
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in sodium secondary batteries utilizing ionic liquid electrolytes†
Kazuhiko
Matsumoto‡
 *abc,
Jinkwang
Hwang‡
a,
Shubham
Kaushik‡
a,
Chih-Yao
Chen‡
b and
Rika
Hagiwara
*abc
*abc,
Jinkwang
Hwang‡
a,
Shubham
Kaushik‡
a,
Chih-Yao
Chen‡
b and
Rika
Hagiwara
*abc
aGraduate School of Energy Science, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: k-matsumoto@energy.kyoto-u.ac.jp; hagiwara@energy.kyoto-u.ac.jp
bAIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Sakyo-ku, Kyoto 606-8501, Japan
cUnit of Elements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8510, Japan
First published on 1st October 2019
Abstract
The development of Na secondary batteries that exhibit both sustainability and high energy density as potential successors to lithium-ion batteries for certain large-scale applications has received considerable research interest in recent years. However, although the importance of the electrolyte in such systems has long been largely overlooked, it is becoming increasingly recognized as a key consideration (along with the electrode material) for the non-incremental improvement of Na secondary batteries. Among the candidate electrolytes in this context, ionic liquids (ILs), which are liquids consisting entirely of ions, offer many unique advantages. In this review, the fundamental properties of ILs and the design strategies employed to facilitate their application in batteries are introduced. Comprehensive summaries of the recent advances in the development of positive and negative electrode materials for Na secondary batteries are then presented. Most of the IL-based systems discussed exhibit remarkably enhanced performances compared to those of batteries based on conventional electrolytes. Furthermore, reversible capacity, rate capability, recyclability, and coulombic efficiency are synergistically enhanced by combining IL electrolytes and elevated temperature conditions. Finally, the practical prospects and future challenges associated with the development of electrode materials fabricated from cheap, abundant elements; the efficient utilisation of Na metal as a negative electrode material; and considerations related to the solid–electrolyte interphase are also discussed.
 Kazuhiko Matsumoto | Kazuhiko Matsumoto received his PhD degree in 2004 from Kyoto University. After his postdoc term at Aichi Institute of Technology, McMaster University, and Kyoto University, he was appointed an Assistant Professor in 2009 at Kyoto University and was promoted as an Associate Professor in 2015. He received the Molten Salt Prize for Young Researchers (Molten Salt Committee, ECSJ) in 2009, the Sano Award for Young Researchers (ECSJ) in 2013, and Commendation for Science and Technology by MEXT (Young Scientist Award) in 2017. His research interests are in ionic liquids, inorganic fluorides, and electrolyte and electrode materials for electrochemical devices. |
 Jinkwang Hwang | Jinkwang Hwang is currently a PhD candidate under the supervision of Professor Hagiwara at Kyoto University, Japan. He is the recipient of research fellowships for young scientist from the Japan Society for the Promotion of Science (JSPS) from 2019. He received his BSc degree in Chemistry from Nagoya University, Japan (2015), and received his Master's degree in Energy Science from Kyoto University, Japan (2017). His current research interests are focused on the synthesis and preparation of electrode materials and electrolytes for Li- and Na secondary batteries and investigation on their electrochemical and physical properties. |
 Shubham Kaushik | Shubham Kaushik is currently a PhD student in the Energy Chemistry lab at the Graduate School of Energy Science, Kyoto University. He received his master's degree in energy science from the same lab under the supervision of Professor Rika Hagiwara. His current research work focuses on the preparation and investigation of novel negative electrode materials for sodium secondary batteries using ionic liquid electrolytes for high power and high energy density applications. |
 Chih-Yao Chen | Chih-Yao Chen received his Master's degree in Materials Science and Engineering from National Cheng Kung University, Taiwan, and PhD degree in Energy Science from Kyoto University under the supervision of Prof. Rika Hagiwara. After working at Osaka University with Prof. Tetsuya Tsuda and Prof. Susumu Kuwabata as a postdoctoral researcher, he joined AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL) directed by Prof. Qiang Xu. His research interests focus on ionic liquids and molten salts and their application in electrochemical energy storage devices. |
 Rika Hagiwara | Rika Hagiwara received his BS, MS, and PhD degrees from Kyoto University. From 1987 to 1991, he worked as a postdoc with Prof. Neil Bartlett at the University of California, Berkeley. In 1991, he moved back to Kyoto University as an Assistant Professor and was promoted to an Associate Professor in 1994, and a full Professor in 2005. He received the Molten Salt Committee Award in 2009 and the Academic Award in 2010 from the Electrochemical Society of Japan. His current research interests include fluorine compounds including ionic liquids and their derivatives as functional materials to be applied for energy storage and conversion. |
Broader contextRechargeable batteries are beginning to enter much wider use owing to the fact that their positive social and environmental impacts in terms of renewable energy integration, the proliferation of electric vehicles, energy security, and sustainable development are becoming increasingly understood and appreciated. The requirements for batteries intended for emerging applications include, but are not limited to, a high-level of safety, wide-temperature durability, and high power output, which are quite dissimilar from those intended for use under ambient conditions. Thus, unprecedented opportunities exist for materials science and technology research in this field. The use of Na secondary batteries with appropriate electrolytes, such as ionic liquids (ILs), is one of the most promising strategies to meet the demands for sustainability, safety, and performance, without increasing manufacturing difficulty or necessitating special cell designs. In this review, we provide an introduction to the fundamentals of ILs themselves as well as a variety of case studies demonstrating their feasibility as electrolytes for Na secondary batteries in concert with a variety of electrode materials. We hope that this review will provide a convenient basis for further improvement and optimisation of Na secondary batteries and help make them a commercial reality. |
1. Introduction
A stable supply of energy is a vital global issue given the continuous increase in the demand for fossil fuels and the finiteness of these non-renewable resources. The present energy economy relies heavily on fossil fuels and is thus subject to problems in terms of future exhaustion as well as climate change caused by increasing atmospheric concentrations of greenhouse gases. Accordingly, increasing environmental awareness combined with energy safety concerns have led society to seek methods of generating, harnessing, and storing energy in a more sustainable and efficient manner.Energy storage systems (ESSs) are starting to play a transformative role in this context. Their application scope spans from personal and residential end-use to broader applications such as improving grid reliability and ameliorating the negative effects of insufficient power plants, thus fundamentally changing our utilisation of traditional centralised grids.1,2 ESSs can also compensate for the intermittency and variability of renewable energy sources and support their integration by providing power on demand and to schedule. The electrification of transportation represents an additional opportunity for utilising ESSs, minimising carbon footprints and reducing fossil fuel consumption. Thus, it is clear that the requirements of ESSs are diverse and more specialised than ever before. However, satisfying all these demands requires further advances and new combinations of different ESS technologies.3–6
Lithium-ion batteries (LIBs) based on non-aqueous electrolytes are currently at the forefront of ESS science owing to their unmatched energy densities, energy efficiencies, and the vast range of power-to-energy ratios they can supply.7,8 The ongoing development of LIBs has realised commercially viable electric vehicles and a continuous reduction of cost, which fell below $300 (kW h)−1 for the first time in 2014.8,9 This cost reduction is primarily attributed to the economy of scale associated with increased production and engineering improvements at the cell and pack levels.8,9 Scaling up of contemporary LIBs for grid-scale applications, however, still entails daunting challenges owing to the anticipated astronomic demand for Li and other critical elements such as Co, potentially leading to shortages much like those faced today with fossil fuels.10–12 It may also entail races to exploit new reserves and geopolitical concerns owing to the uneven distribution of Li resources around the world.10,11 However, it is widely acknowledged that non-incremental improvements in battery performance parameters including capacity, lifetime, affordability, safety, and fast-charging capability must be fulfilled at the active-material level because the performances of current LIBs are approaching their theoretical limits.12 From the materials perspective, battery systems utilising earth-abundant elements as charge carriers and/or electrode materials have attracted considerable interest in recent years.13–22Fig. 1 shows the crust and sea abundances of elements that can potentially act as charge carriers for secondary batteries, with each having its own advantages and disadvantages in scientific terms.
