in literature.27,34,36 PSCs with V1036 ((2-{3,6-bis[bis(4-methoxyphenyl)amino]-9H-carbazol-9-yl}ethyl)phosphonic acid) were already investigated in our previous work.44MeO-2PACz ([2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl]phosphonic acid) and 2PACz ([2-(9H-carbazol-9-yl)ethyl]phosphonic acid) are new molecules based on a carbazole moiety.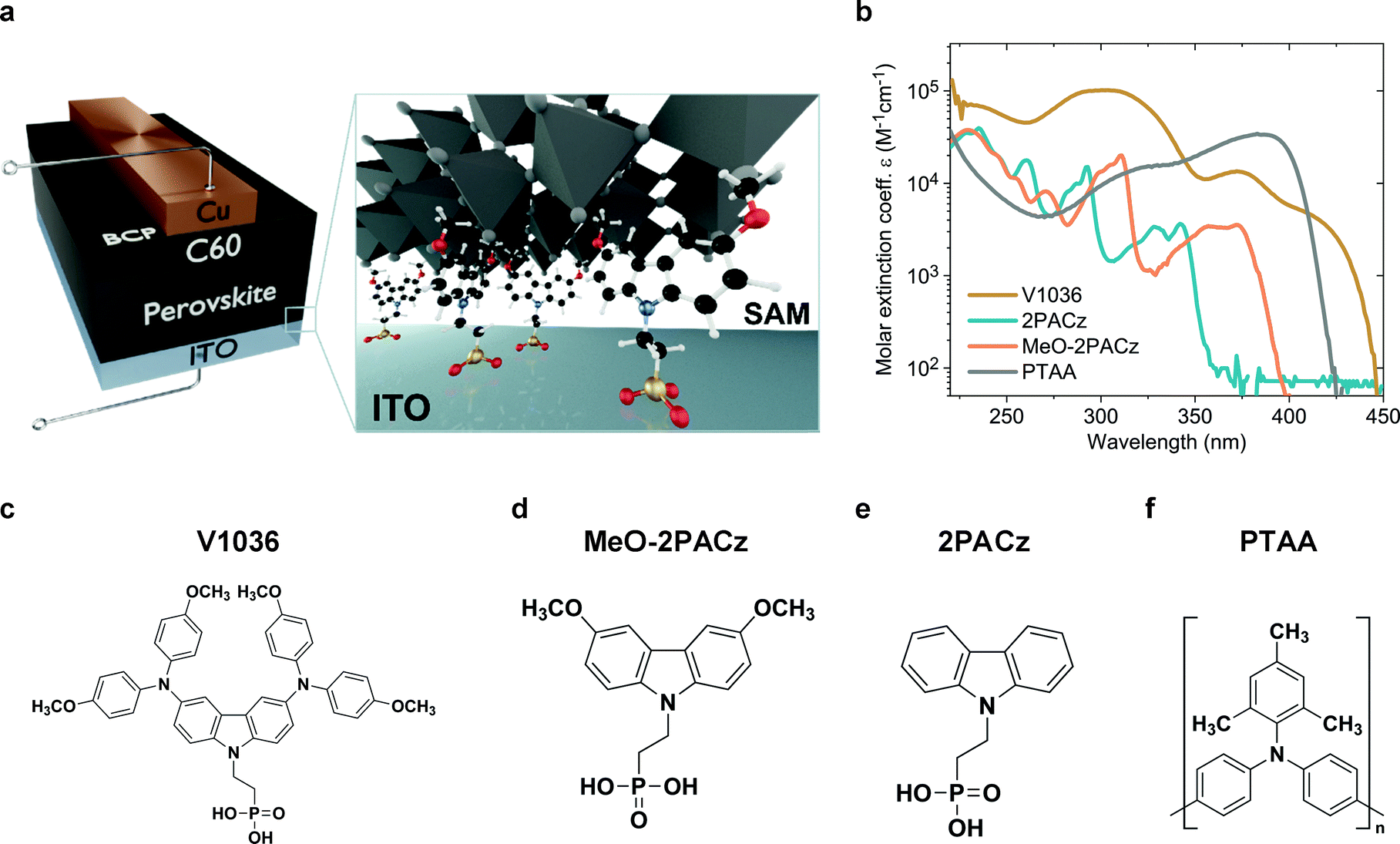 | ||
| Fig. 1 Solar cell device architecture and molecule structures investigated in this work. (a) Schematic of the investigated device structure. The zoom-in visualizes how the SAM molecules attach to the ITO surface and therefore enable the hole selective contact to the perovskite above. (b) Molar extinction coefficient of solutions in tetrahydrofuran containing the different hole-selective contact materials at a concentration of 0.1 mmol l−1. (c) Chemical structure of the SAM molecules V1036,44MeO-2PACz (d) and 2PACz (e). (f) Chemical structure of the typically used polymer PTAA. | ||
Carbazole derivatives have been studied, e.g., for their electron-localizing and thus hole-selective properties,48 starting from first applications in electro-photographic devices. Since then, a huge variety of carbazole-based conductive polymers and molecular glasses has been synthesized and characterized.49 Currently, the carbazole fragment is widely adapted in the synthesis of new materials used in organic light-emitting diodes,50 and, more recently, in PSCs.51 Organic phosphonic acids (PA) are known to form strong and stable bonds on, e.g., ITO surfaces,52–54 enable reliable work function modifications and can principally form bonds to any oxide surface.55–57 In particular, it was calculated for the case of TiO2 that PA has the strongest binding energy among all studied anchoring groups.58 Strong bonds like these enable exceptional stability of the formed monolayers.59 In the frame of perovskite photovoltaics, it has been shown that organic PAs on ITO are stable under continuous solar cell operation for at least 1000 h.60Fig. 1b presents the absorption spectra of all used molecules in a tetrahydrofuran solution. The new SAMs 2PACz and MeO-2PACz show reduced absorption in the visible wavelength regime as compared to PTAA or V1036. The synthesis of these carbazole derivatives was conducted following a simplified version of the previously published synthesis procedure used for V1036 (see Fig. S1 in the ESI†).44 In comparison to V1036, the reaction scheme is one step shorter, no metal-based catalysis was required, and inexpensive, commercially available starting materials were used.
The classic method to coat oxide surfaces with a SAM is to immerse the substrates for several hours into a solution containing the material, optionally under heating of the solution to accelerate binding to the surface. Some molecules, such as the ones used in this work, can also form a dense monolayer simply by spin-coating the solution with a suitable concentration, as was previously described by Nie et al.61 The process is intrinsically self-limiting, since the PA groups only attach to sites on the surface where there is still blank oxide. Following previous studies, we assume that the self-assembly is ordered and stabilized by π–π interactions between adjacent carbazole fragments, in contrast to an ordering that is dominated by van der Waals forces in long-chain aliphatic monolayers.62–64 We investigated solar cells with SAMs formed both by classical dip-coating and spin-coating and did not observe significant differences in solar cell performance between both methods (Fig. S2 in the ESI†). This indicates SAMs of similar surface coverage in both cases. Dip-coating is more suitable for large-area application and conformal coating of textured or rough substrates, while spin-coating is useful for high-throughput optimization in laboratory workflows.
The SAM films obtained from both methods show similar properties in reflection–absorption infrared spectra (RAIRS) measured on ITO substrates as presented in Fig. 2. RAIRS is a molecule-specific, surface-sensitive technique, which allows for probing the structure and bonding of adsorbed molecules on metallic substrates with sub-monolayer sensitivity.65 Here we use the reflection signal (R), normalized to the signal of a bare ITO substrate (R0), to detect the absorption bands of the molecular vibrational modes of the SAM components. By comparing to density functional theory (DFT) calculations (see Fig. S4 and S5, ESI†) and previous reports,66–70 we assign the individual absorption bands to the specific molecular bonds. In general, the observed bands fit to the ones expected from the molecular structure of the SAM molecules. For instance, in the V1036 spectrum in Fig. 2a, the strong band near 1511 cm−1 can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in-plane stretching vibrations of aromatic rings of the carbazole structure with some contribution from CC in-plane stretching vibrations of p-methoxy-phenyl groups.66–68,70 The second strongest band of V1036 near 1246 cm−1 can be associated with C–N stretching vibrations.67,68 Both MeO-2PACz and 2PACz exhibit two bands located near 1490–1494 cm−1 and 1466–1483 cm−1 which are associated with carbazole ring stretching vibrations. Characteristically, 2PACz exhibits two carbazole ring stretching modes at 1242 and 1347 cm−1 and MeO-2PACz a frequency mode at 1582 cm−1 that is associated with the asymmetric stretching vibration of rings with adjacent methoxy groups.69
C in-plane stretching vibrations of aromatic rings of the carbazole structure with some contribution from CC in-plane stretching vibrations of p-methoxy-phenyl groups.66–68,70 The second strongest band of V1036 near 1246 cm−1 can be associated with C–N stretching vibrations.67,68 Both MeO-2PACz and 2PACz exhibit two bands located near 1490–1494 cm−1 and 1466–1483 cm−1 which are associated with carbazole ring stretching vibrations. Characteristically, 2PACz exhibits two carbazole ring stretching modes at 1242 and 1347 cm−1 and MeO-2PACz a frequency mode at 1582 cm−1 that is associated with the asymmetric stretching vibration of rings with adjacent methoxy groups.69
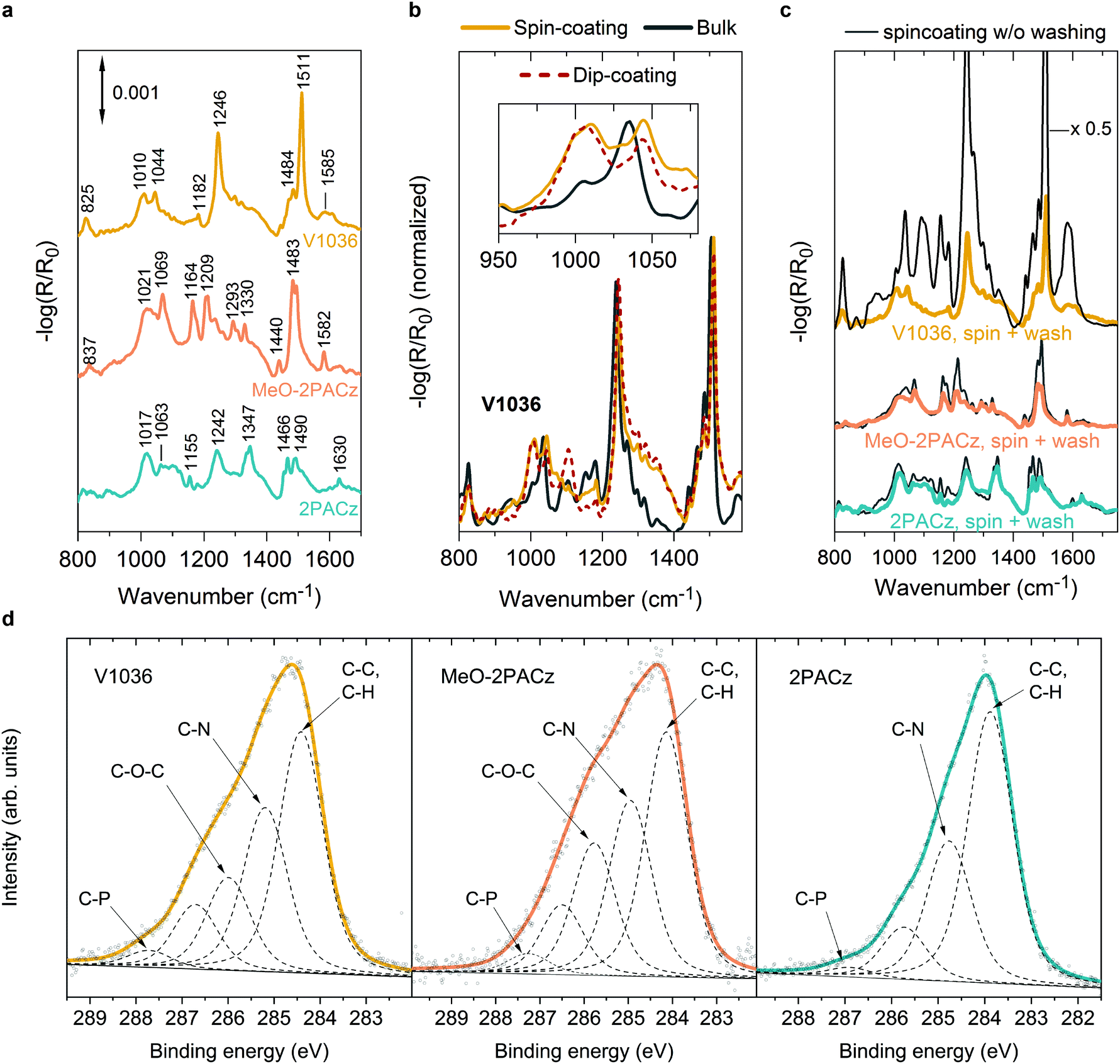 | ||
| Fig. 2 Infrared and X-ray spectroscopic characterizations of SAM-coated ITO substrates. (a–c) FTIR spectrum of the SAM molecule bulk material and reflection–absorption infrared spectra (RAIRS) of monolayers on Si/ITO substrates. (a) Spectra of V1036, MeO-2PACz and 2PACz from spin-coating on Si/ITO substrates, after washing with ethanol and chlorobenzene. (b) Comparison between V1036 bulk material vs. SAM formation from spin-coating and dip-coating. Inset: Detail spectrum in which the monolayer fingerprint (P–O to metal bond) is visible as a broad peak at 1010 cm−1. (c) Effect of the washing step on the RAIRS spectra on spin-coated SAMs. MeO-2PACz and 2PACz already show the monolayer fingerprint without washing. (d) X-ray photoelectron spectroscopy (XPS) spectra of the C1s region, in which the solid line shows the fit to the data and the dotted lines show the components thereof. The additional methoxy group that defines MeO-2PACz in comparison to 2PACz is visible as an additional peak near 286 eV that is assigned to carbon species in C–O–C bonds. | ||
