
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceI2 vapor-induced degradation of formamidinium lead iodide based perovskite solar cells under heat–light soaking conditions†
Fan
Fu
 *ab,
Stefano
Pisoni
b,
Quentin
Jeangros
a,
Jordi
Sastre-Pellicer
b,
Maciej
Kawecki
cd,
Adriana
Paracchino
e,
Thierry
Moser
b,
Jérémie
Werner
a,
Christian
Andres
b,
Léo
Duchêne
f,
Peter
Fiala
a,
Michael
Rawlence
b,
Sylvain
Nicolay
e,
Christophe
Ballif
ae,
Ayodhya N.
Tiwari
b and
Stephan
Buecheler
b
*ab,
Stefano
Pisoni
b,
Quentin
Jeangros
a,
Jordi
Sastre-Pellicer
b,
Maciej
Kawecki
cd,
Adriana
Paracchino
e,
Thierry
Moser
b,
Jérémie
Werner
a,
Christian
Andres
b,
Léo
Duchêne
f,
Peter
Fiala
a,
Michael
Rawlence
b,
Sylvain
Nicolay
e,
Christophe
Ballif
ae,
Ayodhya N.
Tiwari
b and
Stephan
Buecheler
b
aEcole Polytechnique Fédérale de Lausanne (EPFL), Institute of Microengineering (IMT), Photovoltaics and Thin-Film Electronics Laboratory (PV-Lab), Rue de la Maladière 71b, 2002 Neuchâtel, Switzerland. E-mail: Fan.Fu@epfl.ch
bLaboratory for Thin Films and Photovoltaics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Ueberlandstrasse 129, CH-8600 Duebendorf, Switzerland. E-mail: Fan.Fu@empa.ch
cLaboratory for Nanoscale Materials Science, Empa – Swiss Federal Laboratories for Materials Science and Technology, Ueberlandstrasse 129, CH-8600 Duebendorf, Switzerland
dDepartment of Physics, University of Basel, CH-4056 Basel, Switzerland
eCSEM, PV-Center, Jaquet-Droz 1, 2002 Neuchâtel, Switzerland
fMaterials for Energy Conversion, Empa – Swiss Federal Laboratories for Materials Science and Technology, Ueberlandstrasse 129, CH-8600 Duebendorf, Switzerland
First published on 12th August 2019
Abstract
Over the last years, the operational stability of perovskite solar cells has been significantly improved by compositional engineering, interface modification, and improved encapsulation techniques. However, irreversible degradation is still ubiquitously observed during the first 1000 hours of operation, particularly at elevated temperatures. In this work, we elucidate a major mechanism controlling this degradation. For that, formamidinium lead iodide-based perovskite solar cells were stressed under continuous 1 sun illumination at 80 °C in N2, before extensive characterization of their microstructure and composition. The cell active area and hence the photocurrent are found to decrease with time due to the growth of porous PbI2-rich regions. This degradation was observed to originate from a few seed points in the perovskite bulk rather than from the interfaces with the charge-selective layers or from the cell edges. I2 vapor, first released at these defective points and then further released from the decomposition of the perovskite exposed to this vapor, controls the degradation process. Furthermore, this autocatalytic degradation process is shown to locally rupture the top electrode due to vapor pressure build-up. In addition to highlighting the detrimental influence of residual PbI2, we show that such a degradation pathway can be alleviated by reducing the methylammonium and/or iodine content, providing a path to more stable perovskite solar cells.
Broader contextThe promise offered by perovskite solar cells, i.e., high power conversion efficiency at low manufacturing costs, is starting to materialize. In their fresh, as-fabricated state, lab-scale (∼0.1 cm2) perovskite solar cells have reached 24.2%, which is a greater efficiency than that of alternative polycrystalline thin-film photovoltaic technologies, and their combination with crystalline-Si or Cu(In,Ga)Se2 in tandem devices surpasses the bottom cell performance. However, the key parameter that will eventually dictate the economic viability of perovskite-based photovoltaics (single-junction or tandem) is the long-term stability of perovskite solar cells. While significant improvements have been reported, perovskite solar cells usually degrade irreversibly during the first 1000 hours of operation in field-relevant temperatures. Moreover, the underlying mechanism of how the degradation initiates, propagates, and eventually leads to a catastrophic failure of the device remains largely elusive. In this work, we reveal a previously unknown but critical degradation mechanism of formamidinium–lead-iodide based perovskite solar cells operating at the maximum power point under combined heat and light illumination, close to real-world field conditions. We show that the degradation mechanism seems to be universal to state-of-the-art perovskite absorbers and device architectures, which calls for intensive investigations from the whole community to fully address this challenging issue. |
Introduction
Metal halide perovskite (ABX3, A = Cs+, [CH3NH3]+ (MA+), [CH(NH2)2]+ (FA+); B = Pb, Sn; X = Cl, Br, I) absorber materials have been receiving increased attention due to their high-performance and potentially low-costs when implemented in thin film solar cells.1 Although light-to-power conversion efficiencies have rapidly increased to a certified record of 24.2%2 (∼0.1 cm2, initial efficiency), perovskite solar cells still suffer from relatively poor operational stability compared to commercial photovoltaic technologies.3,4It is well documented that extrinsic parameters such as moisture,5 oxygen,6 light,7 temperature,8 electric fields,9 reverse bias,10 strain,11etc. strongly influence the stability of perovskite solar cells. For example, MAPbI3-based perovskite absorbers undergo severe degradation upon exposure to moisture and/or oxygen12–17 or moderate heat due to the volatile MA+ cation.18 Various studies have suggested that degradation mainly initiates from regions with higher defect densities such as grain boundaries,16,19,20 interfaces with charge selective layers21,22 and cell edges.23 As encapsulation techniques improve,24–26 other factors such as the device architecture,14,27 charge selective layers,28,29 interfaces,17,30 electrodes,31,32 and perovskite quality/composition33–35 eventually start to control the device stability. Among the various intrinsic degradation mechanisms proposed for MAPbI3 films, those induced by I2 vapor released by the decomposition of the absorber itself have been the subject of several studies.18,36–38 Particularly, Wang et al.36 demonstrated that MAPbI3 degrades quickly when intentionally exposed to I2. In devices, Ag metal electrodes were found to oxidize when in contact with such vapor (even when encapsulated), which also led to a loss in performance.39–41
Engineering the perovskite absorber by switching to more robust and efficient FAPbI3-based compositions,42,43 and introducing inorganic cations/anions (such as Cs, Rb, and Br),33,44–47 large organic 2D cations,31,48 or molecular additives49,50 has significantly improved the operational stability of perovskite solar cells. Thanks to these modifications, several groups have shown that perovskite solar cells can retain more than 90% of their initial performance after long-term (500–1000 hours) stability tests.26,29,44,51,52 Although the stress conditions (illumination source, atmosphere, temperature, time, etc.) may differ between studies, and some of the degradation can be recovered by leaving the cells in the dark,53,54 these devices typically exhibit irreversible degradation, as summarized in the ESI,† Table S1. But little information is provided on how the degradation initiates, propagates, and eventually leads to a catastrophic failure of the device. In other words, the mechanisms that control this irreversible degradation in solar cells remain unknown.
In this work, we attempt to elucidate these underlying mechanisms. Here, un-encapsulated (FA,MA)Pb(I,Br)3-based perovskite solar cells were continuously operated at the maximum power point at 80 °C in N2 and then characterized in detail. The results highlight the deleterious role of I2, which is generated during cell degradation and acts as a source of further degradation. At the microstructural level, this I2-driven degradation manifests itself in the form of circular regions that are rich in PbI2 and exhibit a porous morphology. These circular regions nucleate from a small number of seeds that appear during heat–light soaking testing and grow with time during operation at 80 °C. We demonstrate that the degradation pattern is independent of the perovskite composition and preparation method, charge selective layers, and transparent conducting oxide, suggesting the generality of the I2 vapor-assisted degradation mechanism.