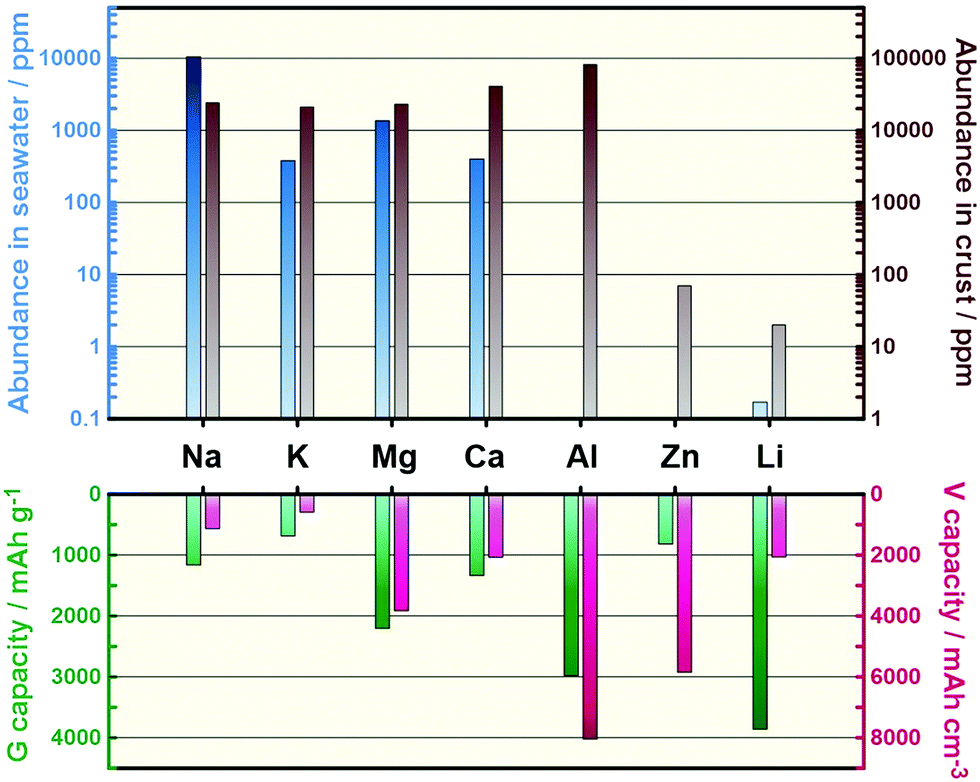 | ||
| Fig. 1 Seawater and crust abundances of potential elements for use as charge carriers in secondary batteries and their theoretical gravimetric and volumetric capacities.31 | ||
The chemical similarity of alkali metals allows the rational extrapolation of the huge body of knowledge pertaining to LIB technology accrued over the past decades, which is mainly applicable to the use and development of Na and potassium secondary batteries. For instance, dendrite-suppressing strategies developed for LIBs have been demonstrated to be applicable to Na23–25 and K26 analogues. With respect to electrode materials, it is known that small alkali metal ions like Li+ tend to occupy octahedral sites, and larger ones prefer trigonal prismatic coordination.27 The difference in preferred coordination and structural competition present an opportunity for exploring interesting new Na- or K-intercalation hosts.14,15 Rechargeable batteries fabricated using multivalent charge carriers (Mg,18,19 Ca,20 Al,21 and Zn22) have the potential to outperform current LIBs owing to their superior theoretical volumetric energy densities. However, despite several exciting advances achieved recently,28–30 research in this field remains at a nascent stage for the most part due to the lack of electrolytes that permit efficient metal deposition/dissolution combined with sufficient operating voltage windows and the lack of positive electrode materials with high capacities.18–21
Na secondary batteries show enormous promise as candidates to meet the sustainability and performance requirements demanded by broad-scale applications for the following reasons:10 (i) Na is the most common alkali metal and the sixth most abundant element on Earth. The abundance of Na is approximately 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm in Earth's crust (20 ppm for Li) and 10500 ppm in seawater (0.17 ppm for Li), making it a practically unlimited resource.31 The price of the Na-containing raw material typically used, Na2CO3, is therefore over 100-times lower than that of Li2CO3, and this gap is constantly expanding (Table S1, ESI†). (ii) Na is the lightest and smallest alkali metal next to Li, and it has a low redox potential (−2.71 V versus a standard hydrogen electrode (SHE)), resulting in reasonably high cell voltages. (iii) The gravimetric capacity penalties (∼15%)14 caused by the higher atomic mass of Na can be compensated for at the cell level by replacing the conventional and heavier Cu foil negative electrode current collector with an Al one, because Na does not alloy with Al.32 This replacement contributes to additional cost advantages for Na secondary batteries (USD 0.3 m−2 and USD 1.2 m−2 for Al and Cu foils, respectively).33 (iv) The larger ionic radius of Na compared to that of Li leads to weaker solvation in polar solvents. Desolvation energetics are known to govern interfacial alkali metal transport, so this weaker interaction facilitates the intercalation of Na+.34
000 ppm in Earth's crust (20 ppm for Li) and 10500 ppm in seawater (0.17 ppm for Li), making it a practically unlimited resource.31 The price of the Na-containing raw material typically used, Na2CO3, is therefore over 100-times lower than that of Li2CO3, and this gap is constantly expanding (Table S1, ESI†). (ii) Na is the lightest and smallest alkali metal next to Li, and it has a low redox potential (−2.71 V versus a standard hydrogen electrode (SHE)), resulting in reasonably high cell voltages. (iii) The gravimetric capacity penalties (∼15%)14 caused by the higher atomic mass of Na can be compensated for at the cell level by replacing the conventional and heavier Cu foil negative electrode current collector with an Al one, because Na does not alloy with Al.32 This replacement contributes to additional cost advantages for Na secondary batteries (USD 0.3 m−2 and USD 1.2 m−2 for Al and Cu foils, respectively).33 (iv) The larger ionic radius of Na compared to that of Li leads to weaker solvation in polar solvents. Desolvation energetics are known to govern interfacial alkali metal transport, so this weaker interaction facilitates the intercalation of Na+.34
Historically, ambient-temperature Na+ and Li+ intercalation chemistry was investigated in parallel in the 1970s,35 but the higher energy density and subsequent commercial success of the latter led to research efforts being predominantly concentrated on LIBs.13 Furthermore, high-temperature Na–S batteries (along with the related Na–NiCl2 batteries) employing Na metal negative electrodes and beta-alumina solid electrolytes (BASEs) have been explored worldwide.36,37 Both cells operate at 270–350 °C to ensure facile Na ion transport between the BASE and electrode materials and eliminate concerns over Na dendrite formation, as Na is molten in this temperature range (melting point, 98 °C). Since the 2000s, megawatt-hour scale Na–S cells have been widely used in Japan and other countries as a means to stabilise energy supply from renewables and thus improve supply reliability.36,37 Accordingly, Na secondary batteries do not fall into the category of “post LIBs” in the historical sense. Through intensive efforts in recent years and as a result of leveraging knowledge of LIBs, phenomenal progress has been realised in the development of electrode and electrolyte materials for Na secondary batteries,38–45 which can now achieve comparable, or even better, performance characteristics than LIBs.46–50
An electrolyte is an indispensable constituent of any battery, transferring and balancing charges in the form of ions between the electrodes.51,52 The electrolyte is positioned between, and in close interaction with, the positive and negative electrodes, and thus it has become increasingly recognised that electrolytes profoundly affect battery behaviour, instead of merely being a medium through which shuttle ions are transported. Of particular importance is the formation of a solid–electrolyte interphase (SEI) on the electrode surface upon decomposition of the electrolyte as the electrode works at a potential over the thermodynamic limit. Because every charge carrier must pass through this interphase to enter the electrode for electron transfer, the nature and thickness of the SEI largely dictates the reversibility and kinetics of the given electrode reactions.52 It has long been widely believed that such a passivation layer is indispensable, as once it is formed on the electrode, the electrolyte is prevented from continuous decomposition and the system is kinetically stabilised.53 The SEI concept was thoroughly elucidated for the successful operation of graphite, the universal negative electrode for LIBs, and it is now applied to Na analogues.
Organic-based electrolytes made from Na salts and organic solvents are most widely used for Na battery studies by virtue of their high ionic conductivity and good wettability, which parallel those featured in well-developed LIB technology. However, several studies have revealed that simply duplicating the strategies used in LIBs leads to SEIs with less homogeneity and inferior stability upon cycling.54–57 In addition, some established additives used in LIBs have been found to be ineffective or even detrimental to Na cell performance.58 While a complete explanation for this is currently unavailable, the underlying factors are52,59 (i) differences in solvation structures and strengths, which affect the SEI evolution process; (ii) the higher redox potential (∼0.3 V) of Na, which alleviates the reductive decomposition of the electrolyte; (iii) poorer polymerisation ability of Na; and (iv) more pronounced dissolution of SEI components due to the milder Lewis acidity of Na. The SEI properties are further complicated by the inherent reactivity of Na metal toward many electrolytes, highlighting the importance of choosing an appropriate electrolyte for the accurate evaluation of new electrode materials in half-cell configurations.52,59 Theoretical calculations of the ionic conductivity within the main SEI components have shown that Na ions must overcome a higher energy barrier to migrate across as compared to the case of Li ions.58,60,61 Based on this understanding, it is clear that Na batteries require their own optimised electrolytes, and the development of electrolytes that provide higher electrochemical stability could be key to solving SEI issues and thus realising high-performance Na batteries.
While LIBs function well for consumer electronics at room temperature, the demand for batteries that withstand and operate under harsh environmental conditions is growing.62,63 The larger spectrum of applications expected for next-generation batteries includes all-climate electric cars, high-altitude drones, and robotics, as well as task-specific areas such as the aerospace industry, thermal reactors, oil and gas exploration, all of which call for batteries that can work safely over a wide temperature range and/or withstand dramatically dissimilar temperatures during charge and discharge. Apart from the flammability and volatility of organic electrolytes,8,63 their high susceptibility to thermal history and temperature fluctuation limits their use in these emerging applications.64,65 Indeed, they have been the cause of many battery faults and even explosions.66 Other cell concepts, such as water-based technologies like lead-acid and nickel–metal hydride batteries, are also limited to operation between −20 and 55 °C.67
For Na secondary batteries to be truly competent as a complementary technology or successor in some applications to LIBs, it is imperative to address the needs of these unprecedented sectors. Accordingly, alternative electrolytes are thought to be a key enabler for expanding the operational temperature window.62,63,68–74 The exploitation of electrolytes with greater chemical stabilities also has a further benefit, i.e., the feasibility of Na metal negative electrodes. These have been intensively researched in the past few years as a means to achieve higher energy densities.25,73,74 Na metal outperforms other negative electrode candidates in terms of redox potential and specific capacity and plays an essential role in combination with many high-capacity but initially unsodiated positive electrode materials to form Na–S, Na–Se, Na–O2, and Na–CO2 batteries. Nevertheless, uncontrolled dendrite growth of Na metal upon cycling and continuous reforming of SEIs, which are interrelated, lead to rapid battery degradation and severe safety hazards.
Among the numerous candidates for next-generation electrolytes, ionic liquids (ILs), which are liquids consisting entirely of ions,75–78 have attracted considerable interest for energy storage devices owing to their numerous unique properties such as negligible vapour pressure, low flammability, high thermal and chemical stabilities, wide liquid-temperature range, broad electrochemical window, and intrinsic ionic conductivity. These properties provide a strong platform for developing batteries with additional functionalities and/or enhanced performances, i.e., high-level safety and wide-temperature durability, for the above-mentioned large-scale battery systems and strategic applications. The absence of molecular species from IL electrolytes leads to elimination of parasitic reactions and their complicated interplay with shuttle ions and other cell components.52 This could contribute to lessening SEI formation and improving coulombic efficiency, which are prerequisites for developing long-lasting batteries. For example, an average coulombic efficiency greater than 99.95% is required to achieve reasonable capacity retention (>60%) over 1000 cycles. Furthermore, it has been demonstrated that Na metal and alloy-based negative electrode materials can provide stable cycling in IL electrolytes but not in conventional electrolytes.79–81 Moreover, the tunable “solvent” properties of ILs allow the use of strategies to suppress the corrosion of Al current collectors and the dissolution of active materials. In conjunction with their electrochemical stability, this could enable the practical operation of high-voltage cell systems.82–84 In addition, the potentially high charge carrier population in ILs could mitigate depletion concerns during operation, which may be of benefit to the energy efficiencies and power capabilities of batteries.85 Other advantages of IL electrolytes for batteries are the reduction of charge-transfer resistance at the electrode,86 their high thermal abuse tolerance,87 and their applicability to various types of electrode materials.79–84,86,87
Although ILs may initially appear to be an uneconomical choice of electrolyte, the amount of electrolyte required in a cell is limited (1–1.3-times the total pore volume of the composite electrodes and separator) and thus its contribution to the entire cost is not always significant.88,89 Furthermore, their cost is expected to decrease as more experience and knowledge pertaining to their synthesis, manufacture, and recycling are gained.10,90,91 It is also important to bear in mind that the enhanced durability and/or additional functionalities of a battery can, in terms of performance-to-cost ratio, compensate for or even eliminate cost concerns arising from the use of IL electrolytes. Fig. 2 shows a Ragone plot for various ESSs. It can be seen that Na secondary batteries with IL electrolytes simultaneously exhibit good specific energy (approaching that of LIBs) and power (exceeding that of a supercapacitor) at optimal temperatures, which is difficult for conventional devices.