Importantly, the RAIR spectra show a signature of monolayer formation by detection of the bound PA functional group, which is the covalent link between hole-transporting fragment and metal oxide. In our previous work, we concluded the absence of multilayers by comparing absorption measurements to optical simulations.44 This conclusion is further supported by the shown RAIRS analysis. Fig. 2b presents the RAIR spectra for molecules of V1036, comparing monolayers on ITO that are derived from spin- and dip-coating, versus the bulk Fourier-transform infrared (FTIR) spectrum obtained from the powder pressed into a KBr tablet. While the main spectral features are the same for all three materials, the monolayers, in contrast to the bulk material, exhibit a broad feature in the RAIR spectrum at 1010 cm−1 (see inset in Fig. 2b). Both monolayers formed from spin- and dip-coating of V1036 show this band, while the bulk material of V1036 only shows a small shoulder and a slightly shifted spectrum compared to the monolayer spectrum. In conjunction with previous reports,71–74 we can assign the peak at 1010 cm−1 to P–O species bound to ITO. The appearance of this peak, together with the disappearance of the P–OH peak that is prominent in the bulk material at ∼950 cm−1 (see Fig. S4 and S5, ESI†), provides evidence for deprotonation of the phosphonic anchoring group and monolayer formation.
After spin-coating the SAM solution and heating the substrates at 100 °C for 10 min, the substrates are typically washed with the solvent that dissolves the molecules (here ethanol) to remove any molecules that did not bind to the oxide surface. The effect of this is visible in Fig. 2c. Here, the RAIR spectra are shown for ITO samples on which the different SAM solutions were spin-coated, with and without washing the substrates afterwards. For V1036 (upmost curve), the intensity drops by a factor of ∼7 after the washing procedure, and the characteristic P–O absorption shoulder, i.e., the monolayer fingerprint, appears at 1010 cm−1. However, with MeO-2PACz and 2PACz, we notice that the washing step only slightly decreases the intensity of the absorption bands and the monolayer fingerprint is already present without washing. Thus, we conclude that simply spin-coating MeO-2PACz and 2PACz solution with a concentration of roughly 0.5–1 mmol l−1 and subsequent heating of the substrate is sufficient for obtaining a monolayer of the material. Indeed, a wide window of concentrations (at least between 0.5 mmol l−1 and 3 mmol l−1, see Fig. S3, ESI†) of the solutions is found for which no extra rinsing step is required to obtain equivalently performing PSCs. This large processing window further adds to the simplicity of the here presented process strategy and highlights the robustness of monolayer formation with the new SAMs.
As another surface-sensitive technique, we utilized X-ray photoelectron spectroscopy (XPS) to detect the atomic species on the SAM-coated substrates. Fig. 2d shows the X-ray photoelectron spectra in the C1s binding energy region of the investigated SAMs on glass/ITO substrates. While the bare ITO substrate shows almost no signal in this region (see Fig. S14, ESI†), the SAM-coated substrates show characteristic signals that can be fitted with 4–5 peaks with a mixed Lorentzian/Gaussian lineshape and a linear background. The strongest peak can be assigned to aromatic carbon (C–C, C–H) with relative peak areas of 0.57, 0.38 and 0.42 compared to the area of the sum of all peaks for 2PACz, MeO-2PACz and V1036 respectively, each indicating the ratio of the atomic specie to the sum of atoms in the molecule structure. The second strongest peak arises from carbon atoms bonded to nitrogen (C–N), with relative peak areas of around 0.3, 0.27 and 0.28 for 2PACz, MeO-2PACz and V1036, respectively. For MeO-2PACz and V1036, an additional peak is present compared to 2PACz (0.21 relative peak area for MeO-2PACz and 0.16 for V1036), at an energy corresponding to ether functional groups.75 In this case, it can be assigned to C atoms in C–O–C bonds, since methoxy groups are present only for MeO-2PACz and V1036. This is in conjunction with an additional analysis of the Oxygen specie in Fig. S12 of the ESI.† Regarding the peak between the respective C–O–C and C–P assignments, we hypothesize that it might stem from the C atoms bonded to three other C atoms in the carbazole fragment (4a and 4b positions). Overall, the trend of relative peak areas compared between the different SAMs is in line with the counts of atoms in the molecule structures depicted in Fig. 1c–e.
Perovskite solar cell performance
For comparing the performance and device-relevant characteristics of SAM-based solar cells, we chose to focus our analysis on the so called “triple cation” perovskite absorber76 Cs5(MA17FA83)95Pb(I83Br17)3 (CsMAFA), which is widely used due to its high reproducibility. The various hole-selective contacts (HSCs) are compared using the device design as shown in Fig. 1a. Since the polymeric hole transport material PTAA is currently being used in the highest-performing p–i–n PSCs,27,34,36 we compare the SAM-based cells to PTAA-based PSCs and analyze the perovskite film and device properties. To keep the devices as simple as possible, the SAM and PTAA cells do not contain any interfacial compatibilizers, additives or doping. As such, our PTAA control cells are comparable to state-of-the art ones as found in literature.27,77Fig. 3a shows J–V characteristics under simulated AM 1.5G illumination of best PSCs obtained on the respective HSCs in forward (JSC to VOC) and reverse scan (VOC to JSC) direction, with continuous maximum power point (MPP) tracks in the inset. Their photovoltaic parameters are summarized in Table 1. A statistical comparison of the PCEs is plotted in Fig. 3c with 41–53 solar cells per HSC (other device metrics in Fig. S7 in the ESI†). From the J–V curves, we obtain that hysteresis is overall negligible with MPP-tracked efficiencies close to the respective J–V scan values and the fill factor (FF) is overall comparable at around 80% between all PSCs. Fig. 3b displays the external quantum efficiencies (EQEs) of the best devices as well as the integrated product of EQE and AM 1.5G spectrum. The JSC values from EQE integration have a negligible difference to the JSC values obtained from the J–V scans (∼1%). The most striking difference between the HSCs is visible in the open-circuit voltage (VOC), with a difference of 63 mV between PTAA and 2PACz. For the most efficient 2PACz solar cell, a VOC of ∼1.19 V is measured, which is among the highest for this perovskite composition and device architecture, and the highest for CsMAFA cells without interlayers, dopants or additives. Overall, the PCE trend resembles the increase in VOC and both MeO-2PACz and 2PACz solar cells surpass the efficiency of PTAA cells, with 2PACz yielding the highest efficiency of 20.9% in J–V scan and 20.8% in the maximum power point (MPP) track. One of the 2PACz cells was masked, encapsulated and sent to Fraunhofer ISE for certification (see Fig. S23–S25 (ESI†) and red star in Fig. 3c). The certified MPP performance of 20.44% is close to our in-house measurement of 20.7% of that specific cell, validating our analysis. Interestingly, all SAM-based cells show a lower leakage current compared to a champion PTAA cell with a ∼10 nm thick PTAA polymer film (see Fig. 3d). This finding demonstrates that the formed SAMs are dense enough (with regard to number of molecules per surface area) to provide efficient rectification, even though this is just one molecular layer covering the ITO surface.