Accelerated degradation testing
The perovskite solar cells investigated here are assessed in a substrate configuration55 and feature a planar stack of soda lime glass substrate/indium tin oxide (ITO)/poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA)/perovskite/phenyl-C61-butyric acid methyl ester (PCBM)/ZnO nanoparticles/aluminum-doped zinc oxide (AZO)/Ni–Al grid/MgF2 anti-reflection coating. The perovskite absorbers were prepared by a partial ion-exchange (PIE) approach,47 where a MABr solution was spin coated onto a crystallized FAPbI3 absorber to form a mixed cation lead mixed halide perovskite absorber. The absorber composition prepared by PIE is referred to as FAPbI3 (starting absorber):MABr (exchanged precursor, concentration). Compared to the anti-solvent method,56 the hybrid method allows the growth of a high-quality perovskite absorber on very rough substrates, such as micrometer-sized pyramids of a textured Si wafer41 and wavy flexible polymer foil.57 The second post-deposition treatment (MABr in this case) introduces bandgap grading and/or surface passivation, facilitating charge extraction and reducing interfacial recombination.47 This additional post-deposition treatment has been implemented in solution processed perovskite solar cells, and significantly improved the VOC as demonstrated by several groups.58,59 Moreover, the PIE method is compatible with novel post-deposition treatments, such as those including guanidinium bromide,60n-hexyl trimethyl ammonium bromide,61 and phenethylammonium iodide,62 to further improve the device performance.As shown in Fig. 1a, accelerated heat–light soaking (HLS) stability tests were performed on these devices until significant degradation could be observed. Fig. 1b displays a cross-sectional scanning electron microscopy (SEM) image of the as-prepared perovskite solar cell investigated in this study. The cell was first tested under maximum power point (MPP) conditions for 1000 hours at 60 °C under continuous 1 sun illumination intensity in a 500 mbar N2 atmosphere, as reported in previous work.47 The cell was subjected to full area illumination, i.e. the cell edges, where the perovskite absorber is exposed to the environment, were also illuminated. Using the same device, illumination, and atmospheric conditions, the stress temperature was then increased to 80 °C to accelerate the degradation process. Fig. 1c presents the normalized power output, VOC, JSC, and FF of a perovskite cell during 320 hours of MPP operation at 80 °C under continuous 1 sun illumination. As can be seen from Fig. 1c, the catastrophic degradation regime is mainly dominated by a lower short-circuit current density (JSC) and a slightly decreased fill factor (FF), while the open-circuit voltage (VOC) even increased initially and then remained almost constant. The HLS experiment was stopped at around 320 h to avoid the complete degradation of the device and enable further characterization to understand the underlying degradation mechanisms.
 | ||
| Fig. 1 Operational stability under heat–light soaking (HLS). (a) Schematic illustration of the HLS experiments. A non-encapsulated NIR-transparent perovskite solar cell was kept at the maximum power point (MPP) under continuous 1 sun illumination intensity. The cell was placed on a copper plate heated to 80 °C and subjected to full-area illumination (scribe line included) under a 500 mbar N2 atmosphere. (b) Cross-sectional SEM image of the perovskite solar cell before HLS. The device structure is glass/ITO/PTAA/perovskite/PCBM/ZnO nanoparticles/AZO/Ni–Al grid/MgF2. (c) Normalized power output, VOC, JSC, and FF of the perovskite cell under HLS. (d and e) J–V curves (d) and EQE spectra (e) of the perovskite solar cell after different stress conditions. The J–V curves were measured under standard test conditions (STC: 100 mW cm−2, AM 1.5G, 25 °C). (f–h) Photographs of the perovskite cell taken at different stages, i.e., before any HLS stress (f), after 1000 hours of HLS at 60 °C (g), and after 320 hours of HLS at 80 °C (h). (i) Electroluminescence image of the cell after HLS at 80 °C for 320 hours. | ||
To accurately evaluate the dominant degradation losses, the photovoltaic performance of the FAPbI3:MABr (10 mg ml−1) perovskite device was measured immediately after HLS under standard test conditions (STC: 100 mW cm−2, simulated AM 1.5G, 25 °C). As shown in Fig. 1d, the cell started from a steady-state efficiency of 13.6%, which dropped to 12.7% after 1000 hours at 60 °C in N2. After a further 320 hours of HLS at 80 °C, the cell delivered a stabilized efficiency of 4.1%, with this value increasing to 4.7% after 10 months of subsequent storage in a vacuum (see the MPP measurements in the ESI,† Fig. S1a). Fig. 1e compares the external quantum efficiency (EQE) measured before degradation, and after 1000 hours at 60 °C, 320 hours at 80 °C and vacuum storage for 10 months (the last two curves were acquired at the position of a remaining dark brown area of the cell). The EQE after degradation did not decrease significantly compared to before, which indicates that the measured JSC loss mainly originates from the reduction of the cell active area as shown in the photographs (Fig. 1f–h). Indeed, two distinct areas can be observed in Fig. 1h: an inactive yellow area and an active dark brown area, as confirmed by electroluminescence imaging (Fig. 1i). To verify that the decrease in JSC indeed stems from the loss in the active area, the JSC of the cell after 320 hours of HLS was re-calculated using only the brown region area. As shown in the ESI,† Table S2, the re-calculated JSC is close to the value calculated from the EQE, confirming this hypothesis.
Steady-state photoluminescence (PL) measured in the brown area shows a PL peak position of 781 nm (corresponding to an optical bandgap of 1.59 eV), which indicates that the bandgap of this region did not change throughout HLS at 60 °C and 80 °C (ESI,† Fig. S2). From Fig. 1f–h, the inactive regions appear to originate from yellow spots that appeared during HLS at 60 °C (see e.g. the red arrow in Fig. 1g) and then these domains grew outwards during HLS at 80 °C. Intriguingly, the majority of the cell edges did not act as degradation centers even though the perovskite is directly exposed to the N2 atmosphere at the scribe lines. In that regard, Alberti et al. demonstrated that exposure to an N2 atmosphere can recover lattice disorder (in MAPbI3) and stabilize undercoordinated Pb2+ and MA+ cations through weak electrostatic interactions.63
Chemistry, crystallography, and microstructure of the degraded cells
To gain further insights into the initiation and propagation of the degradation, X-ray diffraction (XRD) measurements were performed to examine the crystallography of the cell stressed at 80 °C (referred to as “after HLS” hereafter, Fig. 2a). The initial XRD pattern of the device before any long-term stability test shows that the absorber is mainly composed of α-FAPbI3:MABr (10 mg ml−1) without any detectable trace of δ-FAPbI3. After HLS, the intensity of the α-FAPbI3:MABr (10 mg ml−1) main peak decreases significantly, while the dominant phase becomes PbI2. A small peak of metallic Pb is detected at 31.28°. These results complement the previous EL and PL measurements: yellow areas are composed mainly of PbI2, while the dark brown area is still α-FAPbI3:MABr (10 mg ml−1). Interestingly, a small δ-FAPbI3 peak emerges after HLS. Unlike the MAPbI3 absorber, where the perovskite directly decomposes into PbI2, the degradation pathway of α-FAPbI3 appears more complex due to this phase transition.64 As shown in the ESI,† Fig. S3, bare layers of α-FAPbI3 on glass tend to transform into δ-FAPbI3 instead of directly decomposing into PbI2 when stored in ambient air, as also observed in the literature.36,42,65 The phase transition between α- and δ-FAPbI3 is reversible by re-annealing the absorber in the air (ESI,† Fig. S4). | ||
| Fig. 2 Phase composition analysis. (a) X-ray diffraction (XRD) patterns of the NIR-transparent perovskite cell FAPbI3:MABr (10 mg ml−1) before and after 320 hours of HLS at 80 °C. (b and c) In situ XRD of FAPbI3:MABr (10 mg ml−1) NIR-transparent perovskite solar cells measured at 80 °C (b) and 100 °C (c). The sample was flushed with N2 at a flow rate of 50 sccm. | ||
To find out the exact degradation pathway of α-FAPbI3-based solar cells during HLS, i.e. whether α-FAPbI3:MABr (10 mg ml−1)-based absorbers directly decompose into PbI2 or first undergo a phase transition and then decompose into PbI2, full solar cells were analyzed by variable temperature X-ray diffraction. As observed in Fig. 2b, α-FAPbI3:MABr (10 mg ml−1) directly decomposed into PbI2 without generating detectable δ-FAPbI3 during 42 hours of in situ measurements at 80 °C. To facilitate the degradation process, the temperature was further raised to 100 °C in another measurement. As shown in Fig. 2c, the first 10 hours of measurements exhibit a similar degradation pathway to that of the cell measured at 80 °C. However, δ-FAPbI3 was detected after 13 hours at 100 °C and the XRD peak intensity increased gradually with time. These results indicate that α-FAPbI3:MABr (10 mg ml−1) directly decomposed into PbI2 and the δ-FAPbI3 yellow perovskite phase formed at a later stage. The formation of δ-FAPbI3 is likely to stem from a reaction of PbI2 with gaseous species (FAI, FA, I2, etc.) generated during the decomposition of the perovskite and/or mobile ions (FA+, I−, etc.) that may be already present in the perovskite absorber. Concerning the latter case, Yuan et al.38 demonstrated reversible conversion between MAPbI3 and PbI2 by migrating MA+ and I− ions under an electrical bias at elevated temperature. To find out the main δ-FAPbI3 formation route, the XRD data of full devices was compared to that of a bare perovskite absorber (FAPbI3:MABr (10 mg ml−1)). As shown in the ESI,† Fig. S5, unlike in the full devices, no δ-FAPbI3 is detected during HLS treatment of the bare perovskite films: only an increase in PbI2 intensity is observed, even after maintaining the films for 48 h at 100 °C (in an N2 flow). This discrepancy between the bare layers and full devices can be explained as follows. In a bare film configuration, gaseous species released by the perovskite during its degradation escape and do not react with PbI2 to form the δ-FAPbI3 phase. The story changes in full devices, where the contact layers hinder the release of these gaseous species to the atmosphere, hence favoring their reaction with PbI2 to form the δ-FAPbI3 phase. Furthermore, it is worth noting that the decrease of the perovskite main peak intensity was slower in the case of a bare film compared to a full device, suggesting that the presence (and stagnation) of gaseous species (once generated) could accelerate the degradation of the perovskite.