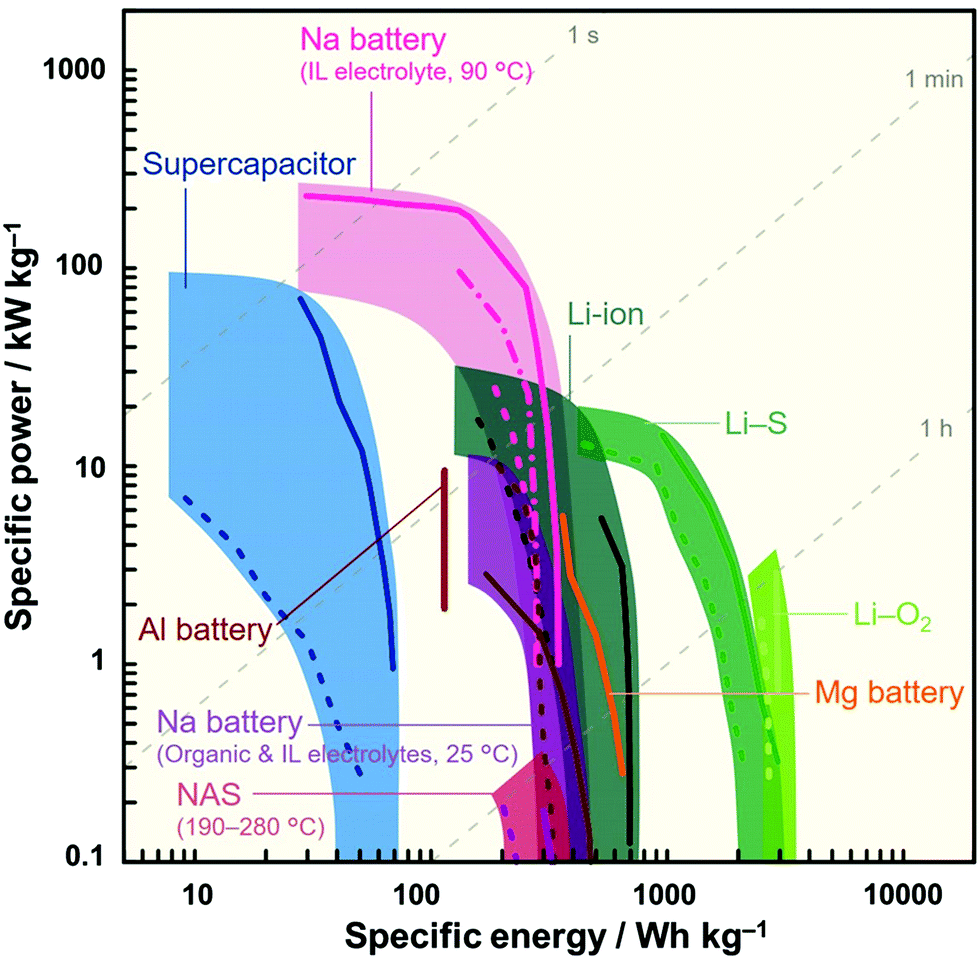 | ||
| Fig. 2 Ragone plot showing sodium secondary batteries with IL electrolytes164,180 in comparison with various energy storage systems (Li-ion,428,429 Li–S,430,431 Li–O2,432,433 Na-ion,49,50 Na–S,434 Al and Mg batteries,29,435 and supercapacitors436,437). The operation temperature of each device is room temperature unless otherwise stated. Specific energy and power densities were calculated based on the weight of the positive electrode material. | ||
Several reviews on the versatile roles of ILs in energy-related fields have been published.92–101 In this review, we focus on the development of IL electrolyte materials for Na secondary batteries. The first section presents an introduction to the fundamental properties and design strategies of IL electrolytes. We then present a comprehensive summary on the negative and positive electrode materials investigated. By exploiting the thermal and chemical stabilities of ILs, the operational window of Na batteries has been effectively extended to the intermediate temperature range (90–150 °C), accompanied by greatly facilitated electrode kinetics without sacrificing stable cycleability. Elevated temperature operation has also been demonstrated to be beneficial for overcoming the electrochemical inactivities of certain electrode materials previously considered inapplicable under ambient conditions.102,103 In addition to the above-mentioned specific applications involving intermediate temperature range, recovering waste heat generated from a battery could be valid to create such an environment. The practical prospects and future challenges associated with the development of electrode materials for Na secondary batteries, the efficient utilisation of Na metal as a negative electrode material, and specific considerations pertaining to the SEI are also discussed.
2. IL electrolytes
ILs can comprise a wide variety of compounds, and thus the possible number of combinations of cation and anion for ILs is virtually countless.104–108 In this section, some performance-relevant properties of ILs as electrolytes for Na secondary batteries are summarised by sectionalising them into physicochemical, thermal, and electrochemical properties. The handling of ILs, especially for electrochemical applications, is also summarised here. Comparisons with organic electrolytes or analogous Li systems will be frequently highlighted for a better understanding of Na systems.Details on other ILs, including protic ILs, deep eutectic solvents, and task-specific ILs, all of which have great research and application importance but fall outside of the scope of this review, can be found elsewhere in the literature.93,104,107–110
2.1 Ionic species for IL electrolytes
Fig. 3 summarises the ionic species used for ILs, especially those of interest for application to Na secondary batteries, along with their abbreviations, which will be used hereafter.79,84,105–107,111–135 Although a number of ILs appear to be of value for secondary batteries, the number of truly useful ILs for this purpose are actually quite limited. In early studies of ILs, fluorocomplex anions (PF6− and BF4−) were widely used to obtain salts with low melting points.136,137 PF6-based ILs are still extensively used for organic synthesis, but their low stability against hydrolysis and high viscosities prevent their use as electrolytes for modern ESSs. Although there are some examples of research into the hydrolytically more stable BF4− in Na secondary batteries, the synthesis of highly pure BF4-based ILs is highly expensive and time-consuming because of their hydrophilicity, and thus they are less attractive for practical use. Sulfonylamide-based (also known as sulfonylimide-based) ILs are widely applied as electrolytes owing to their facile synthesis (they are hydrophobic in most cases) and relatively high ionic conductivities (e.g., 8.8 mS cm−1 for [C2C1im][TFSA]138 and 15.4 mS cm−1 for [C2C1im][FSA]139,140).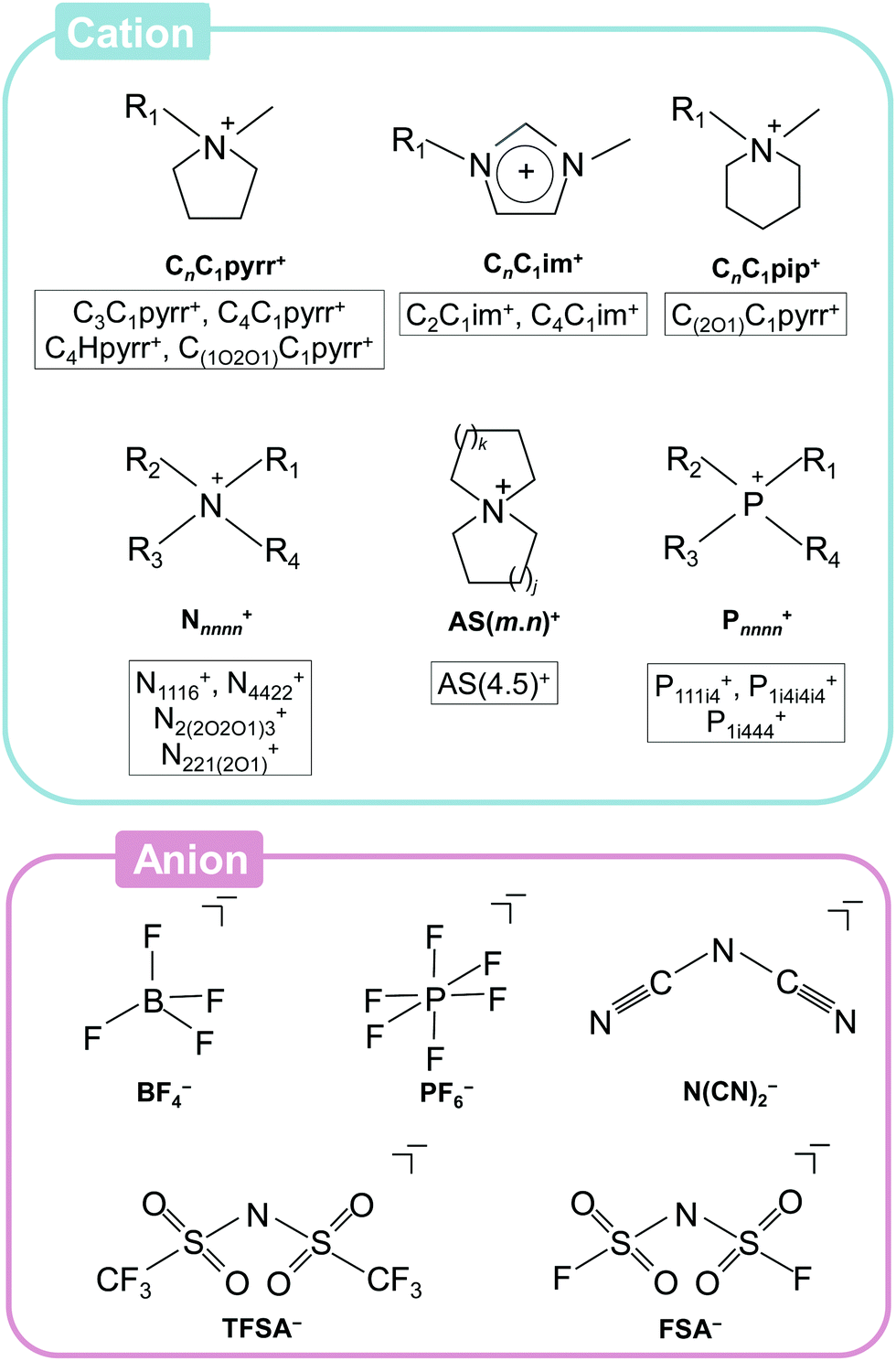 | ||
| Fig. 3 Ionic species in ILs of interest for sodium secondary battery applications. Abbreviations for specific ions are also shown. Cations: CnC1im+ = 1-alkyl-3-methylimidazolium, CnC1pyrr+ = N-alkyl-N-methylpyrrolidinium, CnC1pip+ = N-alkyl-N-methylpiperidinium, Nnnnn+ = tetraalkylammonium, AS[m·n]+ = azoniaspiro[m,n]nonane, and Pnnnn+ = tetraalkylphosphonium. The symbols “n” and “m” in the abbreviations indicate the number of carbon atoms in the alkyl chain (“H” and “i4” denote hydrogen and isobutyl groups, respectively). The alkoxy group is described in the form of “nOn” (e.g. “(1O1)” means methoxymethyl group). Anions: BF4− = tetrafluoroborate, PF6− = hexafluorophosphate, N(CN)2− = dicyanamide, FSA− = bis(fluorosulfonyl)amide, and TFSA− = bis(trifluoromethylsulfonyl)amide. | ||
The cationic species in ILs can be varied more than the anionic species by extending their organic frames. Although melting points typically decrease upon the introduction of bulky or long substituent groups, viscosity and ionic conductivity usually increases and decreases, respectively. Thus, asymmetric cations with short alkyl chains, such as C3C1pyrr+ and C2C1im+, are generally preferred. Non-aromatic pyrrolidinium-based cations are reductively more stable than imidazolium-based cations, and thus widely utilised for secondary battery applications.141 In a recent study, the physicochemical properties of a range of cationic structures (ammonium, pyrrolidinium, piperidinium, and morpholinium) in the forms of FSA- and TFSA-based ILs were systematically studied, revealing the preferable features of ether-functionalised cations.131
Two additional categories are briefly considered here; inorganic ILs and solvate ILs. Inorganic ILs are also electrochemically attractive media at elevated temperatures. In a series of Na[TFSA]–M[TFSA] (M = Li+, K+, Rb+, and Cs+) binary systems, Na[TFSA]–Cs[TFSA] exhibited the lowest eutectic temperature of 110 °C at a Na[TFSA]/M[TFSA] molar ratio of 0.07:0.93.142 Introduction of FSA− further reduces the eutectic temperatures to between 52 and 76 °C with the FSA− salts of Li+, K+, Rb+, and Cs+.143 This system realises even lower eutectic temperatures approaching ambient conditions upon introduction of a third component to form a tertiary system. The lowest eutectic temperature of 36 °C was observed for the Na[FSA]/K[FSA]/Cs[FSA] system at a molar ratio of 40:25:35.144