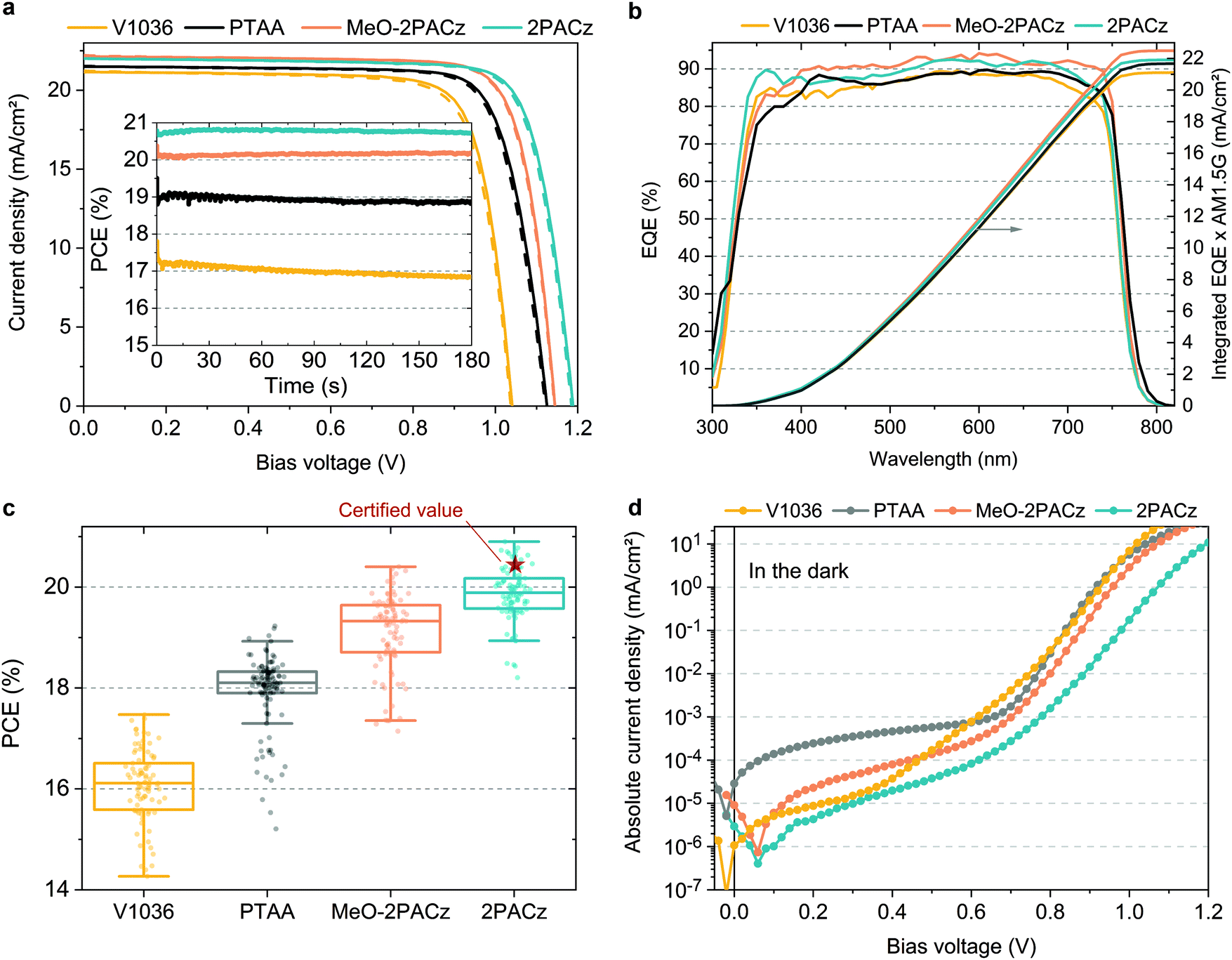 | ||
| Fig. 3 Device-related analysis of SAM-based solar cells in comparison to state-of-the-art PTAA solar cells with triple cation perovskite absorber. (a) J–V curves under simulated AM 1.5G illumination at a scan rate of 250 mV s−1 in forward (JSC to VOC, dashed) and reverse scan (VOC to JSC, solid) with respective MPP tracks in the inset. (b) External quantum efficiency (EQE) spectra of best solar cells and corresponding integration of the product of EQE and AM 1.5G spectrum (right axis). (c) Box plot of power conversion efficiency (PCE) values for 41 V1036, 53 PTAA, 47 MeO-2PACz and 46 2PACz solar cells. (d) Typical J–V curves measured under dark conditions of the respective hole-selective contacts. | ||
| HSC | J SC (mA cm−2) | J SC_EQE (mA cm−2) | V OC (V) | FF (%) | PCE (J–V) (%) | PCE (MPP) (%) |
|---|---|---|---|---|---|---|
| V1036 | 21.2 | 21.1 | 1.041 | 79.3 | 17.5 | 16.9 |
| PTAA | 21.5 | 21.7 | 1.125 | 79.3 | 19.2 | 18.9 |
| MeO-2PACz | 22.2 | 22.5 | 1.144 | 80.5 | 20.4 | 20.2 |
| 2PACz | 21.9 | 21.9 | 1.188 | 80.2 | 20.9 | 20.8 |
Energetic alignment
As shown above, both 2PACz and MeO-2PACz enable higher VOC values in solar cells compared to PTAA. Changes in VOC can have a variety of origins, most importantly changes of the bulk properties and of the non-radiative recombination velocities at one or both interfaces. The latter can be caused by either a higher selectivity due to more favorable energetic alignment and/or less defect states at one or both interfaces to the respective charge-selective contacts. Changes in the bulk properties (e.g., density of trap states) could be caused by altered crystallization of the perovskite film. Since the perovskite film crystallizes on top of various HSCs here, the morphology of the perovskite is analyzed with scanning electron microscopy (SEM) images and X-ray diffraction (XRD) patterns (Fig. S9 and S10, ESI†). No obvious differences in the grain morphology and X-ray diffractograms are observed. Moreover, we estimate the so-called Urbach energy, which is given by the slope of the exponential increase of the absorption edge,2,78 to be around 16 ± 2 meV measured on PSCs based on all four HSCs (obtained from EQE, see Fig. S11, ESI†). The Urbach energy is a measure of electronic disorder in the absorber material, and has been associated with the crystalline quality of lead halide perovskite thin films.79,80 Thus, since we observe neither significant differences in the Urbach energy, XRD, nor coarse grain morphology between the perovskites grown on the investigated HSCs, we conclude that the HSCs do not significantly alter the bulk film properties of the herein used CsMAFA perovskite.To assess the energetic properties of the studied HSCs in relation to the perovskite absorber, we performed ultra-violet photoelectron spectroscopy (UPS) on ITO/HSC and on CsMAFA samples. We can thus compare the positions of the HSC's HOMO (highest occupied molecular orbital) to the perovskite's valence band maximum (VBM). Furthermore, adding the band gaps of the materials estimated from the absorption edge (Fig. 1b), we can also calculate the positions of the HSC's LUMO (lowest unoccupied molecular orbital) and compare it to the perovskite's conduction band minimum, CBM. The spectra are shown in Fig. S18 and S19 (ESI†) and a summary of the results is schematically displayed in Fig. 4a, referenced to the vacuum level. The valence band or HOMO onset values are given in the lowermost row and the work function (i.e., difference between vacuum and Fermi level) values in the upmost row.