The formation of δ-FAPbI3 after long-term light-soaking (1000 hours at 60 °C) is also observed in pure FAPbI3 solar cells as shown in the ESI,† Fig. S6. Previously, it was frequently reported that the α-FAPbI3 phase tends to transform into δ-FAPbI3 even after storage at room temperature. However, we observe that once α-FAPbI3 is incorporated into a full device covered with a TCO, this absorber remains stable for 1000 hours under 1 sun light intensity at 60 °C (ESI,† Fig. S6) and long-term storage in a vacuum at room temperature. As shown in the ESI,† Fig. S7, δ-FAPbI3 was not observed in a α-FAPbI3 solar cell after 460 days of storage in a vacuum.
Fig. 3 shows cross-sectional SEM images taken across the brown/yellow transition area of the full devices after 320 hours HLS at the MPP. Three distinct regions can be observed and are classified into a degraded area (left), a transition area (middle) and an intact area (right). Higher magnification SEM images (Fig. 3b–d) show that the compact morphology with large grains of the absorber is maintained in the intact area (as in Fig. 1b), while the degraded area exhibits a porous morphology indicative of a loss of material from the absorber. The transition area features voids of different sizes primarily within the absorber bulk instead of its interfaces. In that regard, the degradation mechanism of the FAPbI3:MABr (10 mg ml−1) cells investigated here at 80 °C in an N2 atmosphere differs from previous reports: the perovskite bulk rather than the perovskite/charge transport layer interfaces seems to degrade first. The AZO layer appears broken and bent at certain positions in the degraded region away from the transition area (Fig. 3e). Cross-sectional SEM images (ESI,† Fig. S8) of perovskite cells after variable temperature XRD exhibit a similar microstructure. A large number of holes are observed in the absorber and the AZO is locally ruptured.
 | ||
| Fig. 3 Microstructural analysis. (a) Low magnification cross-sectional SEM image spanning from the yellow to the brown areas of the perovskite cell after 320 hours HLS at 80 °C. Three different morphologies can be observed, which are assigned to the degraded area, transition area, and intact area. (b–d) High magnification cross-sectional SEM images of the degraded (b), transition (c), and intact areas (d). (e) Microstructure of a degraded area that features a broken top electrode. | ||
Extracting a thin lamella of material using the conventional focused ion beam (FIB) method and performing a scanning transmission electron microscopy (STEM) energy-dispersive X-ray spectroscopy (EDX) analysis of one of the cracked regions (Fig. 4a–c) reveals that Pb, I, and Br were expelled from the absorber and redeposited on the crack top surface (Fig. 4d). This transport of material is indicative of pressure building up in the absorber, a pressure that eventually leads to a rupture of the top electrode. Regions richer in Pb suggestive of metallic Pb are marked by arrowheads at the crack edge. Other Pb-rich clusters are also observed (Fig. 4e) inside the cell. These are surrounded by PbI2 regions, in accordance with the XRD data shown in Fig. 2a, and large voids, in line with the SEM data presented in Fig. 3d (note that the fraction of voids is higher in the FIB-prepared cross-section due to preferential sputtering of perovskite regions not protected by other layers in the line of sight of the gallium ions). On the other hand, a FIB cross-section extracted from the non-degraded dark brown area exhibited a more compact microstructure and a distribution of elements as anticipated for a functional perovskite (Fig. 4f, the presence of In and Mg within the absorber is an artifact of the quantification procedure). This preserved microstructure is in accordance with the SEM data shown in Fig. 1b and 3d and the EL data of Fig. 1i.
 | ||
| Fig. 4 Chemical composition analysis. (a) Photograph of a perovskite cell after 320 h HLS indicating the region of interest for TEM characterization. (b) The top-view SEM image of a crack at the degradation nucleation seed. (c) STEM high-angle annular dark-field (HAADF) image of the cross-section of the area labeled in (b). (d and e) STEM EDX maps of the regions at the crack tip and within the cell, respectively. (f) STEM HAADF micrograph, STEM EDX map, and corresponding quantified line-scan of the cross-section of one dark brown area (area labeled f in (a)). | ||
To assess which compounds are more likely to escape from the perovskite layer and lead to the top electrode fracture, a thermogravimetric analysis/mass spectrometry (TGA-MS) measurement was performed by heating a powder of the same FAPbI3:MABr (10 mg ml−1) perovskite material up to 400 °C. As presented in the ESI,† Fig. S9, I2 and CH3I are the main gas products generated during the decomposition of the perovskite powder. The generation of other gaseous species such as NH3 and HI molecules66,67 cannot be excluded, but their concentration was outside the detection range of the instrument. Various studies have shown that I2 vapor can be released from both perovskite films and full solar cells under illumination.37,39,41,68,69 For instance, using time-resolved mass spectrometry, Song et al.70 and Juarez-Perez et al.67 independently confirmed that I2 vapor is one of the main volatile compounds when degrading perovskites at a temperature between 60 and 80 °C under illumination (the conditions used here).
To summarize the XRD, electron microscopy and TGA-MS findings so far, the α-FAPbI3:MABr (10 mg ml−1) perovskite layer decomposed mainly to PbI2 but also to δ-FAPbI3 and Pb as minor decomposition products. This then left large PbI2-rich regions exhibiting a porous microstructure, and cracks in the top electrode. These effects were attributed to the volatilization of gaseous products, mainly I2 and other organic compounds, directly from the perovskite bulk. Importantly, I2 has been shown to degrade the perovskite layer to PbI2 in a previous report.71
Degradation mechanism study