Solvate ILs are another category in this field,95,145,146 although the number of solvate ILs investigated for Na secondary batteries is currently limited. Solvate ILs are formed by Lewis basic species such as oligoethers interacting with metal salts at equimolar concentrations. A representative example is [Li(G3)][TFSA] (G3 = triethylene glycol dimethyl ether, also called triglyme) which has a melting point of 23 °C.147 The glyme molecules in such solvate ILs coordinate to the metal centre and their behaviour is different from those of pure glymes or diluted solutions, with the coordinating glymes having considerably higher oxidation resistances and thermal stabilities than free glymes. The Na+ analogues [Na(G3)][TFSA], [Na(G4)][TFSA], and [Na(G5)][TFSA] (G4 = tetraglyme and G5 = pentaglyme) are not room-temperature ILs,148 but their electrochemical application to Na secondary batteries has been studied in both neat and diluted forms with several electrode materials.149
2.2 Thermal properties
The thermal properties of IL electrolytes are typically investigated using thermogravimetry (TG) and differential scanning calorimetry (DSC). In general, ILs have higher thermal stabilities than typical organic solvents,141,150 but the determination of IL thermal decomposition temperatures by TG requires special attention because the results are highly dependent on the atmosphere, cell material, scan rate, sample amount, impurities, and method. This is related to the fact that the measured thermal decomposition of ILs is often kinetically controlled (see details on short-term and long-term stability in a previous review150). Thus, comparisons of thermal decomposition temperatures determined under different conditions are usually meaningless. In contrast to organic solvents, which boil at relatively low temperatures, ILs can extend the upper operating temperatures of batteries. In general, ILs are less stable in air than in inert atmospheres, as water and O2 gas facilitate decomposition.150Systematic studies on the thermal decomposition temperatures of ILs containing Na salts have revealed that the addition of Na[BF4], Na[ClO4], Na[N(CN)2], and Na[TFSA] salts to [C4C1pyrr][TFSA] IL somewhat decreases the decomposition temperature to 330–360 °C (∼400 °C for the neat salt).119,127 A clearly different case is that for ILs supplemented with Na[PF6], which decompose at a lower temperature than the others (weight loss starts around 100 °C). The Na[BF4]–[C2C1im][BF4] IL system does not ignite even upon contact with a flame and is thermally stable up to 380 °C regardless of Na[BF4] concentration (0.10–0.75 mol dm−3).118 The thermal stabilities of FSA-salts are somewhat controversial as they are unstable against hydrolysis, even for a trace amount of water. Slow decomposition rates sometimes enable observation of melting, even when the melting point is higher than the decomposition temperature (e.g. Td = 70 °C and Tm = 130 °C for neat Li[FSA]).143 A systematic thermogravimetric study has shown that the thermal decomposition temperatures of alkali metal FSA salts increase with increasing size of the alkali metal cation.143 The thermal decomposition temperatures of some FSA-based ILs, such as [N4411][FSA], [AS(4.5)][FSA], and [N6111][FSA], containing Na[FSA] are dominated by that of Na[FSA] (∼130 °C).122 Another study revealed the higher thermal decomposition temperatures but essentially the same trend (493 °C for neat [N2(2O2O1)3][FSA] and 271 °C for Na[FSA]–[N2(2O2O1)3][FSA] with 55 mol% Na[FSA]).
Furthermore, the decomposition temperature in the presence of the electrode material is more important in view of battery applications. Electrode materials in the charged state are highly reductive (negative electrode) or oxidative (positive electrode) and thus their contact with electrolytes at high temperature can cause thermal runaway. For example, 1.0 mol dm−3 Li[PF6]-EC/DMC exothermically decomposes in the presence of Li metal and a charged positive electrode, Li1−xCoO2, whereas 0.32 mol kg−1 Li[TFSA]–[C3C1pip][TFSA] exhibits negligible heat evolution under the same conditions, demonstrating the advantage of IL electrolytes in terms of thermal stability.151 Although few studies on the thermal properties of ILs for Na secondary batteries are available, some studies on organic electrolytes with various electrode materials (hard carbon (HC),152–155 NaxSn,156 Na0.5Ni0.5Mn0.5O2,157 Na0.35CrO2,158 Na0.58FeO2,159) revealed the high reactivity of charged electrodes toward organic electrolytes (and that the presence of Na salts affects decomposition temperature). The thermal stability of a Na3V2(PO4)3/Na3V2(PO4)3 symmetric cell with a Na[BF4]–[C2C1im][BF4] IL electrolyte was compared with that with a 1 M Na[ClO4]-PC electrolyte after charging,87 and the resulting DSC curves demonstrated the high stability of the IL system. The thermal stability of electrolytes is also related to the stability of the SEI, and the formation of a high-stability SEI at elevated temperatures was demonstrated in Li-containing ILs.160 These factors must be taken into account in future studies on the application of IL electrolytes for Na secondary batteries.156
Fig. 4 shows a phase diagram for a Na[FSA]–[C3C1pyrr][FSA] system.116 Mixing of a Na salt with organic salts usually leads to a certain type of eutectic system (or one that appears to be eutectic) such as Na[FSA]–[C2C1im][FSA],124 Na[FSA]–[N4411][FSA],122 Na[FSA]–[N6111][FSA],122 Na[FSA]–[AS(4.5)][FSA],122 Na[TFSA]–[C3C1pyrr][FSA],130 Na[TFSA]–[C2C1im][TFSA],117 Na[TFSA]–[C4C1im][TFSA],117 and Na[TFSA]–[C4C1pyrr][TFSA].132,134 The wide liquid-phase range of Na[FSA]–[C2C1im][FSA] (0 ≤ Na[FSA] ≤ 50 mol%) contrasts with that of Na[TFSA]–[C2C1im][TFSA] at room temperature (0 ≤ Na[TFSA] ≤ ∼20 mol%), indicating the ability of FSA− to provide low melting points. In such diagrams, the drop in the liquidus line is often steep, which is partly explained by the existence of a crystallinity gap, where only the glass transition is observed and melting behaviour disappears (see the 0.2 ≤ x(Na[FSA]) ≤ 0.5 range in Fig. 4). The origin of this crystallinity gap is not yet fully understood; it may be either thermodynamically or kinetically stable. However, long-term ageing at low temperatures (in the liquid range) often does not lead to crystallisation of ILs.161 ILs are structurally frozen into a glassy state at the glass transition temperature, and the change in heat capacity can be observed by DSC analysis. The glass transition temperature is correlated with the fluidity (i.e., the reciprocal of viscosity) of ILs,162,163 and thus fluid ILs tend to have low glass transition temperatures. The glass transition temperatures in IL systems containing Na-salts typically increase as the amount of Na salt is increased, as shown in Fig. 4. This is related to the low fluidity observed at high Na[FSA] molar ratios. An increase in glass transition temperature tends to be observed upon increasing the organic cation size (e.g. −77.3 °C vs. −75.8 °C for Na[TFSA]–[C2C1im][TFSA]117 and Na[TFSA]–[C4C1im][TFSA] (20 mol% Na[TFSA])) or replacing FSA− with TFSA− (−91 °C for Na[FSA]–[C2C1im][FSA]124vs. −77.3 °C for Na[TFSA]–[C2C1im][TFSA]117 20 mol% Na salt).