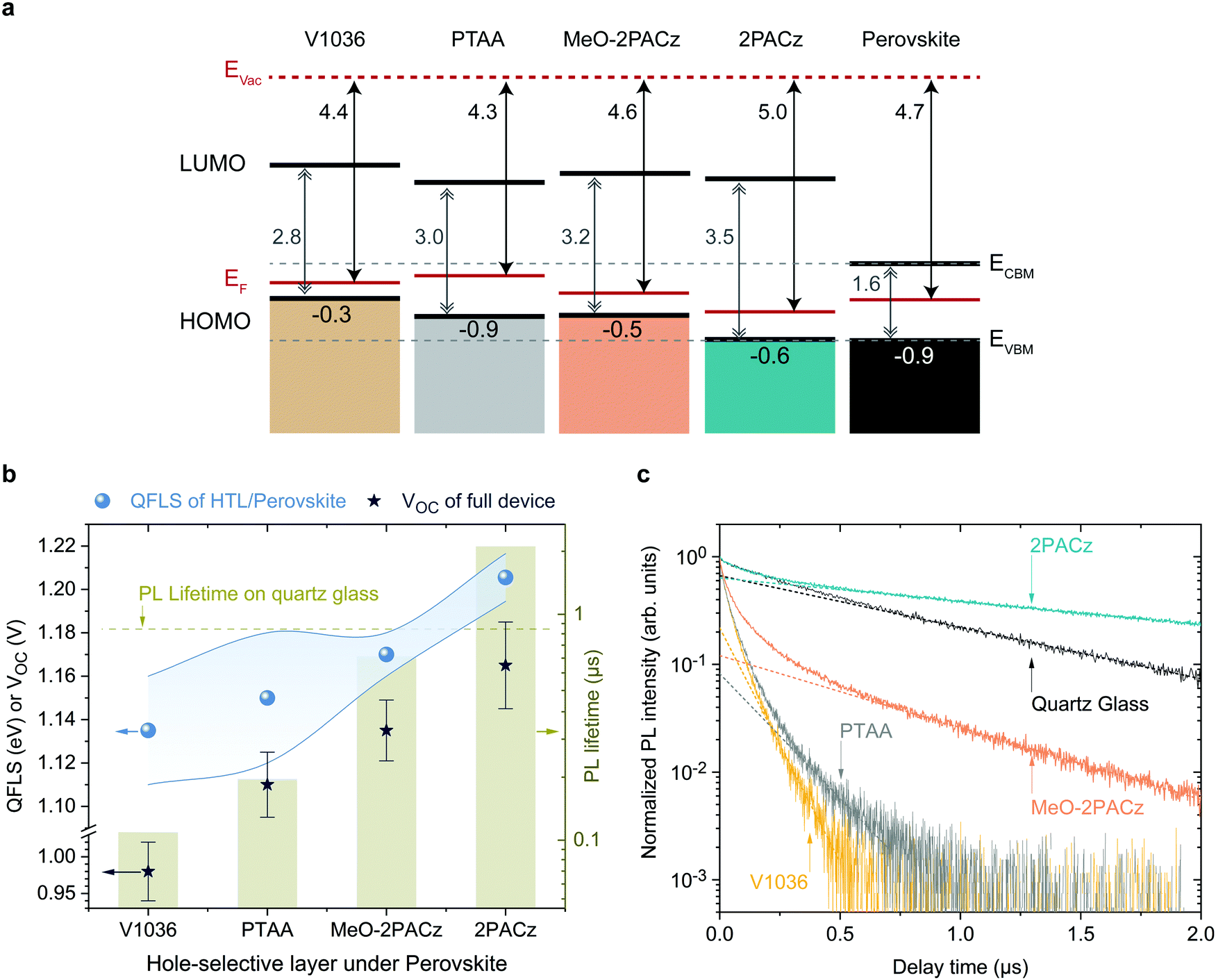 | ||
| Fig. 4 Energetic alignment and Photoluminescence analysis on CsMAFA perovskite. (a) Schematic representation of the band edge positions of the investigated HSCs based on values from UPS measurements, referenced to the vacuum level. The lowermost numbers indicate the difference between Fermi level (EF) of the ITO substrate and HOMO level or valence band maximum (EVBM) (in eV, global error of ∼0.1 eV). The energetic distance between conduction band minimum (ECBM) or LUMO and EVBM was estimated from the onset of optical absorption. The grey, dashed lines are guides to the eye that mark the CBM and VBM levels of the perovskite absorber. (b) Summary of absPL and TrPL measurements. Left axis: average VOC values of solar cells (stars) based on the different HSCs, together with the average quasi Fermi level splitting values (QFLSs, blue spheres) obtained from perovskite films grown on the respective HSCs. The blue filling indicates the span between maximum and minimum QFLS values obtained from several samples (3 V1036, 5 PTAA, 8 MeO-2PACz and 9 2PACz samples). The VOC error bars show the standard deviation from values of 38 V1036 cells, 56 PTAA cells, 42 MeO-2PACz cells and 40 2PACz cells. Right axis: the light-green bars represent the highest obtained PL decay time; the decay time of the perovskite on quartz glass is indicated as a dashed line. (c) Photoluminescence transients of perovskite films deposited on the respective HSCs. The dotted lines are extrapolated fits to the mono-exponential tail of the transients, from which the PL decay time values are obtained. | ||
All SAMs show a p-type character in the energetic diagram. Comparing the valence band onset of the perovskite absorber to the HOMO levels of the HSC layers, it is apparent that MeO-2PACz and 2PACz are energetically more hole-selective than PTAA, due to the higher energetic barrier for electrons, while still allowing for an efficient extraction of holes (no barrier). 2PACz shows the closest alignment to the valence band maximum (VBM) of the perovskite, whereas V1036 shows the strongest offset. In this respect, MeO-2PACz is similar to PTAA, while the presence of methoxy groups in MeO-2PACz suggests a passivating function, as reported in earlier works.81,82 Changes in ITO work function with PA-based SAMs have been thoroughly analyzed in literature.71,83,84 In our case, the binding type between the studied SAMs is the same, thus differences between the work functions can be assigned to differences of the molecular dipole moments of the hole-selective fragments.85,86 The shift in work function between bare ITO (4.6 eV, see Fig. S18a, ESI†) and the SAM-modified surface is higher for 2PACz as compared to MeO-2PACz, being 5.0 and 4.6 eV, respectively. This is in line with the larger molecular dipole moment of 2PACz of +2 D compared to +0.2 D for MeO-2PACz (calculated by DFT following a previously published procedure,56 more details in the ESI†). V1036 has a negative calculated dipole moment of −2.4 D, reducing the ITO work function to 4.4 eV. Judging from this energetic picture, the observed trend in VOC could potentially be explained with how close the HOMO of the HSCs is aligned to the perovskite's VBM.87V1036 devices show the smallest VOC and V1036 the largest offset in EVBM (0.9 eV), whereas 2PACz yields the highest VOC and shows almost no offset to the perovskite's VBM.
Photoluminescence studies
The energetic alignment discussed above already provides a first hint to why PSCs based on the new SAMs outperform those on PTAA. However, it remains unclear how the bands align to each other at the buried interface between the HSC and perovskite itself. Recent reports also point to an insensitivity of the energetic difference in EVBM between perovskite and HSC for moderate misalignment.88 Thus, we further investigate the differences with photoluminescence (PL) studies, using time-resolved PL (trPL) to study the behavior of the charge carriers on short time scales and absolute PL (absPL) to estimate the “implied VOC” or quasi Fermi level splitting (QFLS)89 of the bare absorber computed by the high-energy tail fit method27,90,91 for a temperature of 300 K. The PL measurements were conducted on glass/ITO/HSC/CsMAFA samples without the C60 overlayer. The QFLS was also determined from the full 2PACz solar cell that is shown in Fig. 3a, with negligible difference to the eVOC value obtained from a J–V scan (see Fig. S20, ESI†).Fig. 4b shows a summary of the QFLS values from absPL (blue spheres) on the left axis, together with average values of measured VOC (stars) of the full devices. The right axis presents PL decay times from TrPL (bars), calculated from Fig. 4c, in comparison to the PL decay time for a perovskite film on quartz glass (dashed line), which is known as a highly passivated surface.27,92 The rising trend in average QFLS from V1036 over PTAA to MeO-2PACz and 2PACz fits to the trend in VOC. Compared to V1036 and PTAA, the spread of QFLS values is smaller with MeO-2PACz and 2PACz. The TrPL transients are plotted in Fig. 4b and were recorded at an excitation fluence that is relevant for device operation at 1 sun illumination (fluence ∼15–30 nJ cm−2, see PL section in ESI†). The PL measured on the perovskite film on glass decays mono-exponentially. The deviation from mono-molecular decay in the measurements with HSCs could be attributed to charge transfer effects.93MeO-2PACz allows for PL decay times of over 650 ns, which approaches the decay time of the same perovskite on quartz glass of ∼860 ns, a comparable value to the ones previously reported for the same perovskite in record-efficiency PSCs.27 Interestingly, the decay time on 2PACz with a value of 2 μs even surpasses the one on quartz glass by a factor of over 2. Since the 2PACz PL transient shows signs of slow charge transfer, the significantly longer decay time cannot be attributed to a mere reduction of majority carriers at the interface. We thus conclude that the interface defect density must be negligibly low, highlighting that bare Carbazole is chemically compatible to the perovskite, forming a well-passivated surface. A direct comparison to MeO-2PACz with regard to interface defect density is not possible from the TrPL data alone, since an energetic offset can independently affect interfacial recombination.94 However, the faster decay at early times suggests faster hole extraction. With the only difference between MeO-2PACz and 2PACz being the termination with a methoxy group, it is interesting to observe such an intrinsically different behavior in the charge carrier dynamics.
Comparing the values obtained on PTAA and on 2PACz, the decay times differ by an order of magnitude, and both the QFLS and VOC values are around 60 mV higher with 2PACz. This fits to the thermodynamically expected increase of 60 mV when increasing the photoluminescence yield by a factor of 10, with kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10) ≈ 60 meV, where k is the Boltzmann constant and T the temperature (300 K).95 We emphasize that the FF values of the full solar cells are comparable among all HSCs, suggesting similarly efficient charge extraction. Thus, we conclude that the trend in PL decay time is set by the non-radiative recombination velocity at the interface. Assuming that interface recombination is dominating, the recombination velocities can be estimated to range from 193 cm s−1 (V1036) to a lowest value of 12 cm s−1 for 2PACz (upper estimate, see Section 8 for details, ESI†). The clear correlation between QFLS, PL decay time and VOC provides a strong indication that the differences in VOC of the solar cells are originating from differences in the compatibility of the HSC interface to the perovskite. This is either governed by the interface defect density, energetic alignment as addressed above, or a combination of both. It remains open whether the molecular dipole moments play a role other than the mentioned work function modifications. The energetic difference between QFLS and eVOC with all HSCs is induced by non-radiative recombination at the perovskite/C60 interface (see also Fig. S20, ESI†). Previously it was identified that the perovskite/C60 interface limits the VOC to a fixed value in the used PSC architecture, independent of the perovskite bulk quality.27 Indeed, our PTAA devices achieve max. ∼1.13 V, even though the perovskite on PTAA can reach a QFLS of up to 1.18 eV. It is thus interesting that by increasing the hole-selectivity and decreasing the interface recombination velocity, the 1.13 V limitation can be overcome although the perovskite morphology stays the same. It is furthermore worth noting that a recent study that compared a wide variety of typically used charge-selective layers identified that every single layer introduces non-radiative losses compared to the bare perovskite bulk, lowering the PL decay time and QFLS.94 In sharp contrast, the herein introduced 2PACz HSC is an important demonstration that the opposite is possible as well, rendering a “lossless” hole-selective interface. With SAM contacts, further enhancement of the VOC to a level of the QFLS of the bare perovskite film or above is expected upon mitigating the losses introduced at the C60 interface.
ln(10) ≈ 60 meV, where k is the Boltzmann constant and T the temperature (300 K).95 We emphasize that the FF values of the full solar cells are comparable among all HSCs, suggesting similarly efficient charge extraction. Thus, we conclude that the trend in PL decay time is set by the non-radiative recombination velocity at the interface. Assuming that interface recombination is dominating, the recombination velocities can be estimated to range from 193 cm s−1 (V1036) to a lowest value of 12 cm s−1 for 2PACz (upper estimate, see Section 8 for details, ESI†). The clear correlation between QFLS, PL decay time and VOC provides a strong indication that the differences in VOC of the solar cells are originating from differences in the compatibility of the HSC interface to the perovskite. This is either governed by the interface defect density, energetic alignment as addressed above, or a combination of both. It remains open whether the molecular dipole moments play a role other than the mentioned work function modifications. The energetic difference between QFLS and eVOC with all HSCs is induced by non-radiative recombination at the perovskite/C60 interface (see also Fig. S20, ESI†). Previously it was identified that the perovskite/C60 interface limits the VOC to a fixed value in the used PSC architecture, independent of the perovskite bulk quality.27 Indeed, our PTAA devices achieve max. ∼1.13 V, even though the perovskite on PTAA can reach a QFLS of up to 1.18 eV. It is thus interesting that by increasing the hole-selectivity and decreasing the interface recombination velocity, the 1.13 V limitation can be overcome although the perovskite morphology stays the same. It is furthermore worth noting that a recent study that compared a wide variety of typically used charge-selective layers identified that every single layer introduces non-radiative losses compared to the bare perovskite bulk, lowering the PL decay time and QFLS.94 In sharp contrast, the herein introduced 2PACz HSC is an important demonstration that the opposite is possible as well, rendering a “lossless” hole-selective interface. With SAM contacts, further enhancement of the VOC to a level of the QFLS of the bare perovskite film or above is expected upon mitigating the losses introduced at the C60 interface.