The degradation process observed here differs from moisture- and/or oxygen-induced degradation mechanisms as reported previously5,6,20,37 and highlighted in the ESI,† Fig. S10, which investigates the degradation of cells in ambient air. The data presented above tend to indicate that the release of I2 vapor from the cell, an effect that has been shown to decompose perovskite layers,36 may be the main cause of degradation during these HLS experiments. As explained below, the release of I2 vapor from the decomposition of the absorber may trigger further decomposition of the neighboring perovskite regions.Now we attempt to probe the factors that trigger the release of I2 in the first place. PbI2 and perovskite (FAPbI3:MABr (10 mg ml−1)) films (deposited on PTAA/ITO/glass substrates) were illuminated and heated on a hotplate inside an N2-filled glovebox (water and oxygen content below 0.1 ppm, ∼0.1 sun illumination intensity, 80 °C, ESI,† Fig. S11a). PbI2 was found to be more prone to degradation than the perovskite absorber under these conditions as shown in the XRD patterns of Fig. 5a and b. The PbI2(011) peak shifted towards higher angles, indicating a lattice contraction. The (111) and (002) peaks of metallic Pb (31.28° and 36.27°, respectively) appeared and increased with time, indicating a loss of iodine. Control XRD experiments performed in the dark indicate that illumination, even under a 0.1 sun intensity, appears to be the main driver for iodine loss, not temperature (ESI,† Fig. S12). Moreover, the PbI2 film started to change color from yellow to dark grey after 1 hour in these conditions (ESI,† Fig. S11b). The microstructure of the PbI2 films also changed, with the density of pinholes increasing with time (ESI,† Fig. S11c). Contrary to the PbI2 samples, metallic Pb was not detected by XRD in the perovskite films even after 10 hours in these conditions, and the small initial PbI2 peak disappeared (Fig. 5b). Overall, the color and the microstructure of the perovskite film did not change substantially in these conditions (ESI,† Fig. S11b and f). Based on these data, I2 appears to be released easily from PbI2 under illumination, suggesting that PbI2-rich regions induced by the decomposition of the perovskite or already present in the as-deposited state (Fig. 2a) may act as a source of I2 vapor under illumination. Even though residual PbI2 has been reported to improve the initial cell efficiency,72–75 these findings highlight its detrimental role in the long term stability, as hinted at in a recent study.76
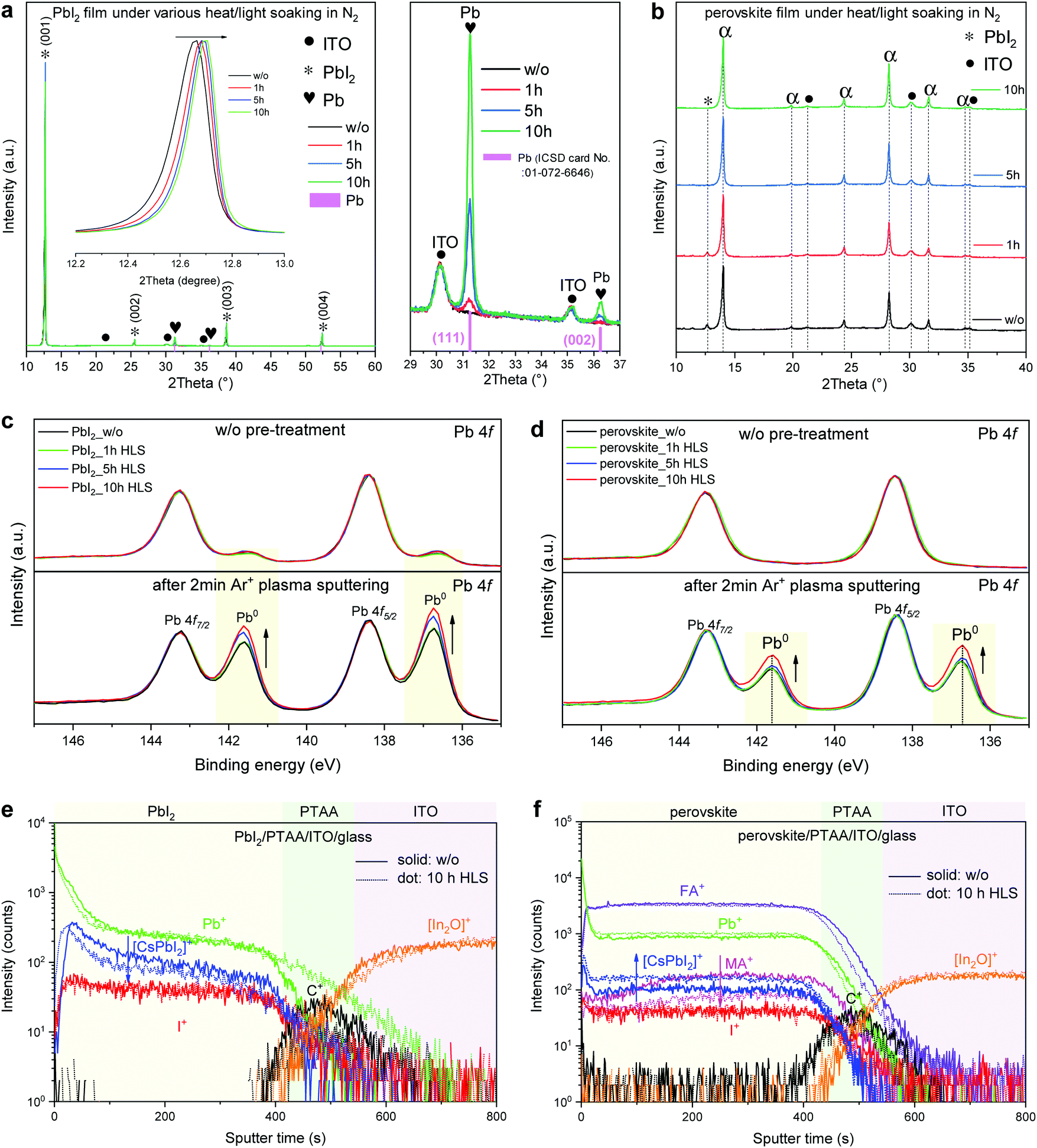 | ||
| Fig. 5 Degradation behavior of PbI2 and the perovskite under HLS. (a and b) XRD patterns of PbI2 (a) and perovskite films (b) subjected to a different period of heat light soaking (HLS) treatment. The HLS treatment was conducted in an N2 filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm) and the investigated films were placed on a hotplate heated to 80 °C under a ∼0.1 sun illumination intensity using a white LED array. (c and d) X-ray photoelectron spectroscopy (XPS) Pb 4f core-level spectra of PbI2 (c) and perovskite (d) films after HLS treatment. The spectra were taken from both the surface (without any pre-treatment) and after 2 min Ar+ plasma sputtering to reveal the subsurface. (e and f) Time-of-flight secondary ions mass spectrometry (ToF-SIMS) depth profiles of fresh and aged (10 hours HLS treatment) PbI2 (e) and perovskite (f) films. The solid and dot lines represent fresh and 10 h HLS treated samples, respectively. | ||
X-ray photoelectron spectroscopy (XPS) was performed to examine the compositional changes upon illumination (Fig. 5c and d). A small Pb0 peak of constant intensity is observed on the top surface in all the PbI2 films (irrespective of the HLS treatment). On the other hand, such a peak is not observed for the as-prepared and stressed perovskite films under the same conditions. While the Ar+ plasma used to reveal the bulk composition of the layers artificially damaged both PbI2 and the perovskite, the magnitude of the Pb0 peak in the bulk increases with the HLS duration in both cases. This difference tends to indicate that the HLS treatment modifies mainly the perovskite interior rather than its top surface, which is consistent with previous findings at the device level (SEM in Fig. 3). To complement this data, time-of-flight secondary ion mass spectrometry (ToF-SIMS) depth profiling was performed using a Cs+ sputter beam and the positive secondary ion signals are shown in Fig. 5e and f. The [CsPbI2]+ signal, which was used to probe the variation in concentration of PbI2, decreased in the PbI2 samples during HLS (Fig. 5e), which is in accordance with partial decomposition of PbI2 to Pb and I2 under illumination. Concerning the perovskite layers, the [CsPbI2]+ signal increased while the MA+ intensity decreased after 10 hours of HLS (Fig. 5f). Meanwhile, the FA+, I+, Pb+, C+, and [In2O]+ signals remained almost constant.
Overall, these XRD, XPS and ToF-SIMS results indicate that the perovskite degradation initiated from the perovskite bulk, in agreement with microstructural observations. Particularly, the loss of MA+ from the perovskite absorber under HLS followed by subsequent perovskite decomposition could be a critical step to trigger the release of I2 vapor. On the basis of previous literature, other routes for I2 vapor generation are also possible. Kim et al.69 observed that light excitation enhances by several orders of magnitude the halide conductivity due to an increased population of halide vacancies. Photogenerated holes react with the iodine sub-lattice, which pushes neutral iodine into the nearby interstitial lattice.69 Recent ab initio molecular dynamics also showed the formation of neutral iodine interstitials via a trap filling reaction, and two filled traps can further form an I2 molecule.77 Both paths (loss of an organic molecule and loss of a neutral iodine atom) could contribute to initial I2 generation under heat/light soaking conditions. In addition, the presence of PbI2, either induced by the migration/loss of ions such as MA+ (Fig. 5f) or excess PbI2 (Fig. 2a) already present in the as-deposited state, may accelerate the cell degradation by dissociating slowly into Pb and the deleterious I2 under illumination.
Fig. 6 schematically illustrates the proposed degradation pathway inferred from the experimental data presented above. An I2 vapor-triggered and -assisted degradation mechanism is proposed to account for the degradation behavior under HLS. First principles calculations77 have shown that the defect chemistry in lead halide perovskites is dominated by lead vacancies (VPb) and interstitial halogen (such as Ii) defects (Fig. 6a). Following a trap filling reaction, the defect Ii+/Ii− pair could react to form an I2 molecule in the bulk under illumination.77 Additionally, the loss of volatile MA+ could result in local decomposition of the perovskite associated with a release of I2 vapor (Fig. 6b). The I2 would then further deteriorate the neighboring perovskite and induce the formation of more I2 vapor and PbI2 (Fig. 6c). This process is autocatalytic and dominates the degradation of the perovskite in these HLS conditions.36 Meanwhile, the formation of PbI2-rich regions, which could then decompose into metallic Pb and I2 vapor under illumination, further accelerates the degradation process. In addition, PbI2 can react in some places with the generated gas to form δ-FAPbI3, as observed by XRD. The release of gaseous degradation products gives rise to a pressure buildup inside the perovskite (Fig. 6d). Once the pressure is high enough, the top electrode eventually breaks (Fig. 6e). It seems that degradation nucleates from only a few points from the experimental observations. But once it starts, it proceeds auto-catalytically as long as I2 remains trapped in the cell, which highlights the influence of the permeability of the top electrode on the degradation mechanism reported here.