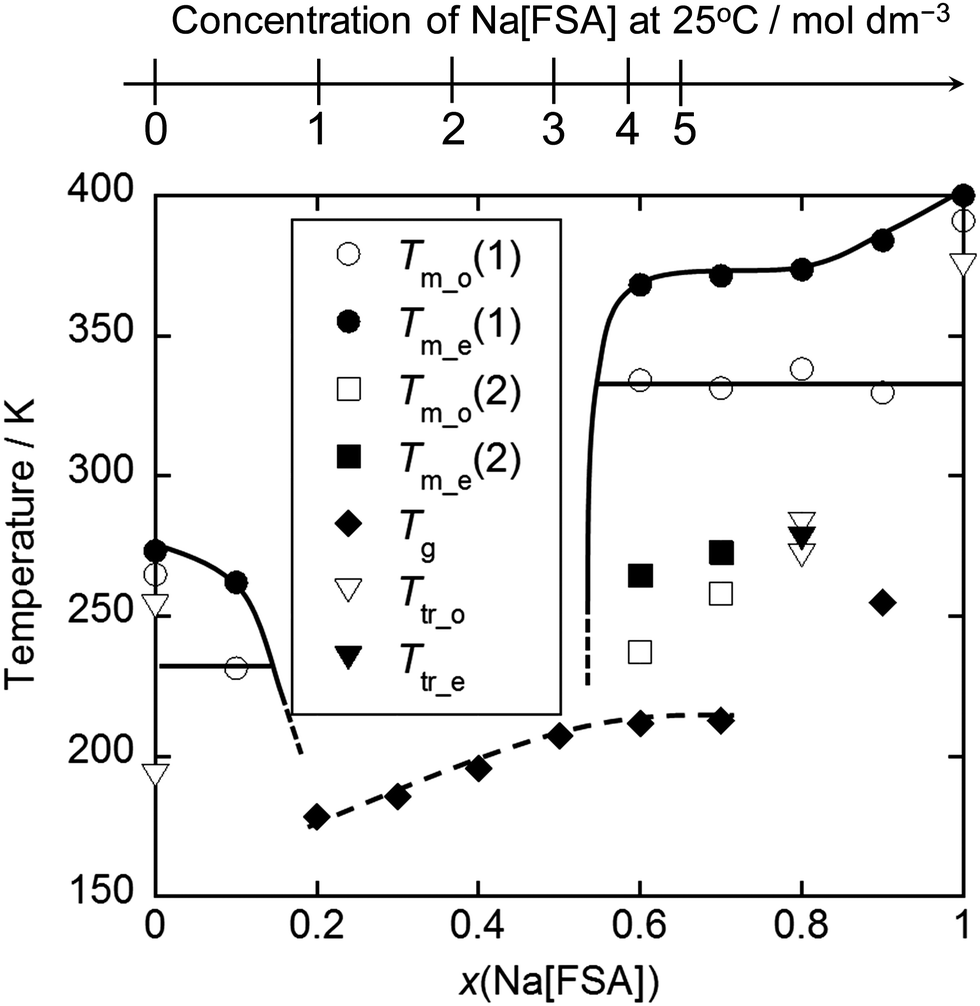 | ||
| Fig. 4 Phase diagram for the Na[FSA]–[C3C1pyrr][FSA] system. Tm_o(1) and Tm_e(1) are the melting onset and final temperatures, respectively; Tm_o(2) and Tm_o(2) are the melting onset and final temperatures for the metastable state, respectively; Tg is the glass transition temperature. Ttr_o and Ttr_e are the onset and final temperatures for the solid-solid phase transition. The scale at the top is the concentration of Na[FSA] at 25 °C. Reproduced with permission.116 Copyright 2015, American Chemical Society. | ||
Fig. 5 summarises the verified temperature range of IL electrolytes for Na secondary batteries (the highest limit of each IL system is not its thermal decomposition temperature) and the operating temperature ranges of various applications using secondary batteries. The available temperature range of commercial LIBs is included for comparison. Although several studies on FSA-based ILs have explored low-temperature operation (−30 °C),164–166 most works on Na secondary batteries were performed at room temperature (25 °C). However, several studies have been performed above room temperature, with many cases concerning electrochemical tests above the melting point of the IL or improving the performances of electrode materials that do not work well at room temperature. Intermediate-temperature operation (up to 150 °C) has also been attempted with ILs, exploiting the use of waste heat and hot environments, in order to enhance Na+ ion transport and electrode reactions.63,164,167 For example, an increase in temperature from 25 to 85 °C leads to an increase in ionic conductivity from 1.9 to 17.3 mS cm−1 for Na[FSA]–[C3C1pyrr][FSA] (30 mol% Na[FSA]). Further details of performance at intermediate temperatures will be presented in the following sections on positive and negative electrodes.
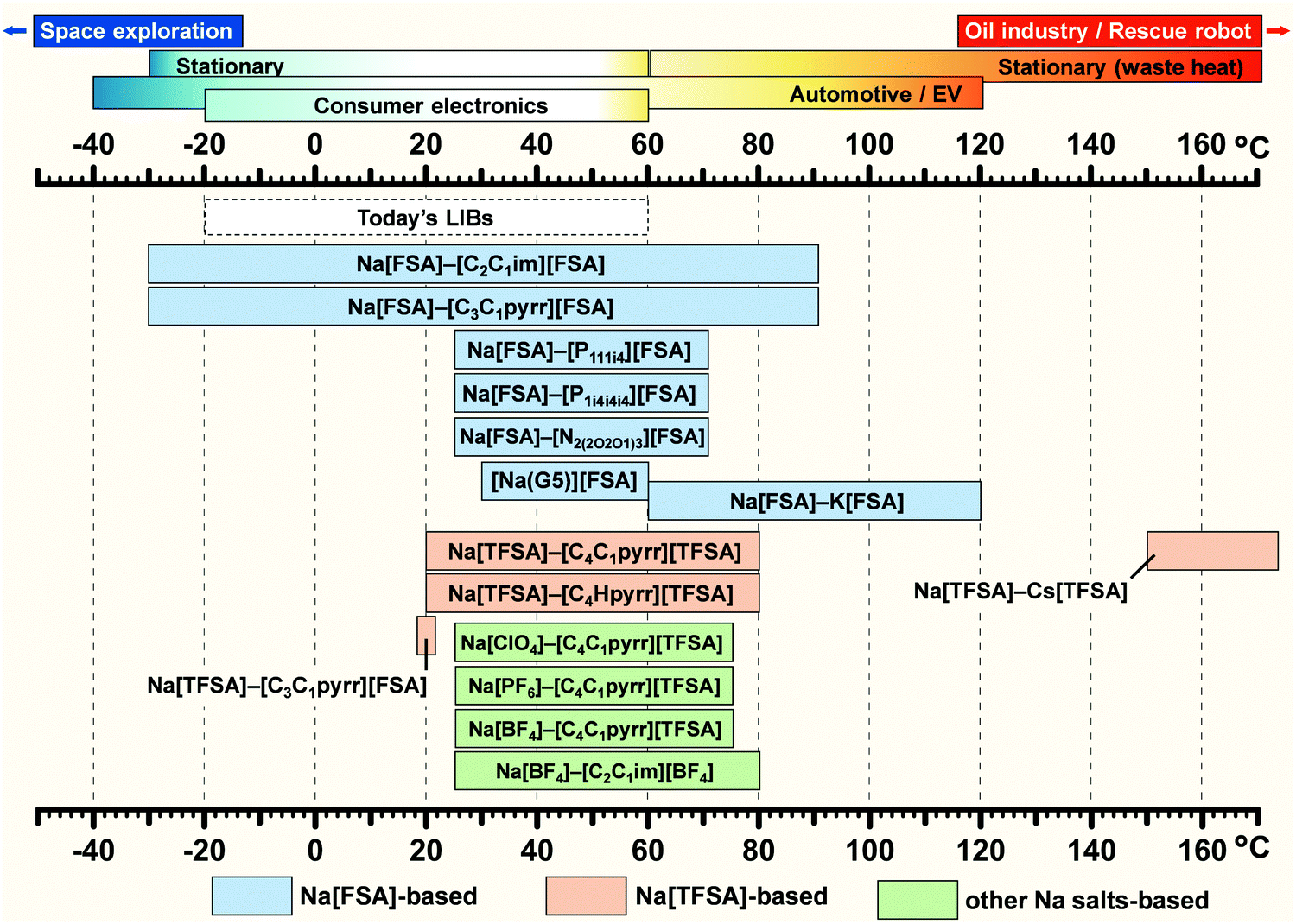 | ||
| Fig. 5 Temperature range of ILs verified for sodium secondary batteries. The highest limit of each IL system does not indicate its thermal decomposition temperature. The operating temperature ranges of various applications and commercial LIBs are included for comparison. See Fig. 3 for abbreviations of each ionic species.62,63,87,127,128,149,164,166,237,268,289,316,321,438 | ||
2.3 Physicochemical properties
Fig. 6 summarises the molar concentration dependency of viscosity, ionic conductivity, and Na+ ion transport number for selected ILs used for Na secondary batteries (see Table S2, ESI†). In general, ILs exhibit higher viscosities than organic electrolytes at similar Na contents. This arises from the strong coulombic interactions between cations and anions, and thus dilution of ILs with a solvent leads to a decrease in viscosity.168–170 Statistical data have shown that an increase in ion volume increases the viscosity of ILs,171 whereas the introduction of ether groups tends to decrease viscosity.131,172 High viscosity is physically not preferable as it makes the handling of IL electrolytes, such as their impregnation into composite electrodes and separators, difficult. In addition, viscosity (η) is related to molar ionic conductivity (λ) via the Walden rule (or fractional Walden rule with the α parameter between zero and unity corresponding to the slope of the Walden plot (eqn (1)) (log(η−1) vs. log(λ))):162,173–175| η·λ = const. (or ηα·λ = const.) | (1) |
152). Na ion transport in the steady state is governed by Na ion conductivity (σNa+), as expressed in eqn (2):| σNa+ = σ·tNa+ | (2) |
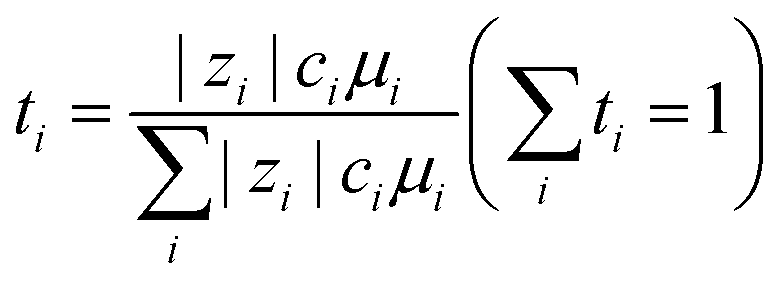 | (3) |