To conclude our analysis, we identified several aspects as to why the herein introduced molecules lead to high-performance PSCs, besides the already discussed dense film-forming properties. Consequently, a clear guidance could be drawn from our findings to develop other lossless contact materials in the near future: on the one hand, the structure of these small molecules leads to a small density of interfacial trap states as seen by the comparison of quartz glass to 2PACz-covered ITO and by the comparison of PTAA to MeO-2PACz. On the other hand, as shown by simulations of a similar device stack in a recent work,94 our own energetic alignment data and other experimental studies,96–100 energetic alignment for majority carrier extraction influences interfacial recombination losses and is a crucial factor for achieving high VOC values. The strong correlation in our SAM model system (layers with similar carbazole chemistry and similar thickness, but different HOMO levels) indicates that by using carbazole derivatives and tuning the work function by dipole moment engineering, ideal hole-selective layers could be rationally designed for specific perovskite absorbers. Further investigations on the exact hole extraction mechanisms at a SAM interface might include simulations of the electric field in atomic scales,84 tunneling of charge carriers and atomistic simulations on the interaction between dipole moment and perovskite interface.
Stability assessment
In addition to the increased efficiencies of both MeO-2PACz and 2PACz-based solar cells compared to PTAA, we also observe an increased stability. Fig. 5a shows the time evolution of PCE under continuous MPP tracking for the investigated HSCs at simulated 1 sun AM 1.5G illumination without active sample cooling. The samples reach temperatures of at least 40 °C under operation. Only small differences are visible between cells with the investigated HSCs, while a slight advantage is evident for the MeO-2PACz and 2PACz-based devices after 11 h of operation (<3% PCE loss with 2PACz with a stable value after an initial drop, ∼3% loss with MeO-2PACz, ∼6% loss with PTAA and almost 12% loss with V1036). A stronger difference is visible when increasing the stress on the solar cells, which is done here by light soaking at open-circuit condition under 1 sun illumination, a condition at which a high average density of charge-carriers is present in the device that can induce a quick degradation of the PSC.101 The most notable differences occur in the time evolution of VOC, as displayed in Fig. 5b. PTAA-based solar cells show a substantial drop (60 mV amplitude) after two hours, while the VOCs of all SAM-based cells remain virtually stable after an initial drop caused by increasing temperature. Interestingly, light soaking steadily improves the VOC of V1036-based samples. The difference in stability between PTAA and SAMs under illumination cannot simply be explained with the differences in non-radiative recombination rates at the interface, since judging by QFLS and PL decay time, V1036 should then show the weakest VOC stability. We attribute the quick degradation of PTAA-based cells to be a material-specific characteristic of the CsMAFA/PTAA contact that occurs under conditions with a high number of excess charge carriers and direct illumination of the PTAA, as also observed in a recent work.102 A previous study identified that a large number of excess charge carriers, as under illumination at open-circuit, leads to a lowered energetic threshold of ionic movement.101 We hypothesize that the diffusion of iodine to the PTAA interface leads to structural damage of the PTAA, as recently argued by Sekimoto et al.103 In contrast, a SAM, being a chemically robust electrode modification with virtually no volume, is neither susceptible to structural damages nor to an accumulation of ions.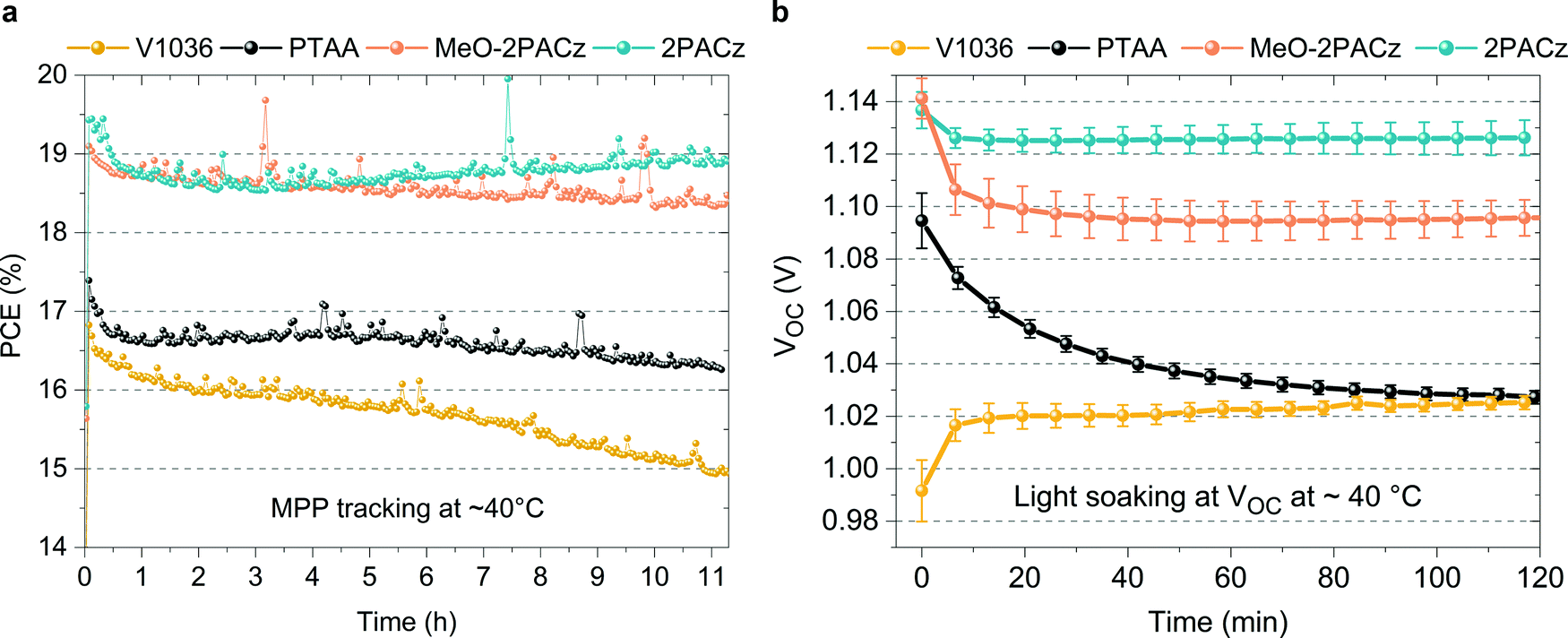 | ||
| Fig. 5 Stability assessments of PSCs based on the investigated HSCs in N2 atmosphere. (a) MPP tracking under continuous, simulated 1 sun AM 1.5G illumination of uncooled devices (reaching a temperature of ∼40 °C in operation). (b) Time evolution of VOC values of solar cells kept at open-circuit conditions under light-soaking at full sun illumination at ∼40 °C cell temperature (no active cooling). The values were extracted from J–V-scans every 6–8 min. The error bars show the standard deviation of these values across the individual cells (4 V1036, 6 PTAA, 3 MeO-2PACz and 10 2PACz cells). | ||
Versatility of a SAM contact and tandem solar cell integration
In order to show versatility of the new HSC molecules, we demonstrate in Fig. 6 that the SAMs also yield highly efficient perovskite solar cells with perovskite compositions other than CsMAFA. In addition to the “triple cation” composition utilized for the analysis above, we investigate here a “double cation” MA5FA95Pb(I95Br5)3 (MAFA)8 absorber with a lower band gap of ca. 1.55 eV and a “single cation” (MAPbI3) absorber layer that is fabricated by direct co-evaporation with an optical band gap of ca. 1.60 eV (see Fig. S26 for the absorption onset, ESI†). Table 2 shows the corresponding performance metrics. We noticed that on MeO-2PACz, the MAFA and MAPbI3 perovskites tend to crystallize more reproducibly than on 2PACz, thus we chose MeO-2PACz for the purpose of demonstrating that a SAM enables a broad spectrum of applications.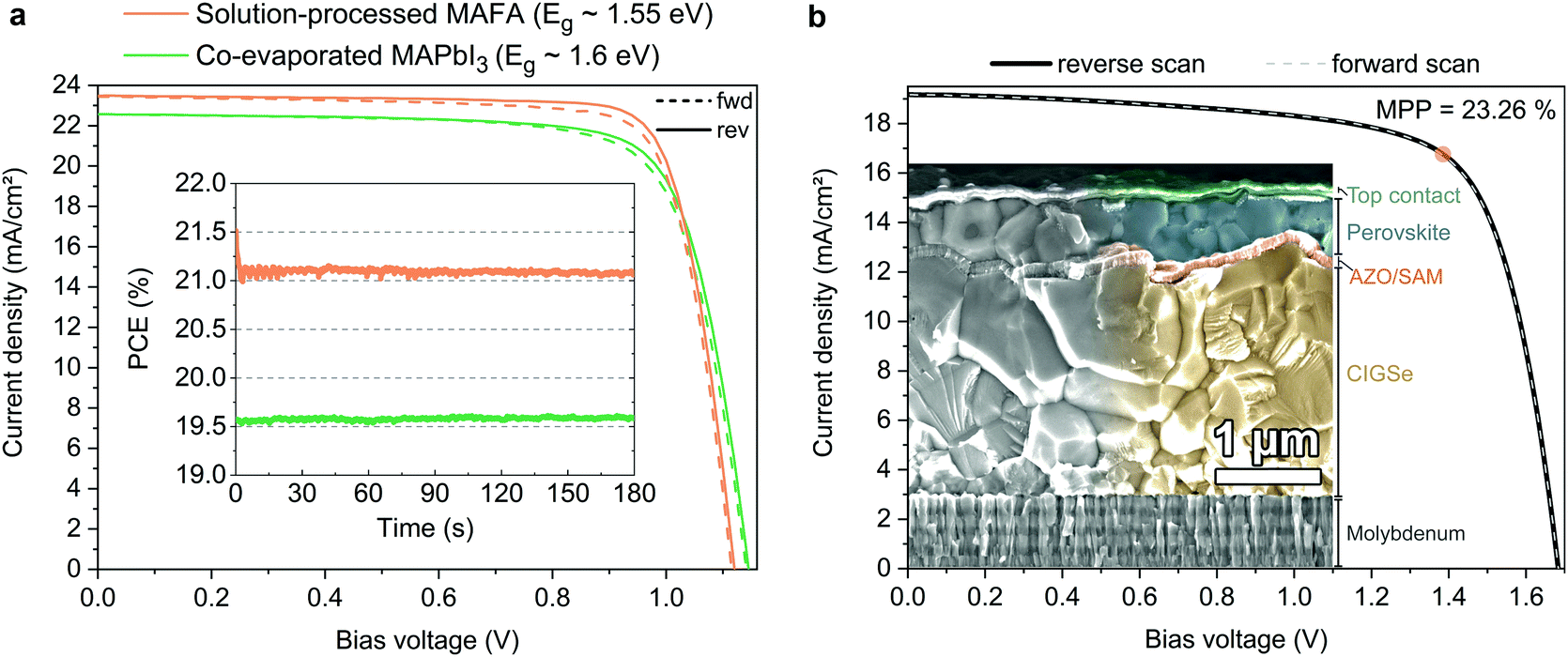 | ||
| Fig. 6 Display of the versatility of SAM contacts and tandem solar cell integration. (a) J–V curves under illumination of solar cells based on a solution processed “double cation” perovskite absorber (FA95MA5Pb(I95Br5)3, orange line) and co-evaporated perovskite absorber (MAPbI3, green line). Inset: Continuous MPP tracks of these cells. (b) J–V curve of a monolithic CIGSe/perovskite tandem solar cell (active area of 1.034 cm2), with MeO-2PACz2PACz as HSC conformally covering the rough CIGSe bottom cell. The orange circle indicates the MPP at 23.26% PCE (see certified MPP track by Fraunhofer ISE in Fig. S27, ESI†). Inset: SEM image of the cross-section of a representative tandem device. The recombination contact consists of aluminum-doped zinc oxide, sputtered onto the CIGSe surface and covered by MeO-2PACz. | ||
| Perovskite | Device type | J SC (mA cm−2) | J SC_EQE (mA cm−2) | V OC (V) | FF (%) | PCE (J–V) (%) | PCE (MPP) (%) |
|---|---|---|---|---|---|---|---|
| a First value corresponds to the perovskite top cell and second value to the CIGSe bottom cell. | |||||||
| Co-evaporated MAPbI3 | Single | 22.6 | 22.5 | 1.145 | 76.8 | 19.8 | 19.6 |
| Double cation MAFA | Single | 23.5 | 23.4 | 1.120 | 80.6 | 21.2 | 21.1 |
| Triple cation CsMAFA | Monolithic Tandem | 19.17 (certified) | 20.2/19.1a (in-house) | 1.68 (certified) | 71.9 (certified) | 23.16 (certified) | 23.26 ± 0.75 (certified) |
Perovskite processing by co-evaporation is highly attractive due to large-area compatibility and absence of potentially harmful solvents for fabrication. Utilizing MeO-2PACz as the HSC also works well with the co-evaporated MAPbI3 perovskite absorber. The herein shown stabilized PCE of 19.6% with over 1.14 V VOC is approaching the PCE of the best co-evaporated PSC to date and represents the highest reported value for p–i–n type devices utilizing co-evaporation for the perovskite absorber.104,105
Recently, highest PSC performances were reported for the MAFA double cation composition.15,106 Compared to the CsMAFA solar cells shown before, the MAFA cell has an advantage in higher current density due to the lower bandgap of the composition, but still enables a relatively high VOC. Without detailed optimization, the MAFA absorber enables a stabilized PCE of 21.1% when utilizing MeO-2PACz as the HSC. As seen in Table 2, for all three solar cells the JSC values measured in the J–V curve closely fit to the values obtained by integrating the product of EQE measurement and AM 1.5G spectrum (Fig. S26a, ESI†). 2PACz enables an over 21%-efficient MAFA PSC as well, as shown in Fig. S27 (ESI†).