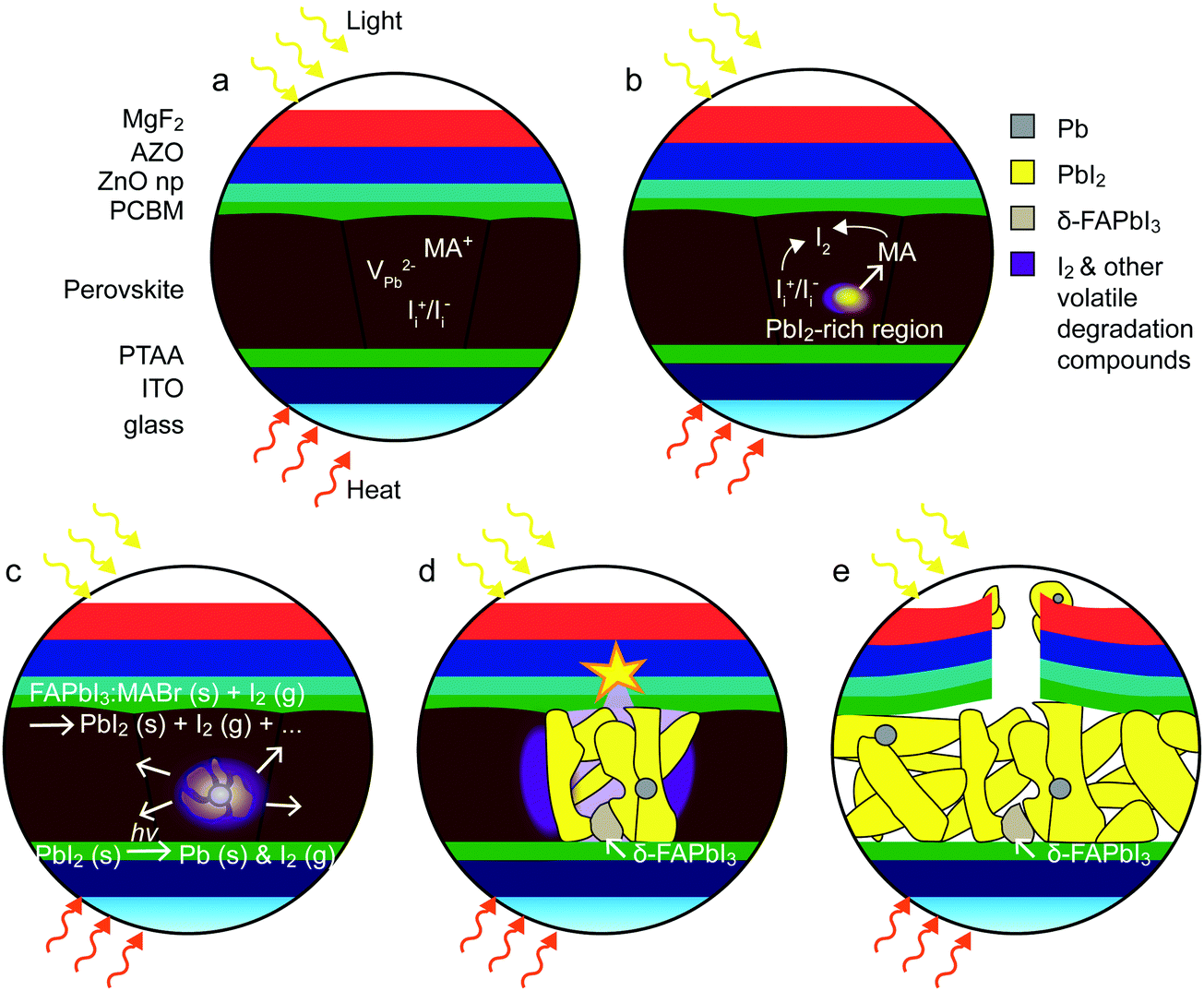 | ||
| Fig. 6 Schematic illustration of the I2 vapor-assisted degradation mechanism. (a) Device structure of a starting perovskite solar cell. (b) PbI2-rich regions and I2 vapor start to appear after long-term HLS soaking in certain locations due to the loss of MA and/or I. (c) I2 vapor triggers the degradation of the neighboring perovskite absorber to release more detrimental I2 vapor; meanwhile, the degradation product PbI2 could easily decompose into metallic Pb and release I2 vapor, which accelerates the degradation process. (d) The perovskite decomposition process continues auto-catalytically, giving rise to a rise in pressure within the cell, with δ-FAPbI3 forming locally due to the reaction of PbI2 with the decomposition gases. (e) The top electrode eventually ruptures when the pressure is high enough to break the TCO. | ||
Compositional dependent stability
From the above discussion, it is desirable to reduce the iodine and residual PbI2 content in order to achieve better operational stability at high temperature. As shown in Fig. 7, the starting point of the catastrophic degradation was retarded to around 250 hours at 80 °C by increasing the Br content in the FAPbI3:MABr (20 mg ml−1) absorber. As before, circular degradation patterns were observed (Fig. 7a–c). The J–V curves and EQE spectra (ESI,† Fig. S13 and S14) of the cell measured after HLS confirm that the degradation was mainly due to a loss of active area. The top-view morphology of the cell after degradation is shown in the ESI,† Fig. S15, where a degradation center is highlighted. In addition to the Br/I ratio, the high-temperature operational stability also depends on the composition of the organic cations.78 For instance, the catastrophic degradation onset began earlier for the MAPbI3:MABr (2.5 mg ml−1) cell stressed at 80 °C, and the degradation features are similar to what was observed for FAPbI3:MABr (10 mg ml−1) (ESI,† Fig. S16 and S17).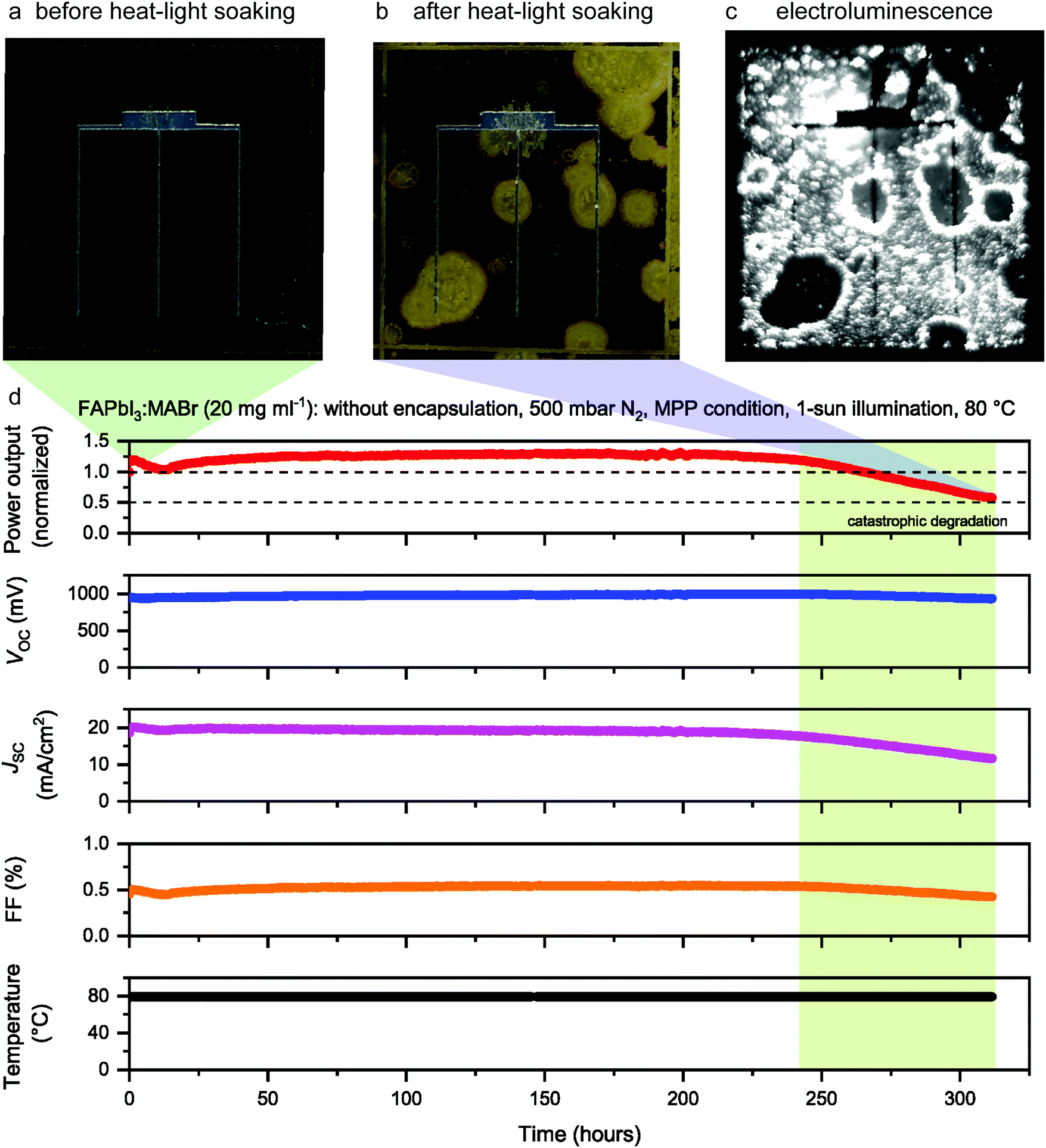 | ||
| Fig. 7 Operational stability of FAPbI3:MABr (20 mg ml−1) solar cells. (a and b) Photographs of the cell before (a) and after (b) 320 hours MPP operation at 80 °C under a 500 mbar N2 atmosphere. (c) The corresponding electroluminescence (EL) image of the same cell. (d) The photovoltaic parameters of the cell during 320 hours MPP operation at 80 °C under a 500 mbar N2 atmosphere. | ||
Since the operational stability depends on both the organic cations and halide anions,46,78,79 simultaneous engineering of organic cations and halide anions is expected to yield more stable perovskite solar cells. As shown in the ESI,† Fig. S18–S20, (Cs,FA)Pb(I,Br)3 based perovskite solar cells with absorber bandgaps ranging from 1.7 to 1.82 eV demonstrate promising operational stability even in ambient air. The superior operational stability with a bandgap ranging from 1.7 to 1.82 eV is highly desirable for perovskite-based tandem applications.41,80–83 Our results show that proper composition engineering significantly improves the operational stability of perovskite solar cells, which is consistent with previous studies using mixed cation/mixed halide compositions.26,33,45,84
So far, the stability tests are performed on perovskite prepared by a hybrid thermal evaporation/spin coating method. To verify that the observed degradation pattern is independent of the specific device architecture and layer stacks (perovskite composition and preparation method, charge selective layers, and TCO) employed in this study, we conduct a 1000 hour HLS experiment on perovskite solar cells using a Cs0.17FA0.83Pb(I0.83Br0.17)3 absorber prepared by an anti-solvent method, and featuring a different transparent top contact made of C60/SnO2/ITO/MgF2. The detailed preparation procedure can be found elsewhere.85 As shown in the ESI,† Fig. S21, the Cs0.17FA0.83Pb(I0.83Br0.17)3 perovskite absorber is more stable than MAPbI3:MABr (2.5 mg ml−1) and FAPbI3:MABr (10 mg ml−1) under the same HLS conditions. However, we still observe the same circular degradation pattern in Cs0.17FA0.83Pb(I0.83Br0.17)3 perovskite solar cells, particularly in the cell stressed under open circuit voltage conditions. The degradation pattern remains the same when replacing a solution processed PTAA hole transporting layer with magnetron sputtered NiO (ESI,† Fig. S22). These results demonstrate that the degradation pattern reported here is independent of the charge selective contact, perovskite absorber composition and preparation method, and TCO, suggesting the generality of the I2-vapor-assisted degradation mechanism in iodide containing semi-transparent perovskite solar cells.