:1 single salt in a solution (e.g., NaPF6 in propylene carbonate). However, IL electrolytes for Na secondary batteries containing organic cations are usually composed of more than three constituent ions, and thus concentration has to be taken into account. The simple ratio of the diffusion coefficients of constituent ions does not always indicate the transport number in ILs as ILs are highly concentrated systems (i.e., not infinitely diluted systems) and the frame of reference has to be carefully considered.175 The term ‘apparent transport number’ is also used in several reports, and is derived by considering the different environments of Na+ in ILs in the initial and final states during polarisation.116,179,180
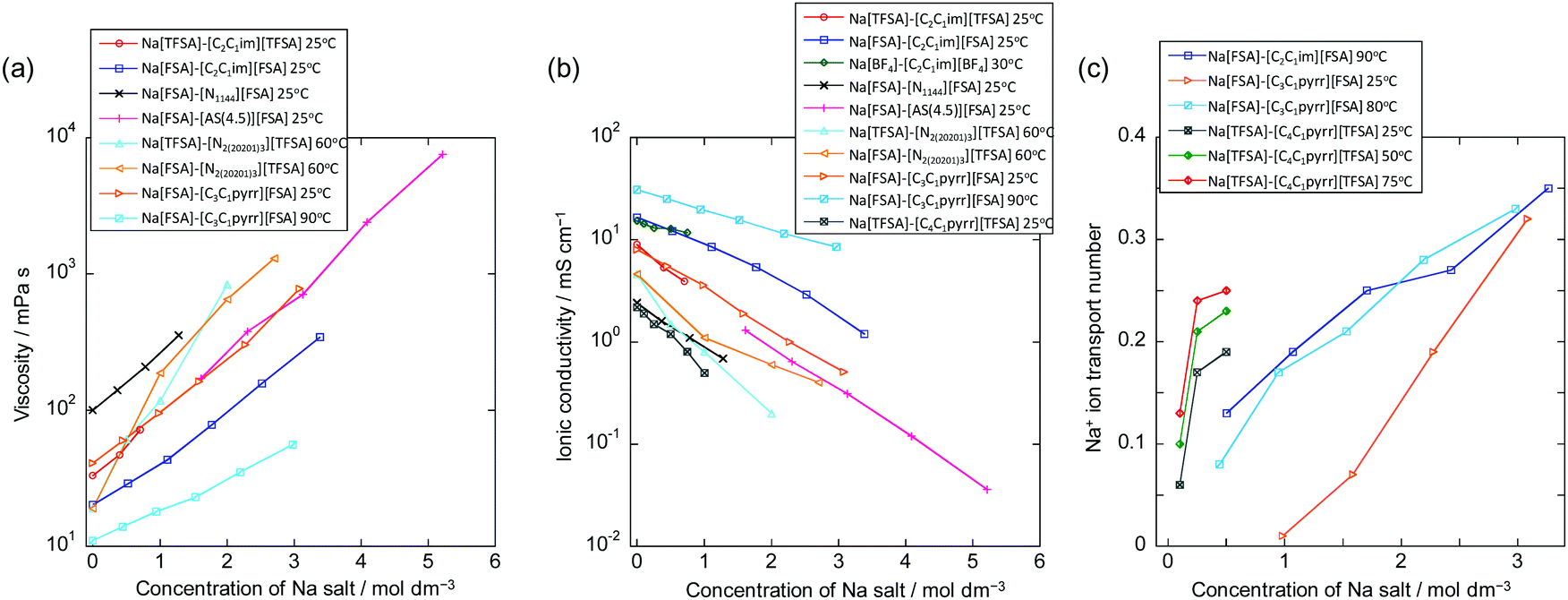 | ||
| Fig. 6 Dependency of (a) viscosity, (b) ionic conductivity, and (c) Na+ ion transport number on Na salt concentration for IL electrolytes used in sodium secondary batteries. See Tables S2 and S3 (ESI†) for the data represented here.79,84,116–124 Other related data are also available in the literature.125–135 | ||
The tNa+ values for several Na+-containing ILs have been reported (see Fig. 6c for Na salt concentration dependency)79,84,116,119,125,126,135,180 and tend to increase with increasing Na salt concentration in the Na[FSA]–[C3C1pyrr][FSA]79,116 and Na[FSA]–[C2C1im][FSA]180 systems (e.g., 0.13 (10 mol% Na[FSA]) to 0.35 (50 mol% Na[FSA]) for Na[FSA]–[C2C1im][FSA]). The Na+ ion transport number also clearly increases with increasing temperature. The resulting Na+ ion conductivity exhibits a maximum at a certain Na[FSA] fraction (e.g. at 20 mol% Na[FSA] for Na[FSA]–[C3C1pyrr][FSA] and 30 mol% Na[FSA] for Na[FSA]–[C2C1im][FSA]).180 Molecular dynamics simulations have indicated that clustering of Na+ and FSA− ions occurs at high Na[FSA] concentrations,79,181 enabling site exchange and/or indicates a structural diffusion mechanism for Na+, leading to high transport numbers. Clustering of Na+ and FSA− ions and rapid exchange of the Na+ ion between different coordination environments have also been suggested in this and analogous IL systems by Raman spectroscopic,180 NMR spectroscopic,79,182 and atomic force microscopic182 studies.
The fluidity (η−1) of an IL usually deviates from Arrhenius behaviour as it approaches the glass transition temperature (Tg), and the rate at which the transport properties change with temperature near the glass transition is termed ‘fragility’.162 In the same way, such temperature dependence of viscosity and ionic conductivity for ILs is best fitted with the VTF equations as follows (eqn (4) and (5)).183–186 Inorganic–organic hybrid IL systems including Na+-containing ILs also follow this behaviour.116,122,124,125,130
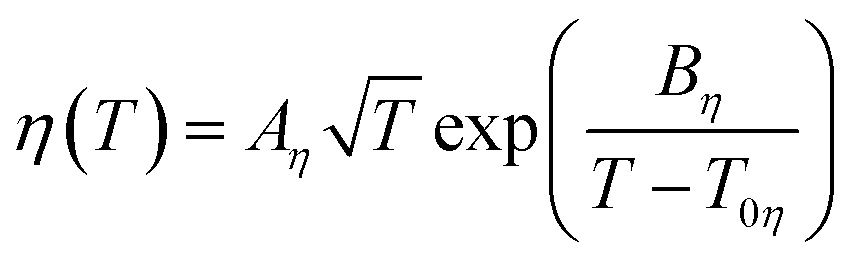 | (4) |
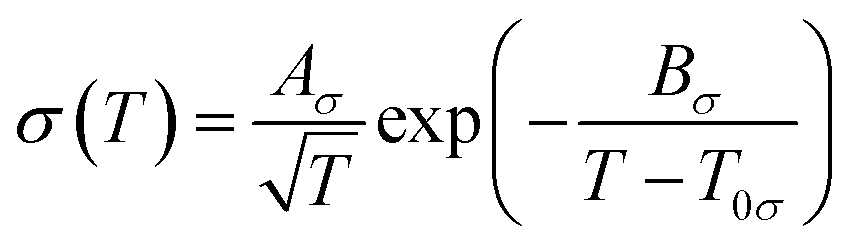 | (5) |
The experimentally observed glass transition temperature is always higher than the ideal glass transition temperature (Tg > T0, where T0 is the ideal glass transition temperature in the VTF equation) by the empirical approximation T0/Tg ≈ 0.75.187
The polarity of a solvent is regarded as an indicator of its ability to dissolve and stabilise ions and is briefly mentioned here because it is an important concept in the design of electrolytes based on ILs for secondary batteries. Polarity is usually discussed based on physical parameters such as relative permittivity, dipole moment, and refractive index. Dielectric spectroscopy has revealed that the relative permittivities of ILs containing BF4−, PF6−, SO3CF3−, and TFSA− fall between 11 and 15.188–191 However, such macroscopic parameters are not always effective for assessing the polarity of ILs because the solubility of inorganic salts is largely different (even though the relative permittivities of the ILs above are similar).