Furthermore, we show in Fig. 6b that a SAM is also a suitable HSC for manufacturing PSCs on rough surfaces, which is essential for e.g., CIGSe/perovskite tandem solar cells. All-thin-film tandem solar cells pose an attractive strategy for cheap, versatile and flexible high-efficiency solar cells and are a promising route for the introduction of halide perovskites into industrial production. CIGSe enables thin-film solar cells with a suitable bandgap for the use in perovskite-based tandem solar cells. However, the rough surface of the CIGSe makes it difficult to process the thin HSCs that currently enable efficient perovskite top cells. Recently, we demonstrated that the use of a NiOx layer processed by atomic layer deposition (ALD) in combination with PTAA represents a CIGSe/perovskite-compatible hole-transport layer that prevents shunting caused by the CIGSe roughness, enabling a 21.6%-efficient tandem cell.47 Here we now show that a SAM removes the need of an ALD step, since the self-assembly process works reliably even on rough surfaces, by dipping the CIGSe bottom cell into a MeO-2PACz solution. The perovskite layer was fabricated by solution-processing and the top contact layers by either evaporation or ALD/sputtering (see ESI† for details). The so prepared tandem solar cell, shown in Fig. 6b, shows a stabilized efficiency of 23.26% on an area of 1.03 cm2 (certified by Fraunhofer ISE, see Fig. S30–S32, ESI†), while the bottom cell alone has a PCE of 15–16% (see Fig. S28, ESI†). The EQEs of both sub-cells measured in the tandem are shown in Fig. S29 (ESI†). Following the VOC trend shown earlier, our SAM-based tandem shows an improved VOC as compared to our previously published one (∼90 mV increase from 1.59 to 1.68 V). However, the SAM-based tandem cell shows a lower FF (72% vs. 76% with the NiO2/PTAA double layer), which here is mainly limited by the shunt resistance. Since the tandem current is limited here by the bottom cell, further optimization could be dedicated to developing a more robust stack design that prevents micro-shunts caused by processing and measuring (here we use the same design as in our previous work47). Nonetheless, the here presented tandem cell surpasses the previous record46 (22.4%) in PCE and area (0.04 cm2vs. 1.03 cm2 here). The simplicity of the tandem stack and the use of as-deposited CIGSe additionally suggests that our approach could be easily adopted in higher throughput fabrication.
In summary, the compatibility of the SAM to three different perovskite compositions, two different processing techniques (solution and vacuum process), two different oxides (ITO in the single junctions and Al-doped zinc oxide in the tandem) and two different substrate morphologies (rough and flat) strongly suggests that SAM hole-selective contacts represent a universal approach for perovskite-based photovoltaics.
Conclusion
Two new simple molecules that form self-assembling monolayers (SAMs), MeO-2PACz and 2PACz, were synthesized and integrated into inverted perovskite solar cells, enabling hole-selective contacts with minimized non-radiative losses. The new SAMs can be deposited on transparent conductive oxides via spin-coating or by dipping the substrate into the solution, both yielding layers of comparable properties, combining high reproducibility and ease of fabrication. Both new SAMs outperform the polymer PTAA, the material that enabled the highest-performing p–i–n PSCs to date, in efficiency, stability and versatility. With a standard triple-cation absorber, a maximum power conversion efficiency of 20.9%, certified efficiency of 20.44% and a VOC of up to 1.19 V were demonstrated. MeO-2PACz further enabled a 21.1%-efficient solar cell with a double-cation absorber, and a stabilized efficiency of 19.6% with a co-evaporated single-cation absorber. Photoelectron spectroscopy and photoluminescence (PL) investigations revealed a well-suited energetic alignment and strongly reduced non-radiative recombination at the interface between absorber and contact, leading to a PL decay time of 2 μs for a perovskite on 2PACz. As deduced from a comparison between the PL transients of perovskite grown on 2PACzversus on quartz glass, the interface defect density at the 2PACz/perovskite interface is minimal. From the trend between surface recombination velocity, VOC and offset between the SAM HOMO level and perovskite valence band edge, our model system provides experimental evidence for the energetic alignment and interface defect density being similarly important for mitigating non-radiative recombination losses. The results highlight that carbazole derivatives can combine all necessary features for lossless interfaces and are thus a compelling material class for future chemical engineering of high-performance hole-selective contacts. In a light-soaking stress test at open circuit conditions, SAM-based PSCs showed a higher stability compared to PTAA-based cells. Finally, by integrating a SAM contact into a monolithic CIGSe/perovskite tandem solar cell, the ability of conformally creating a hole-selective layer on a rough surface was demonstrated. This led to a stabilized, certified PCE of 23.26% with facile device design on an active area of 1.03 cm2, surpassing the values achieved with a complex bilayer47 or mechanical polishing.46 Importantly, the herein demonstrated solar cells are fabricated without additional passivation layers, additives or dopants. Together with the minimal material consumption, manifold substrate compatibility and simplicity during fabrication, the SAM contacts might present a realistic way to further progress perovskite photovoltaics into a low-cost, wide-spread technology.Author contributions
A. A. A., A. M., M. J. and S. A. coordinated the project and designed the experiments. A. M., Er. K. and T. M. designed, synthesized and characterized the SAM molecules. A. A. A. optimized the SAM formation procedures, carried out sample fabrication and solar cell characterization, coordinated data analysis and prepared the manuscript figures. M. T. recorded the RAIRS and FTIR spectra, and calculated the theoretical spectra; M. T., G. N., A. M. and A. A. A. analyzed the data. G. C. measured the UPS and XPS data; A. A. A., A. M. and G. C. analyzed the data. A. A. A. recorded the absolute PL data in a setup designed by J. A. M. and T. U., and, together with S. L., recorded the trPL data in a setup designed by C. J. H., S. L. and T. U. The PL data was analyzed by A. A. A., J. A. M, S. L. and T. U. Fabrication of the co-evaporated solar cell was carried out by M. R.; M. R. and L. G. E. optimized the co-evaporation process. A. A. A. and M. J. processed the CIGSe/perovskite tandem solar cell, with optimization of the top contact by Ei. K. The CIGSe bottom cell was fabricated by T. B. and C. K. All authors contributed to manuscript writing and scientific discussions. B. R., T. U., L. K., G. N., R. S., V. G. and S. A. supervised the project.Conflicts of interest
There are no competing interests to declare.Acknowledgements
The authors thank M. Gabernig, C. Ferber, T. Lußky, H. Heinz, C. Klimm and M. Muske at Institute for Silicon Photovoltaics (HZB), T. Hänel, B. Bunn, K. Mayer-Stillrich, M. Kirsch, S. Stutzke and J. Lauche at PVcomB (HZB) for technical assistance. For XPS and UPS measurements, the Energy Materials In-Situ Laboratory (EMIL), R. Wilks and J. Frisch are acknowledged. Funding was provided by German Federal Ministry for Education and Research (BMBF) (grant no. 03SF0540) within the project “Materialforschung für die Energiewende”, (BMWi) project “EFFCIS” (grant no. 0324076B). A. M. and T. M. acknowledge funding by the Research Council of Lithuania under grant agreement no. S-MIP-19-5/SV3-1079 of the SAM project. G. N. gratefully acknowledges the Center of Spectroscopic Characterization of Materials and Electronic/Molecular Processes (SPECTROVERSUM Infrastructure) for use of their FTIR spectrometer. T. B. acknowledges funding from the BMWi project Speedcigs (grant no. 0324095D). The authors acknowledge the HyPerCells graduate school for support and the funding by the Helmholtz Foundation for the HySPRINT Innovation lab which was supported by the Helmholtz Energy Materials Foundry (HEMF). The research leading to these results has partly received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 763977 of the PerTPV project.References
- Web of Science search for ‘perovskite’ AND ‘solar’ AND ‘cells’ in the title.