Conclusions
We report an I2 vapor-assisted self-propagating degradation mechanism in formamidinium–lead-iodide based perovskite solar cells stressed under operational conditions at 80 °C under an N2 atmosphere. At high operational temperature, the degradation initiates primarily from the perovskite absorber interior instead of the perovskite/charge-selective layer interfaces or cell edges. The release of I2 vapor appears as the main degradation driver. After an initial release either from traces of PbI2 or from the perovskite itself, I2 deteriorates the neighboring perovskite, releasing more I2 and self-sustaining the degradation, which propagates across the active area. We show that the degradation pattern is independent of the perovskite composition and preparation method, charge selective layers, and top transparent contact, suggesting the universality of this I2 vapor-assisted degradation mechanism in iodide containing organometal halide perovskites. Overall, our results present new insights into the degradation mechanisms of perovskite solar cells operating under long-term light soaking conditions at high temperatures. In view of designing photo- and thermally-stable perovskite single junction and perovskite-based tandem photovoltaics, we demonstrate that incorporating more Br and/or Cs to reduce the MA+ and iodine content improves the operational stability of perovskite solar cells.Methods
Perovskite solar cell fabrication
Perovskite solar cells were grown on commercial indium tin oxide (ITO) coated glass (sheet resistance: 8 Ω sq−1, Zhuhai Kaivo Optoelectronics, P. R. China). The ITO glass was washed by hand first and then subjected to a soap and de-ionized water sonication bath at 85 °C, each for 15 min. The ITO glass was then dried by blowing nitrogen and used for solar cell processing without additional ozone treatment. The hole transporting layer was prepared by spin coating 30 μl of PTAA (Sigma-Aldrich) solution (5 mg ml−1 in toluene doped with 1 wt% F4-TCNQ (97%, Sigma-Aldrich)) at 6000 rpm for 45 s on a 2.5 × 2.5 cm2 substrate, followed by thermal annealing at 100 °C for 10 min. Afterwards, a ∼200 nm PbI2 (ultra dry, 99.999%, Alfa Aesar) compact film was thermally evaporated onto the rotating PTAA/ITO/glass without intentional heating. The deposition rate was controlled within 1–1.5 Å s−1, and the deposition pressure was between 3 and 6 × 10−8 mbar. After PbI2 deposition, the samples were transferred into a glovebox for further processing. The mixed-perovskite layers were formed by a partial ion exchange reaction, which contains three main stages. First, the starting absorber FAPbI3 was prepared as follows: 300 μl of FAI solution (65 mg ml−1 in isopropanol) was first spread on the PbI2 surface, and then rotation to 6000 rpm was immediately started, which lasted for 45 s. The as-deposited films were annealed at 150 °C for 10 min. After cooling, the starting absorber FAPbI3 was spin coated with various concentrations of MABr at 6000 rpm for 45 s to induce the exchange of organic cations and halides. Afterwards, the absorbers were annealed at 100 °C for 1 hour under a chlorobenzene vapor atmosphere. For the electron transporting layer, 30 μl of PCBM (PC61BM, 99.5%, Solenne BV, Netherlands) solution (20 mg ml−1 in chlorobenzene) was spin coated at 5000 rpm for 45 s, followed by 60 min annealing at 100 °C with cells covered by a Petri dish in the presence of 10 μl chlorobenzene. After cooling, 30 μl of undoped ZnO nanoparticles (2.7 wt% (crystalline ZnO dissolved in isopropanol), particle size: 10–15 nm, Sigma-Aldrich) were spin coated on top of the PCBM at 4000 rpm for 45 s. The ZnO nanoparticles were dried at 100 °C for 60 s to evaporate the solvent. Finally, the samples were coated with a ZnO:Al front contact by RF-magnetron sputtering and a Ni/Al (50 nm/4000 nm) metallic grid by e-beam evaporation. Finally, all cells were covered with a 105 nm MgF2 anti-reflection coating deposited by e-beam evaporation. Single cells were defined by mechanical scribing down to the back ITO contact.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m/Δm was reached. For depth profiling, a 500 eV Cs+ sputter beam with a current of 28.77 nA was used to remove the material layer-by-layer in interlaced mode from a raster area of 300 μm × 300 μm. The mass-spectrometry was performed on an area of 100 μm × 100 μm in the center of the sputter crater. A low-energy electron flood gun was used for charge compensation.
eV). The measurements were recorded in fixed analyzer transmission mode with an angle of emission of 45° and at an instrument base pressure of below 8 × 10−7Pa. The work function of the instrument is calibrated regularly to a binding energy of 83.95 eV (FWHM = 0.8 eV) for the Au 4f7/2 peak. The linearity of the energy scale is checked according to ISO 15472. Spectra were recorded with a pass energy of Ep = 29.5eV and an energy step size of ΔE = 0.125eV. After probing the pristine layers, Ar+ sputtering for 120 s at 500 eV was performed to obtain information from the subsurface. Due to pronounced charging of the samples, a manual charge correction has been applied to align the metallic Pb0 components of the Pb 4f7/2 core level at 136.8 eV.
000 m/Δm was reached. For depth profiling, a 500 eV Cs+ sputter beam with a current of 28.77 nA was used to remove the material layer-by-layer in interlaced mode from a raster area of 300 μm × 300 μm. The mass-spectrometry was performed on an area of 100 μm × 100 μm in the center of the sputter crater. A low-energy electron flood gun was used for charge compensation.
eV). The measurements were recorded in fixed analyzer transmission mode with an angle of emission of 45° and at an instrument base pressure of below 8 × 10−7Pa. The work function of the instrument is calibrated regularly to a binding energy of 83.95 eV (FWHM = 0.8 eV) for the Au 4f7/2 peak. The linearity of the energy scale is checked according to ISO 15472. Spectra were recorded with a pass energy of Ep = 29.5eV and an energy step size of ΔE = 0.125eV. After probing the pristine layers, Ar+ sputtering for 120 s at 500 eV was performed to obtain information from the subsurface. Due to pronounced charging of the samples, a manual charge correction has been applied to align the metallic Pb0 components of the Pb 4f7/2 core level at 136.8 eV.