ILs are not homogeneous dielectrics and local interactions apparently play more important roles. Polarity based on local interactions is evaluated by spectroscopic methods using solvatochromic dyes.192–197 The empirical ET(30) and λCu parameters are determined by the π–π* absorption band of Reichardt's betaine dye and the d–d absorption band of Cu2+ in [Cu(acac)(tmen)] (acac = acetylacetone and tmen = N,N,N′,N′-tetramethylethylenediamine), respectively. The ET(30) parameter exhibits good correlation with the acceptor number of solvents and generally decreases in ILs as the alkyl chain length on the cation increases and as the form of the cation changes from aromatic (imidazolium and pyridinium) to non-aromatic (pyrrolidinium and tetraalkylammonium).198–203 Anionic structures have a minor effect on the acceptor number of cations and accordingly ET(30). Conversely, the λCu parameter correlates with the donor number of solvents and is thus affected by the anionic structures in ILs. The resulting donor ability has the trend [CF3CO2]− > [SO3CF3]− > [FSA]− ≈ [TFSA]− > [BF4]− > [PF6]−.204,205 There are other indicators known as Kamlet–Taft parameters (π*, α, and β206–209) that are widely applied in discussing the polarity of ILs.197,202,205 The π* parameter is related to the dipolarity/polarisability of solvents, and those for ILs are generally higher than those for typical aprotic organic solvents and increase from nonaromatic to aromatic cation-based ILs. TFSA-based ILs have smaller π* parameter values compared to those of other ILs. The α and β parameters correlate with hydrogen bonding donor and acceptor abilities, respectively, but are less important for electrolytes for secondary batteries. The correlation of polarity and the molar conductivity ratio, Λimp/ΛNMR, has been discussed in previous works.205,210Λimp and ΛNMR are the molar conductivities obtained by AC impedance and NMR spectroscopies, respectively. This correlation provides basic insight for considering ion structures in IL electrolytes for secondary batteries because the Λimp/ΛNMR ratio is the proportion of ions (charged species) that contribute to ionic conduction from all the diffusing species on the time scale of the measurement.210
The λCu parameter, which is predominantly affected by the anionic structure, exhibits good correlation with the Λimp/ΛNMR ratio; an increase in the donor ability of the anion decreases the number of ions contributing to ion conduction. The ET(30) values show a somewhat more complicated correlation with the Λimp/ΛNMR ratio. The major dependency is due to the difference in the cation backbone structures for TFSA-based ILs, and the Λimp/ΛNMR ratio increases with decreasing ET(30), i.e., decreasing the acceptor number of cations. Na ions in ILs are surrounded by the anions therein, and thus polarity values based on anions, such as those of λCu, are considered to reflect IL behaviour. However, direct evidence of correlation between such parameters and Na+ ion properties have not been adequately investigated from static and dynamic viewpoints. Thus, further work in the future, including computational studies, is required in this field.
2.4 Electrochemical properties
The term ‘electrochemical window’ is used to indicate the potential range from the reductive to oxidative limits of electrolytes and is one of the most important properties of electrolytes for secondary batteries. It is theoretically limited by the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) (see the literature211 on the correct use of these terms) of the molecule constituting the electrolyte, but is very often kinetically dominated in practical situations, as is clearly demonstrated by the importance of the SEI.45,212 In terms of Na secondary batteries, an electrochemical window from the Na+/Na redox potential up to ∼5 V is ideal for operation with high energy density, and many ILs containing BF4−, TFSA−, or FSA− satisfy this demand (or provide windows close to this).Fig. 7 shows the electrochemical windows of several ILs. A study combining molecular dynamics (MD) simulations and density functional theory (DFT) calculations indicated that the electrochemical stabilities of ILs should be discussed based on the stabilities of ions combined with counter ions in a certain structural model. Thus, considering the stability of individual ions in a vacuum is not always sufficient when considering the electrochemical stabilities of ILs (Fig. 7a).213 This study also indicated the higher instability to reduction of TFSA− compared to that of C3C1pyrr+, and this has been experimentally confirmed.214 Thermodynamic assessment based on DFT data has demonstrated that the electrochemical window is independent of the alkyl chain length for 1-alkyl-3-methylimidazolium cations and that BF4− and PF6− complex anions provide more electrochemically stable ILs than TfO or TFSA anions.215
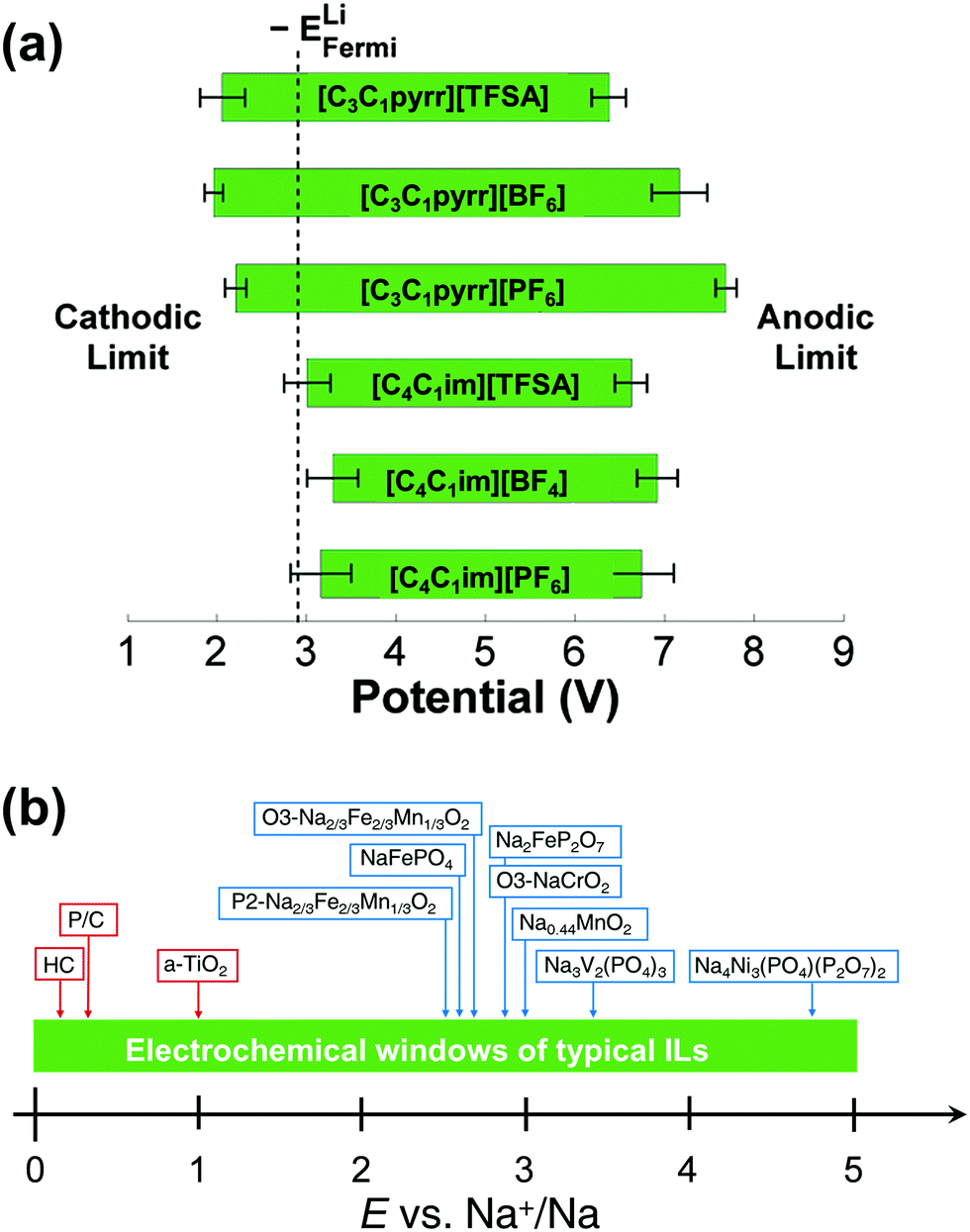 | ||
| Fig. 7 (a) Electrochemical windows for selected ILs as derived by a combination of MD and DFT calculations. Solid green bars indicate the potential range in which the IL is stable. The calculated Li metal Fermi level is also indicated. The 95% confidence interval for each limit is given by the black dumbbells. The potential scale is relative to the vacuum level. Reproduced with permission.213 Copyright 2011, American Chemical Society. (b) Electrochemical windows of typical ILs used for sodium secondary batteries. | ||
X-ray photoelectron spectroscopy (XPS) can provide information for discussing the oxidative and reductive resistances of ILs. The direct comparison of energy gaps revealed by XPS and voltammetry requires special care owing to differences in the properties of the bulk material and the electrode surface as well as differences in the Madelung potentials of the bulk and free surface.216 A study combining ultraviolet photoemission, inverse photoemission, and near-edge X-ray absorption fine structure spectroscopy indicated that mainly cations contribute to the top of the occupied states and the bottom of the unoccupied states of [C2C1im][BF4] and [C2C1im][PF6].217 Additional studies have confirmed that the molecular orbital energies of imidazolium-based ILs with [BF4]−, [PF6]−, [TfO]−, and [TFSA]− are significantly affected by the electrostatic Madelung potential among the ions. For ILs with [BF4]− and [PF6]−, both the highest occupied and lowest unoccupied states are derived from the imidazolium cation, i.e., the cation determines the band gap. Conversely, the highest occupied states for ILs with [TfO]− and [TFSA]− are determined by contributions from both the cation and anion.218,219
Cyclic and linear sweep voltammetry have revealed that pure ILs with typical fluoroanions have electrochemical windows of 4 to 5 V (see Fig. 7b for the relationship between electrochemical windows for typical ILs and the average discharge potentials of selected electrode materials for Na secondary batteries107,111,141,220,221). Although the three-electrode cell is a common setup for this type of measurement (typically, Pt, W, Cu, Al, and glass-like carbon are used as working electrode materials), two-electrode cells (usually coin-type cells) are often used for ILs containing shuttle ions such as Li+ and Na+ combined with Li and Na metal counter electrodes. The limit potentials for pure ILs are often compared with respect to the Ag+/Ag couple or more preferably the Fc+/Fc couple, as based on IUPAC recommendations.222 In the cases of Li+- or Na+-containing ILs, the electrochemical windows are usually discussed based on the Li+/Li or Na+/Na redox couple. Special attention has to be paid for the two-electrode cell because of the possible large polarisation on the Li or Na metal counter electrodes, which causes deviation from the Li+/Li or Na+/Na redox potential (see below). The addition of Li or Na salts to ILs often extends their reductive limits, regardless of the anion structure.121,223–226 Although aromatic cations including alkylimidazolium and alkylpyridinium suffer from instability to reduction in pure IL forms compared to nonaromatic- or linear alkyl-ammonium cations, the use of FSA− as a counter anion greatly improves stability, even for aromatic cases; the difference due to the cationic structure is minimised for FSA-based ILs. This phenomenon has been interpreted in terms of both differences in SEI properties and the double-layer structure model. The former is based on the observation that FSA− and TFSA− reductively produce a radical anion, and the radical anion from FSA− has unusually low reactivity to form a stable surface film.227 The latter is based on a specific double-layer model where FSA− acts as an anchor to pull Li+ toward and exclude organic cations from the interface.224,226
Residual water in ILs can have a significant effect on the electrochemical window.221 A significant decrease in the electrochemical window (over 1 V) is observed with increasing water content (15, 58, and 273 ppm) for a Pt electrode in a [N3111][TFSA] IL, but not for a glass-like carbon electrode. Practical active electrode materials also produce different behaviours compared to Na and Li metal electrodes; both cathodic and anodic decomposition of ILs is enhanced in the presence of certain electrode materials such as nano-sized active materials, especially graphitised carbon, and at elevated temperatures.103,228
An interesting behaviour of IL electrolytes is the low corrosivity of Al electrodes at high potentials. Unlike PF6-based electrolytes, which instantaneously form a fluorine-containing passivation film on Al,229 TFSA− and FSA− corrode Al current collectors in organic solutions.140,230–233 This is detrimental for practical applications because Al is commonly used as a current collector. However, Al corrosion is minimised in Li-based IL electrolytes.124,234–236 The same behaviour has also been observed in Na systems; Al corrosion also occurs in 1 mol dm−3 Na[TFSA]-PC solution,152 but not in FSA- or TFSA-based ILs,237,238 indicating that, in ILs, a stable passivation film is formed on Al electrodes at high potentials.