- S. de Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F. Haug, J. Yum and C. Ballif, J. Phys. Chem. C, 2014, 5, 1035–1139 CAS.
- W. J. Yin, T. Shi and Y. Yan, Appl. Phys. Lett., 2014, 104(6), 063903 CrossRef.
- K. X. Steirer, P. Schulz, G. Teeter, V. Stevanovic, M. Yang, K. Zhu and J. J. Berry, ACS Energy Lett., 2016, 1, 360–366 CrossRef CAS.
- Z. Liu, L. Krückemeier, B. Krogmeier, B. Klingebiel, J. A. Márquez, S. Levcenko, S. Öz, S. Mathur, U. Rau, T. Unold and T. Kirchartz, ACS Energy Lett., 2019, 4, 110–117 CrossRef CAS.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- Z. Wei, H. Chen, K. Yan and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 13239–13243 Search PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- A. De Vos, J. Phys. D: Appl. Phys., 1980, 13, 839–846 CrossRef.
- S. Albrecht, M. Saliba, J. P. Correa Baena, F. Lang, L. Kegelmann, M. Mews, L. Steier, A. Abate, J. Rappich, L. Korte, R. Schlatmann, M. K. Nazeeruddin, A. Hagfeldt, M. Grätzel and B. Rech, Energy Environ. Sci., 2016, 9, 81–88 RSC.
- J. P. Mailoa, C. D. Bailie, E. C. Johlin, E. T. Hoke, A. J. Akey, W. H. Nguyen, M. D. McGehee and T. Buonassisi, Appl. Phys. Lett., 2015, 106(12), 121105 CrossRef.
- T. Todorov, T. Gershon, O. Gunawan, Y. S. Lee, C. Sturdevant, L. Y. Chang and S. Guha, Adv. Energy Mater., 2015, 5, 1–6 Search PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- G. E. Eperon, T. Leijtens, K. A. Bush, R. Prasanna, T. Green, J. T. W. Wang, D. P. McMeekin, G. Volonakis, R. L. Milot, R. May, A. Palmstrom, D. J. Slotcavage, R. A. Belisle, J. B. Patel, E. S. Parrott, R. J. Sutton, W. Ma, F. Moghadam, B. Conings, A. Babayigit, H. G. Boyen, S. Bent, F. Giustino, L. M. Herz, M. B. Johnston, M. D. McGehee and H. J. Snaith, Science, 2016, 354, 861–865 CrossRef CAS PubMed.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13(7), 460–466 CrossRef CAS.
- K. Jäger, L. Korte, B. Rech and S. Albrecht, Opt. Express, 2017, 25, A473 CrossRef PubMed.
- K. Bush, A. F. Palmstrom, Z. J. Yu, M. Boccard, R. Cheacharoen, J. P. Mailoa, D. P. McMeekin, R. L. Z. Hoye, C. D. Bailie, T. Leijtens, I. M. Peters, M. C. Minichetti, N. Rolston, R. Prasanna, S. Sofia, D. Harwood, W. Ma, F. Moghadam, H. J. Snaith, T. Buonassisi, Z. C. Holman, S. F. Bent and M. D. McGehee, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- E. Köhnen, M. Jošt, A. B. Morales-Vilches, P. Tockhorn, A. Al-Ashouri, B. Macco, L. Kegelmann, L. Korte, B. Rech, R. Schlatmann, B. Stannowski and S. Albrecht, Sustainable Energy Fuels, 2019, 3, 1995–2005 RSC.
- J. Y. Jeng, Y. F. Chiang, M. H. Lee, S. R. Peng, T. F. Guo, P. Chen and T. C. Wen, Adv. Mater., 2013, 25, 3727–3732 CrossRef CAS PubMed.
- M. Saliba, M. Stolterfoht, C. M. Wolff, D. Neher and A. Abate, Joule, 2018, 2, 1019–1024 CrossRef CAS.
- A. Magomedov, E. Kasparavičius, K. Rakstys, S. Paek, N. Gasilova, K. Genevičius, G. Juška, T. Malinauskas, M. K. Nazeeruddin and V. Getautis, J. Mater. Chem. C, 2018, 6, 8874–8878 RSC.
- F. Lamberti, T. Gatti, E. Cescon, R. Sorrentino, A. Rizzo, E. Menna, G. Meneghesso, M. Meneghetti, A. Petrozza and L. Franco, Chem, 2019, 1–12 Search PubMed.
- E. Kasparavicius, A. Magomedov, T. Malinauskas and V. Getautis, Chem. – Eur. J., 2018, 24, 9910–9918 CrossRef CAS PubMed.
- J. H. Heo, H. J. Han, D. Kim, T. K. Ahn and S. H. Im, Energy Environ. Sci., 2015, 8, 1602–1608 RSC.
- P. Docampo, J. M. Ball, M. Darwich, G. E. Eperon and H. J. Snaith, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- M. Kaltenbrunner, G. Adam, E. D. Głowacki, M. Drack, R. Schwödiauer, L. Leonat, D. H. Apaydin, H. Groiss, M. C. Scharber, M. S. White, N. S. Sariciftci and S. Bauer, Nat. Mater., 2015, 14, 1032–1039 CrossRef CAS PubMed.
- M. Stolterfoht, C. M. Wolff, J. A. Márquez, S. Zhang, C. J. Hages, D. Rothhardt, S. Albrecht, P. L. Burn, P. Meredith, T. Unold and D. Neher, Nat. Energy, 2018, 3, 847–854 CrossRef CAS.
- J. Seo, S. Park, Y. Chan Kim, N. J. Jeon, J. H. Noh, S. C. Yoon and S. Il Seok, Energy Environ. Sci., 2014, 7, 2642–2646 RSC.
- C. M. Wolff, F. Zu, A. Paulke, L. P. Toro, N. Koch and D. Neher, Adv. Mater., 2017, 29, 1–8 CrossRef PubMed.
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, L. Shi, Z. Liu, J. Huang, J. S. Yun, Y. Zeng, A. Pu, K. Sun, Z. Hameiri, J. A. Stride, J. Seidel, M. A. Green and X. Hao, Adv. Energy Mater., 2018, 1800138 CrossRef.
- J. Lee, H. Kang, G. Kim, H. Back, J. Kim, S. Hong, B. Park, E. Lee and K. Lee, Adv. Mater., 2017, 29, 1–8 Search PubMed.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- D. Luo, W. Yang, Z. Wang, A. Sadhanala, Q. Hu, R. Su, R. Shivanna, G. F. Trindade, J. F. Watts, Z. Xu, T. Liu, K. Chen, F. Ye, P. Wu, L. Zhao, J. Wu, Y. Tu, Y. Zhang, X. Yang, W. Zhang, R. H. Friend, Q. Gong, H. J. Snaith and R. Zhu, Science, 2018, 360(6396), 1442–1446 CrossRef CAS PubMed.
- P. Caprioglio, F. Zu, C. M. Wolff, J. A. Márquez Prieto, M. Stolterfoht, P. Becker, N. Koch, T. Unold, B. Rech, S. Albrecht and D. Neher, Sustainable Energy Fuels, 2019, 3, 550–563 RSC.
- S. Yang, J. Dai, Z. Yu, Y. Shao, Y. Zhou, X. Xiao, X. C. Zeng and J. Huang, J. Am. Chem. Soc., 2019, 141(14), 5781–5787 CrossRef CAS PubMed.
- M. Halik and A. Hirsch, Adv. Mater., 2011, 23, 2689–2695 CrossRef CAS PubMed.
- A. Vilan, D. Aswal and D. Cahen, Chem. Rev., 2017, 117, 4248–4286 CrossRef CAS PubMed.
- W. C. Bigelow, D. L. Pickett and W. A. Zisman, J. Colloid Sci., 1946, 1, 513–538 CrossRef CAS.
- W. B. Russel, Sci. Technol., 2001, 413, 8257–8263 Search PubMed.
- B. Gothe, T. De Roo, J. Will, T. Unruh, S. Mecking and M. Halik, Nanoscale, 2017, 9, 18584–18589 RSC.
- L. Liu, A. Mei, T. Liu, P. Jiang, Y. Sheng, L. Zhang and H. Han, J. Am. Chem. Soc., 2015, 137, 1790–1793 CrossRef CAS PubMed.
- Z. Gu, L. Zuo, T. T. Larsen-Olsen, T. Ye, G. Wu, F. C. Krebs and H. Chen, J. Mater. Chem. A, 2015, 3, 24254–24260 RSC.
- A. Magomedov, A. Al-Ashouri, E. Kasparavičius, S. Strazdaite, G. Niaura, M. Jošt, T. Malinauskas, S. Albrecht and V. Getautis, Adv. Energy Mater., 2018, 8, 1801892 CrossRef.
- E. Yalcin, M. Can, C. Rodriguez-Seco, E. Aktas, R. Pudi, W. Cambarau, S. Demic and E. Palomares, Energy Environ. Sci., 2019, 12, 230–237 RSC.
- Q. Han, Y.-T. Hsieh, L. Meng, J.-L. Wu, P. Sun, E.-P. Yao, S.-Y. Chang, S.-H. Bae, T. Kato, V. Bermudez and Y. Yang, Science, 2018, 361, 904–908 CrossRef CAS PubMed.
- M. Jošt, T. Bertram, D. Koushik, J. A. Marquez, M. A. Verheijen, M. D. Heinemann, E. Köhnen, A. Al-Ashouri, S. Braunger, F. Lang, B. Rech, T. Unold, M. Creatore, I. Lauermann, C. A. Kaufmann, R. Schlatmann and S. Albrecht, ACS Energy Lett., 2019, 4, 583–590 CrossRef.
- H. Hoegl, J. Phys. Chem., 1965, 69, 755–766 CrossRef CAS.