Solar cell characterization
The current density–voltage characteristics of perovskite solar cells were measured using an ABA class solar simulator and Keithley 2400 source meter. The illumination intensity was calibrated to 1000 W m−2 using a certified single crystalline silicon solar cell. The substrate temperature was kept at 25 °C using a Peltier element. The J–V measurements were performed in both forward (from −0.5 V to 1.2 V) and reverse (from 1.2 V to −0.5 V) directions separately without any pre-treatment (e.g. holding at forward bias for a certain time, etc.). The step size was set to 20 mV and the scan velocity varied from 18 to 190 mV s−1. The external quantum efficiency of the cells was measured using a lock-in amplifier. The probing beam was generated by a chopped white source (900 W, halogen lamp, 260 Hz) and a dual grating monochromator. The beam size was adjusted to ensure that the illumination area was fully inside the cell area. The shading effect of the metallic grid was taken into account by including the middle grid line in the illuminated area. A certified single crystalline silicon solar cell was used as the reference cell. White light bias was applied during the measurement with a halogen bias lamp. The steady-state efficiency as a function of time was recorded using a maximum power point tracker, which adjusts the applied voltage in order to reach the maximum power point (perturb and observe algorithm). The starting voltage was set to 0.1 V.Heat light soaking (HLS)
The long-term stability of perovskite solar cells was studied in a home-made stress chamber under controlled illumination intensity, temperature, and atmosphere. Specifically, un-encapsulated perovskite devices were placed on a copper plate at 80 °C and kept at the maximum power point (MPP) under continuous 1 sun illumination intensity using a white LED array. The stress chamber was maintained with a 500 mbar N2 atmosphere during the stability test.Data availability statements
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.Contributions
F. F., C. B., A. N. T., and S. B. designed and coordinated the research and experiments. F. F. fabricated the perovskite solar cells, and performed most of the characterization. S. P. performed the EL and PL measurements. Q. J. performed the TEM measurements and data analysis. J. S. P. and M. R. performed the in situ XRD measurements. M. K. performed the SIMS depth profiling, and C. A. conducted the XPS experiments. J. W. and P. F. helped with the UV-vis spectrometry and SEM top view measurements. L. D. conducted the TGA-MS measurements and analysis. A. P., T. M., and S. N. conducted the long-term stability tests of solution processed perovskite solar cells. F. F. with the help of Q. J. wrote the manuscript with inputs from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial funding from the Swiss National Science Foundation (SNF)-NRP70, PV2050 (project No. 407040_153976 and 407040_153916), the Sinergia Episode (CRSII5_171000), SNF-NanoTera, Bridge Power (176552) and the Swiss Federal Office of Energy (SYNERGY: 20NA21_150950), as well as the Competence Center for Energy and Mobility, is gratefully acknowledged. M. K. acknowledges the funding from the Swiss National Science Foundation (Grant No. CR23I2-162828).References
- M. A. Green and A. Ho-Baillie, Perovskite Solar Cells: The Birth of a New Era in Photovoltaics, ACS Energy Lett., 2017, 2, 822–830 CrossRef CAS.
- M. A. Green, et al., Solar cell efficiency tables (version 54), Prog. Photovoltaics Res. Appl., 2019, 27, 565–575 CrossRef.
- Y. Yang and J. You, Make perovskite solar cells stable, Nature, 2017, 544, 155–156 CrossRef CAS PubMed.
- J.-P. Correa-Baena, et al., Promises and challenges of perovskite solar cells, Science, 2017, 358, 739–744 CrossRef CAS PubMed.
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Impact of H2O on organic-inorganic hybrid perovskite solar cells, Energy Environ. Sci., 2017, 10, 2284–2311 RSC.
- N. Aristidou, et al., Fast oxygen diffusion and iodide defects mediate oxygen-induced degradation of perovskite solar cells, Nat. Commun., 2017, 8, 1–40 CrossRef PubMed.
- Y. Li, et al., Light-Induced Degradation of CH3NH3PbI3 Hybrid Perovskite Thin Film, J. Phys. Chem. C, 2017, 121, 3904–3910 CrossRef CAS.
- G. Divitini, et al., In situ observation of heat-induced degradation of perovskite solar cells, Nat. Energy, 2016, 1, 15012 CrossRef CAS.
- Q. Jeangros, et al., In situ TEM analysis of organic-inorganic metal-halide perovskite solar cells under electrical bias, Nano Lett., 2016, 16, 7013–7018 CrossRef CAS PubMed.
- A. R. Bowring, L. Bertoluzzi, B. C. O. Regan and M. D. Mcgehee, Reverse Bias Behavior of Halide Perovskite Solar Cells, Adv. Energy Mater., 2017, 1702365, 1–7 Search PubMed.
- J. Zhao, et al., Strained hybrid perovskite thin films and their impact on the intrinsic stability of perovskite solar cells, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- J. S. Manser, M. I. Saidaminov, J. A. Christians, O. M. Bakr and P. V. Kamat, Making and Breaking of Lead Halide Perovskites, Acc. Chem. Res., 2016, 49, 330–338 CrossRef CAS PubMed.
- T. Leijtens, et al., Stability of Metal Halide Perovskite Solar Cells, Adv. Energy Mater., 2015, 5, 1–23 Search PubMed.
- T. A. Berhe, et al., Organometal halide perovskite solar cells: degradation and stability, Energy Environ. Sci., 2016, 9, 323–356 RSC.
- G. Niu, X. Guo and L. Wang, Review of recent progress in chemical stability of perovskite solar cells, J. Mater. Chem. A, 2015, 3, 8970–8980 RSC.
- M. Jung, T. J. Shin, J. Seo, G. Kim and S. I. Seok, Structural features and their functions in surfactant-armoured methylammonium lead iodide perovskites for highly efficient and stable solar cells, Energy Environ. Sci., 2018, 0–24, 10.1039/C8EE00995C.
- J. A. Christians, et al., Tailored interfaces of unencapsulated perovskite solar cells for >1.000 hour operational stability, Nat. Energy, 2018, 3, 68–74 CrossRef CAS.
- B. Conings, et al., Intrinsic Thermal Instability of Methylammonium Lead Trihalide Perovskite, Adv. Energy Mater., 2015, 1–8 Search PubMed.
- Y. J. Sung, et al., Humidity-Induced Degradation via Grain Boundaries of HC(NH2)2PbI3 Planar Perovskite Solar Cells, Adv. Funct. Mater., 2018, 28, 1705363 CrossRef.
- Q. Wang, et al., Scaling behavior of moisture-induced grain degradation in polycrystalline hybrid perovskite thin films, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- Z. Fan, et al., Layer-by-Layer Degradation of Methylammonium Lead Tri-iodide Perovskite Microplates, Joule, 2017, 1–15, DOI:10.1016/j.joule.2017.08.005.
- N. Ahn, et al., Trapped charge-driven degradation of perovskite solar cells, Nat. Commun., 2016, 7, 13422 CrossRef CAS PubMed.
- K. A. Bush, et al., Thermal and Environmental Stability of Semi-Transparent Perovskite Solar Cells for Tandems Enabled by a Solution-Processed Nanoparticle Buffer Layer and Sputtered ITO Electrode, Adv. Mater., 2016, 1–7, DOI:10.1002/adma.201505279.
- W. Chen, et al., Efficient and stable large-area perovskite solar cells with inorganic charge extraction layers, Science, 2015, 350, 944–948 CrossRef CAS PubMed.
- F. Bella, et al., Improving efficiency and stability of perovskite solar cells with photocurable fluoropolymers, Science, 2016, 354, 203–206 CrossRef CAS PubMed.
- K. A. Bush, et al., 23.6%-efficient monolithic perovskite/silicon tandem solar cells with improved stability, Nat. Energy, 2017, 2, 17009 CrossRef CAS.
- A. Mei, et al., A hole-conductor-free, fully printable mesoscopic perovskite solar cell with high stability, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- S. S. Shin, et al., Colloidally prepared La-doped BaSnO3 electrodes for efficient, photostable perovskite solar cells, Science, 2017, 356, 167–171 CrossRef CAS PubMed.
- N. Arora, et al., Perovskite solar cells with CuSCN hole extraction layers yield stabilized efficiencies greater than 20%, Science, 2017, 358, 768–771 CrossRef CAS PubMed.
- J. Yang, B. D. Siempelkamp, E. Mosconi, F. De Angelis and T. L. Kelly, Origin of the Thermal Instability in CH3NH3PbI3 Thin Films Deposited on ZnO, Chem. Mater., 2015, 27, 4229–4236 CrossRef CAS.
- G. Grancini, et al., One-Year stable perovskite solar cells by 2D/3D interface engineering, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- M. Kaltenbrunner, et al., Flexible high power-per-weight perovskite solar cells with chromium oxide–metal contacts for improved stability in air, Nat. Mater., 2015, 14, 1032 CrossRef CAS PubMed.
- M. Saliba, et al., Cesium-containing triple cation perovskite solar cells: improved stability, reproducibility and high efficiency, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- E. T. Hoke, et al., Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics, Chem. Sci., 2015, 6, 613–617 RSC.
- A. J. Knight, et al., Electronic Traps and Phase Segregation in Lead Mixed-Halide Perovskite, ACS Energy Lett., 2019, 4, 75–84 CrossRef CAS.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour, Nat. Energy, 2017, 2, 1–8 Search PubMed.
- N. Aristidou, et al., The Role of Oxygen in the Degradation of Methylammonium Lead Trihalide Perovskite Photoactive Layers, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- Y. Yongbo, et al., Electric-Field-Driven Reversible Conversion Between Methylammonium Lead Triiodide Perovskites and Lead Iodide at Elevated Temperatures, Adv. Energy Mater., 2015, 6, 1501803 Search PubMed.
- K. Yuichi, et al., Silver Iodide Formation in Methyl Ammonium Lead Iodide Perovskite Solar Cells with Silver Top Electrodes, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- Y. Han, et al., Degradation observations of encapsulated planar CH3NH3PbI3 perovskite solar cells at high temperatures and humidity, J. Mater. Chem. A, 2015, 3, 8139–8147 RSC.
- F. Sahli, et al., Fully textured monolithic perovskite/silicon tandem solar cells with 25.2% power conversion efficiency, Nat. Mater., 2018, 17, 820–826 CrossRef CAS PubMed.
- N. J. Jeon, et al., Compositional engineering of perovskite materials for high-performance solar cells, Nature, 2015, 517, 476 CrossRef CAS PubMed.
- W. S. Yang, et al., Iodide management in formamidinium–lead–halide-based perovskite layers for efficient solar cells, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- M. Saliba, et al., Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- J.-W. Lee, et al., Formamidinium and Cesium Hybridization for Photo- and Moisture-Stable Perovskite Solar Cell, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells, Nano Lett., 2013, 13, 1764 CrossRef CAS PubMed.
- F. Fu, et al., Compositionally Graded Absorber for Efficient and Stable Near-Infrared-Transparent Perovskite Solar Cells, Adv. Sci., 2018, 1700675, DOI:10.1002/advs.201700675.
- Z. Wang, et al., Efficient ambient-air-stable solar cells with 2D–3D heterostructured butylammonium-caesium-formamidinium lead halide perovskites, Nat. Energy, 2017, 2, 17135 CrossRef CAS.
- X. Li, et al., Improved performance and stability of perovskite solar cells by crystal crosslinking with alkylphosphonic acid ω-ammonium chlorides, Nat. Chem., 2015, 7, 703 CrossRef CAS PubMed.
- Y. Zhao, et al., A polymer scaffold for self-healing perovskite solar cells, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
- H. Tan, et al., Efficient and stable solution-processed planar perovskite solar cells via contact passivation, Science, 2017, 355, 722–726 CrossRef CAS PubMed.
- Y. Hou, et al., A generic interface to reduce the efficiency-stability-cost gap of perovskite solar cells, Science, 2017, 5561, eaao5561 Search PubMed.
- M. Saliba, M. Stolterfoht, C. M. Wolff, D. Neher and A. Abate, Measuring Aging Stability of Perovskite Solar Cells, Joule, 2018, 2, 1019–1024 CrossRef CAS.
- K. Domanski, E. A. Alharbi, A. Hagfeldt, M. Grätzel and W. Tress, Systematic investigation of the impact of operation conditions on the degradation behaviour of perovskite solar cells, Nat. Energy, 2018, 3, 61–67 CrossRef CAS.
- F. Fu, et al., High-efficiency inverted semi-transparent planar perovskite solar cells in substrate configuration, Nat. Energy, 2016, 2, 16190 CrossRef.
- N. J. Jeon, et al., Solvent engineering for high-performance inorganic–organic hybrid perovskite solar cells, Nat. Mater., 2014, 13, 897 CrossRef CAS PubMed.
- S. Pisoni, et al., Impact of interlayer application on band bending for improved electron extraction for efficient flexible perovskite mini-modules, Nano Energy, 2018, 49, 300–307 CrossRef CAS.
- M. Yang, et al., Facile fabrication of large-grain CH3NH3PbI3−xBrx films for high-efficiency solar cells via CH3NH3Br-selective Ostwald ripening, Nat. Commun., 2016, 7, 12305 CrossRef CAS PubMed.
- K. T. Cho, et al., Highly efficient perovskite solar cells with a compositionally engineered perovskite/hole transporting material interface, Energy Environ. Sci., 2017, 10, 621–627 RSC.
- D. Luo, et al., Enhanced photovoltage for inverted planar heterojunction perovskite solar cells, Science, 2018, 360, 1442–1446 CrossRef CAS PubMed.
- E. H. Jung, et al., Efficient, stable and scalable perovskite solar cells using poly(3-hexylthiophene), Nature, 2019, 567, 511–515 CrossRef CAS PubMed.
- Q. Jiang, et al., Surface passivation of perovskite film for efficient solar cells, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- A. Alberti, et al., Nitrogen Soaking Promotes Lattice Recovery in Polycrystalline Hybrid Perovskites, Adv. Energy Mater., 2019, 9, 1803450 CrossRef.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- W.-G. Li, H.-S. Rao, B.-X. Chen, X.-D. Wang and D.-B. Kuang, A formamidinium-methylammonium lead iodide perovskite single crystal exhibiting exceptional optoelectronic properties and long-term stability, J. Mater. Chem. A, 2017, 5, 19431–19438 RSC.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Thermal degradation of CH3NH3PbI3 perovskite into NH3 and CH3I gases observed by coupled thermogravimetry-mass spectrometry analysis, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- E. J. Juarez-Perez, et al., Photodecomposition and thermal decomposition in methylammonium halide lead perovskites and inferred design principles to increase photovoltaic device stability, J. Mater. Chem. A, 2018, 6, 9604–9612 RSC.
- L. Zhao, et al., Ultrasensitive Heterojunctions of Graphene and 2D Perovskites Reveal Spontaneous Iodide Loss, Joule, 2018, 2, 2133–2144 CrossRef CAS.
- G. Y. Kim, et al., Large tunable photoeffect on ion conduction in halide perovskites and implications for photodecomposition, Nat. Mater., 2018, 17, 445–449 CrossRef CAS PubMed.
- Z. Song, et al., Probing the origins of photodegradation in organic–inorganic metal halide perovskites with time-resolved mass spectrometry, Sustainable Energy Fuels, 2018, 2, 2460–2467 RSC.
- S. Wang, Y. Jiang, E. J. Juarez-Perez, L. K. Ono and Y. Qi, Accelerated degradation of methylammonium lead iodide perovskites induced by exposure to iodine vapour, Nat. Energy, 2016, 2, 16195 CrossRef.
- D. Bi, et al., Efficient luminescent solar cells based on tailored mixed-cation perovskites, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- C. Roldán-Carmona, et al., High efficiency methylammonium lead triiodide perovskite solar cells: the relevance of non-stoichiometric precursors, Energy Environ. Sci., 2015, 8, 3550–3556 RSC.
- Y. C. Kim, et al., Beneficial Effects of PbI2 Incorporated in Organo–Lead Halide Perovskite Solar Cells, Adv. Energy Mater., 2016, 6, 1502104 CrossRef.
- B. Park, et al., Understanding how excess lead iodide precursor improves halide perovskite solar cell performance, Nat. Commun., 2018, 9, 3301 CrossRef PubMed.
- T. Matsui, et al., Compositional Engineering for Thermally Stable, Highly Efficient Perovskite Solar Cells Exceeding 20% Power Conversion Efficiency with 85 °C/85% 1000 h Stability, Adv. Mater., 2019, 1806823 CrossRef PubMed.
- S. G. Motti, et al., Controlling competing photochemical reactions stabilizes perovskite solar cells, Nat. Photonics, 2019, 13, 532–539 CrossRef CAS.
- C. Yi, et al., Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells, Energy Environ. Sci., 2016, 9, 656–662 RSC.
- D. W. Ferdani, et al., Partial cation substitution reduces iodide ion transport in lead iodide perovskite solar cells, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- G. E. Eperon, M. T. Hörantner and H. J. Snaith, Metal halide perovskite tandem and multiple-junction photovoltaics, Nat. Rev. Chem., 2017, 1, 0095 CrossRef CAS.
- F. Fu, et al., Low-temperature-processed efficient semi-transparent planar perovskite solar cells for bifacial and tandem applications, Nat. Commun., 2015, 6, 8932 CrossRef CAS PubMed.
- J. Werner, et al., Perovskite/Perovskite/Silicon Monolithic Triple-Junction Solar Cells with a Fully Textured Design, ACS Energy Lett., 2018, 3, 2052–2058 CrossRef CAS.
- D. Zhao, et al., Efficient two-terminal all-perovskite tandem solar cells enabled by high-quality low-bandgap absorber layers, Nat. Energy, 2018, 3, 1093–1100 CrossRef CAS.
- D. P. McMeekin, et al., A mixed-cation lead mixed-halide perovskite absorber for tandem solar cells, Science, 2016, 351, 151–155 CrossRef CAS PubMed.
- B. A. Kamino, et al., Low-Temperature Screen-Printed Metallization for the Scale-Up of Two-Terminal Perovskite–Silicon Tandems, ACS Appl. Energy Mater., 2019, 2, 3815–3821 CrossRef CAS.
- G. Cliff and G. W. Lorimer, The quantitative analysis of thin specimens, J. Microsc., 1975, 103, 203–207 CrossRef.
- M. U. Rothmann, et al., Structural and Chemical Changes to CH3NH3PbI3 Induced by Electron and Gallium Ion Beams, Adv. Mater., 2018, 1800629 CrossRef PubMed.
- Z. Dang, et al., In Situ Transmission Electron Microscopy Study of Electron Beam-Induced Transformations in Colloidal Cesium Lead Halide Perovskite Nanocrystals, ACS Nano, 2017, 11, 2124–2132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02043h |
| This journal is © The Royal Society of Chemistry 2019 |