- J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS.
- B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- M. S. Kang, S. Do Sung, I. T. Choi, H. Kim, M. Hong, J. Kim, W. I. Lee and H. K. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 22213–22217 CrossRef CAS PubMed.
- T. J. Gardner, C. D. Frisbie and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 6927–6933 CrossRef CAS.
- E. L. Hanson, J. Guo, N. Koch, J. Schwartz and S. L. Bernasek, J. Am. Chem. Soc., 2005, 127, 10058–10062 CrossRef CAS PubMed.
- S. A. Paniagua, E. L. Li and S. R. Marder, Phys. Chem. Chem. Phys., 2014, 16, 2874–2881 RSC.
- N. Metoki, L. Liu, E. Beilis, N. Eliaz and D. Mandler, Langmuir, 2014, 30, 6791–6799 CrossRef CAS PubMed.
- I. Lange, S. Reiter, M. Pätzel, A. Zykov, A. Nefedov, J. Hildebrandt, S. Hecht, S. Kowarik, C. Wöll, G. Heimel and D. Neher, Adv. Funct. Mater., 2014, 24, 7014–7024 CrossRef CAS.
- R. Quiñones, A. Raman and E. S. Gawalt, Thin Solid Films, 2008, 516, 8774–8781 CrossRef.
- F. Ambrosio, N. Martsinovich and A. Troisi, J. Phys. Chem. Lett., 2012, 3, 1531–1535 CrossRef CAS PubMed.
- S. A. Paniagua, A. J. Giordano, O. L. Smith, S. Barlow, H. Li, N. R. Armstrong, J. E. Pemberton, J. L. Brédas, D. Ginger and S. R. Marder, Chem. Rev., 2016, 116, 7117–7158 CrossRef CAS PubMed.
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H. P. Steinrück, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358, 1192–1197 CrossRef CAS PubMed.
- H. Y. Nie, M. J. Walzak and N. S. McIntyre, J. Phys. Chem. B, 2006, 110, 21101–21108 CrossRef CAS PubMed.
- S. Casalini, C. A. Bortolotti, F. Leonardi and F. Biscarini, Chem. Soc. Rev., 2017, 46, 40–71 RSC.
- P. Xue, P. Wang, P. Chen, B. Yao, P. Gong, J. Sun, Z. Zhang and R. Lu, Chem. Sci., 2017, 8, 6060–6065 RSC.
- R. F. Dou, X. C. Ma, L. Xi, H. L. Yip, K. Y. Wong, W. M. Lau, J. F. Jia, Q. K. Xue, W. S. Yang, H. Ma and A. K. Y. Jen, Langmuir, 2006, 22, 3049–3056 CrossRef CAS PubMed.
- F. M. Hoffmann, Surf. Sci. Rep., 1983, 3, 107–192 CrossRef CAS.
- O. I. Negru and M. Grigoras, J. Polym. Res., 2015, 22, 1–11 CrossRef CAS.
- A. Botta, S. Pragliola, V. Venditto, A. Rubino, S. Aprano, A. De Girolamo Del Mauro, M. Grazia Maglione and C. Minarini, Polym. Compos., 2015, 36, 1110–1117 CrossRef CAS.
- O. Abbas, G. Compère, Y. Larondelle, D. Pompeu, H. Rogez and V. Baeten, Vib. Spectrosc., 2017, 92, 111–118 CrossRef CAS.
- I. Matulaitiene, Z. Kuodis, A. Matijoška, O. Eicher-Lorka and G. Niaura, J. Phys. Chem. C, 2015, 119, 26481–26492 CrossRef CAS.
- S. Schneider, M. Füser, M. Bolte and A. Terfort, Electrochim. Acta, 2017, 246, 853–863 CrossRef CAS.
- S. A. Paniagua, P. J. Hotchkiss, S. C. Jones, S. R. Marder, A. Mudalige, F. S. Marrikar, J. E. Pemberton and N. R. Armstrong, J. Phys. Chem. C, 2008, 112, 7809–7817 CrossRef CAS.
- M. Bomers, A. Mezy, L. Cerutti, F. Barho, F. Gonzalez-Posada Flores, E. Tournié and T. Taliercio, Appl. Surf. Sci., 2018, 451, 241–249 CrossRef CAS.
- R. Quiñones, S. Garretson, G. Behnke, J. W. Fagan, K. T. Mueller, S. Agarwal and R. K. Gupta, Thin Solid Films, 2017, 642, 195–206 CrossRef.
- W. Zhang, W. Ju, X. Wu, Y. Wang, Q. Wang, H. Zhou, S. Wang and C. Hu, Appl. Surf. Sci., 2016, 367, 542–551 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, 1992 Search PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 17102 CrossRef CAS.
- F. Urbach, Phys. Rev., 1953, 92, 1324 CrossRef CAS.
- J. Kim, S. H. Lee, J. H. Lee and K. H. Hong, J. Phys. Chem. Lett., 2014, 5, 1312–1317 CrossRef CAS PubMed.
- A. Sadhanala, F. Deschler, T. H. Thomas, S. E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 2501–2505 CrossRef CAS PubMed.
- C. Huang, W. Fu, C. Z. Li, Z. Zhang, W. Qiu, M. Shi, P. Heremans, A. K. Y. Jen and H. Chen, J. Am. Chem. Soc., 2016, 138, 2528–2531 CrossRef CAS PubMed.
- S. J. Park, S. Jeon, I. K. Lee, J. Zhang, H. Jeong, J. Y. Park, J. Bang, T. K. Ahn, H. W. Shin, B. G. Kim and H. J. Park, J. Mater. Chem. A, 2017, 5, 13220–13227 RSC.
- P. J. Hotchkiss, H. Li, P. B. Paramonov, S. A. Paniagua, S. C. Jones, N. R. Armstrong, J. L. Brédas and S. R. Marder, Adv. Mater., 2009, 21, 4496–4501 CrossRef CAS.
- M. Timpel, H. Li, M. V. Nardi, B. Wegner, J. Frisch, P. J. Hotchkiss, S. R. Marder, S. Barlow, J. L. Brédas and N. Koch, Adv. Funct. Mater., 2018, 28, 1–12 CrossRef.
- H. Li, P. Paramonov and J. L. Bredas, J. Mater. Chem., 2010, 20, 2630–2637 RSC.
- J. Rittich, S. Jung, J. Siekmann and M. Wuttig, Phys. Status Solidi B, 2018, 255, 27–29 Search PubMed.
- S. Ryu, J. H. Noh, N. J. Jeon, Y. Chan Kim, W. S. Yang, J. Seo and S. Il Seok, Energy Environ. Sci., 2014, 7, 2614–2618 RSC.
- R. A. Belisle, P. Jain, R. Prasanna, T. Leijtens and M. D. McGehee, ACS Energy Lett., 2016, 1, 556–560 CrossRef CAS.
- K. Schick, E. Daub, S. Finkbeiner and P. Würfel, Appl. Phys. A: Solids Surf., 1992, 54, 109–114 CrossRef.
- A. Delamarre, L. Lombez and J. F. Guillemoles, Appl. Phys. Lett., 2012, 100(13), 131108 CrossRef.
- T. Unold and L. Gütay, Advanced Characterization Techniques for Thin Film Solar Cells, John Wiley & Sons, Ltd, 2011, pp. 151–175 Search PubMed.
- D. W. Dequilettes, S. Koch, S. Burke, R. K. Paranji, A. J. Shropshire, M. E. Ziffer and D. S. Ginger, ACS Energy Lett., 2016, 1, 438–444 CrossRef CAS.
- B. Krogmeier, F. Staub, D. Grabowski, U. Rau and T. Kirchartz, Sustainable Energy Fuels, 2018, 2, 1027–1034 RSC.
- M. Stolterfoht, P. Caprioglio, C. M. Wolff, J. A. Márquez, J. Nordmann, S. Zhang, D. Rothhardt, U. Hörmann, Y. Amir, A. Redinger, L. Kegelmann, F. Zu, S. Albrecht, N. Koch, T. Kirchartz, M. Saliba, T. Unold and D. Neher, Energy Environ. Sci., 2019, 12(9), 2778–2788 RSC.
- R. T. Ross, J. Chem. Phys., 1967, 46, 4590–4593 CrossRef CAS.
- P. Schulz, L. L. Whittaker-Brooks, B. A. Macleod, D. C. Olson, Y. L. Loo and A. Kahn, Adv. Mater. Interfaces, 2015, 2(7), 1400532 CrossRef.
- J. P. Correa Baena, L. Steier, W. Tress, M. Saliba, S. Neutzner, T. Matsui, F. Giordano, T. J. Jacobsson, A. R. Srimath Kandada, S. M. Zakeeruddin, A. Petrozza, A. Abate, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Energy Environ. Sci., 2015, 8, 2928–2934 RSC.
- P. Caprioglio, M. Stolterfoht, C. M. Wolff, T. Unold, B. Rech, S. Albrecht and D. Neher, Adv. Energy Mater., 2019, 1901631 CrossRef.
- P. Schulz, D. Cahen and A. Kahn, Chem. Rev., 2019, 119, 3349–3417 CrossRef CAS PubMed.
- P. Schulz, E. Edri, S. Kirmayer, G. Hodes, D. Cahen and A. Kahn, Energy Environ. Sci., 2014, 7, 1377–1381 RSC.
- Y. Yan, P. N. Rudd, J. Zhao, Z. Yu, Y. Fang, Y. Lin, Y. Yuan, Y. Deng, J. Huang, B. Chen and C. Bao, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- B. Chen, J. Song, X. Dai, Y. Liu, P. N. Rudd, X. Hong and J. Huang, Adv. Mater., 2019, 1902413 CrossRef PubMed.
- T. Sekimoto, T. Matsui, T. Nishihara, R. Uchida, T. Sekiguchi and T. Negami, ACS Appl. Energy Mater., 2019, 2, 5039–5049 CrossRef CAS.
- J. Avila, L. Gil-Escrig, P. P. Boix, M. Sessolo, S. Albrecht and H. J. Bolink, Sustainable Energy Fuels, 2018, 2, 2429–2434 RSC.
- C. Momblona, L. Gil-Escrig, E. Bandiello, E. M. Hutter, M. Sessolo, K. Lederer, J. Blochwitz-Nimoth and H. J. Bolink, Energy Environ. Sci., 2016, 9, 3456–3463 RSC.
- N. J. Jeon, H. Na, E. H. Jung, T. Y. Yang, Y. G. Lee, G. Kim, H. W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02268f |
| This journal is © The Royal Society of Chemistry 2019 |