Na metal deposition–dissolution behaviour is another factor to consider when assessing the suitability of electrolytes for Na secondary batteries. In addition to the general importance of the behaviour of negative electrodes,74 this consideration is important for the possible use of Na metal as a negative electrode or counter electrode in two-electrode cells. However, the large polarisation of Na metal sometimes makes the accurate evaluation of target electrodes difficult when using a half-cell configuration.86,124,239–242Fig. 8 summarises some information on Na metal deposition–dissolution.
 | ||
| Fig. 8 (a) Photographs of ILs (top) before and (bottom) after Na metal immersion for four weeks. (A) Na[FSA]–[C2C1im][FSA] (10:90 in mol) and (B) Na[TFSA]–[C2C1im][TFSA] (10:90 in mol). Reproduced with permission.121 Copyright 2016, American Chemical Society. (b) Photographs of Na metal electrodes after alternate 50-cycle Na deposition–dissolution at 0.2 mA cm−2 in Na[FSA]–[C2C1im][FSA] (30 mol% Na[FSA]) at 0, 25, and 90 °C. Reproduced with permission.246 Copyright 2016, The Electrochemical Society. (c) Na/Na symmetric cell polarisation profiles for the Na[FSA]–[C3C1pyrr][FSA] IL at 25 °C (5 mol% Na[FSA], 25 mol% Na[FSA], and 50 mol% Na[FSA]) and a summary of overpotential trends for this system. Reproduced with permission.79 Copyright 2016, American Chemical Society. | ||
The reaction of Na metal in [C2C1im][FSA] and [C2C1im][TFSA] ILs has been investigated.121Fig. 8a shows the appearances of [C2C1im][FSA] and [C2C1im][TFSA] ILs before and after immersion of Na metal for four weeks. The transparent colourless [C2C1im][TFSA] IL turns brown after four weeks, whereas [C2C1im][FSA] shows no change, suggesting the high effectiveness of FSA− against reduction. Just 10 mol% FSA− is effective for suppressing the decomposition of ILs. The continuous increase of interfacial resistance for a TFSA− system has been studied by impedance spectroscopy. As discussed above, both the SEI layer and double-layer structure can contribute to the differences between FSA− and TFSA−,121,223–226 but the time dependence of interfacial resistance seems to correlate with the former in this case. The presence of Na[FSA] is also a key factor for stabilising interfacial resistance.
Table 1 lists selected data for Na metal deposition–dissolution tests, mainly summarising coulombic efficiency (as obtained by cyclic voltammetry and repetitive DC polarisation tests), cycle dependence, and water effects. Cyclic voltammetry tests provide information on Na metal deposition–dissolution behaviour. Repetitive cathodic and anodic polarisation in Na/Na or Al/Na cells with Na metal deposited on the Al is also used to observe long-term behaviour. Na metal dissolution does not occur from deposited Na metal in alkylimidazolium-based ILs without FSA−.121,243 However, it is observed in alkylpyrrolidinium-, tetraalkylammonium-, and tetraalkylphosphonium-based ILs, even those without FSA−, indicating the strong influence of the cation structure on Na metal deposition–dissolution behaviour.132,243 In the presence of FSA−, high coulombic efficiencies are observed in most cases.79,121,122,124,125,130,135,168,169,244 An increase in measurement temperature typically improves the coulombic efficiencies of Na metal deposition–dissolution.122,124,245 The higher coulombic efficiencies at 90 °C are considered to arise from the suppression of dendritic Na metal deposition at temperatures near the melting point of Na metal (98 °C) because diffusion of the Na atoms at the metal surface becomes faster. This is clearly demonstrated by the different appearances of the Na/Na symmetric cell deposition–dissolution test beaker cells operated at 0, 25, and 90 °C shown in Fig. 8b.246 After 50-cycle deposition–dissolution at 0.2 mA cm−2, dendrites are clearly observed on the Na metal electrode surfaces operated at 0 and 25 °C, whereas those operated at 90 °C maintain their shiny flat appearances and show no dendrite formation.
| IL | Na salt concentration, coulombic efficiency, and some comments | Ref. |
|---|---|---|
| a Measurement temperature was room temperature (∼25 °C) unless otherwise mentioned. See Fig. 3 for abbreviation of ionic species in ILs. C4C1C1im+: 1-butyl-2,3-dimethylimidazolium. CV: cyclic voltammetry, DC: DC repetitive polarization for Na metal deposition–dissolution, ηCV and ηDC: coulombic efficiencies of Na metal deposition–dissolution based on cyclic voltammetry and repetitive DC polarization. | ||
| Na[TFSA]–[C4C1pyrr][TFSA] | 0.1 mol dm−3, ηCV = 75% | 243 |
| 0.4 mol dm−3, ηCV ≈ 30% | 425 | |
| Na[TFSA]–[N2133][TFSA] | 0.1 mol dm−3, reversible in CV | 243 |
| Na[TFSA]–[N6222][TFSA] | 0.1 mol dm−3, ηCV = 57% | 243 |
| Na[TFSA]–[N1(2O2O1)3][TFSA] | 2.0 mol kg−1, at 50 and 70 °C; reversible in CV at 80 °C | 120 |
| Na[TFSA]–[C2C1im][TFSA] | 10 mol%, ηCV ≈ 0% | 121 |
| Na[TFSA]–[C4C1im][TFSA] | 0.1 mol dm−3, ηCV ≈ 0% | 243 |
| Na[TFSA]–[C4C1C1im][TFSA] | 0.1 mol dm−3, ηCV ≈ 0% | 243 |
| Na[TFSA]–[C4C1im][SO3CF3] | 0.1 mol dm−3, ηCV ≈ 0% | 243 |
| Na[TFSA]–[C3C1pyrr][FSA] | 0.25, 0.75, and 1.35 mol kg−1, ηCV ≈ 40–60%; ηCV decreases to ∼30% after 5 cycles | 130 |
| Na[FSA]–[C3C1pyrr][FSA] | 20 mol%; ηDC = 94% at 80 °C; reversible in CV at 80 °C | 245 |
| 5–50 mol%, reversible in CV; ηCV = 70% (5 mol%), unstable after cycling; ηCV = 30% (30 mol%), stable after cycling; low overpotential at high Na[FSA] concentrations | 79 | |
| 50 mol%, low overpotential at high H2O contents (up to 2000 ppm) after cycling | 168 | |
| 50 mol%, low overpotential at high H2O contents (500 ppm) | 169 | |
| Na[FSA]–[C2C1im][FSA] | 30 mol%, ηDC = 69% at 25 °C, ηDC = 96% at 90 °C | 124 |
| 10 mol%, reversible in CV | 121 | |
| Na[FSA]–[P1i4i4i4][FSA] | 45 mol%, ηCV = 93% at 50 °C, ηCV decreases to 53% after 20 cycles | 244 |
| 45 mol%, at 50 °C, decrease of polarization after cycling | 125 | |
| Na[FSA]–[P111i4][FSA] | 2.3 mol dm−3, at 50 °C, stable cycling in DC | 135 |
| 42 mol%, at 50 °C, low overpotential at high H2O contents (up to 500 ppm) after cycling | 169 | |
| 42 mol%, at 50 °C, decrease of polarization after cycling | 125 | |
| Na[FSA]–[N2(20201)3][FSA] | 55 mol%, at 50 °C, decrease of polarization after cycling | 125 |
| Na[FSA]–[N4411][FSA] | 30 mol%,ηCV = 32% at 25 °C, ηDC = 67% at 25 °C, ηDC = 92% at 90 °C | 122 |
| Na[FSA]–[N6411][FSA] | 30 mol%,ηCV = 36% at 25 °C, ηDC = 79% at 25 °C, ηDC = 92% at 90 °C | 122 |
| Na[FSA]–[AS(4.5)][FSA] | 30 mol%,ηCV = 35% at 25 °C, ηDC = 71% at 25 °C, ηDC = 90% at 90 °C | 122 |
| Na[DCN]–[C4C1pyrr][DCN] | 0.5 mol dm−3, ηCV = 74% | 247 |
| Na[FSA]–K[FSA] | 56 mol%, 90 °C, reversible in CV | 316 |
| 45 mol%, 67 °C, reversible in CV | 426 | |
| Na[TFSA]–Cs[TFSA] | 10 mol%, 150 °C, reversible in CV | 237 |
The effects of Na salt concentration on Na metal deposition–dissolution behaviour have been investigated in Na[FSA]–[C3C1pyrr][FSA] IL electrolyte.79 Higher salt concentrations result in lower interfacial impedance, leading to a lower polarisation potential and enabling relatively high current charging (Fig. 8c). This study concluded that the interfacial layers formed in the higher-concentration electrolytes are more important to the deposition–dissolution behaviour than the ionic conductivity of the electrolyte itself. The influence of water on Na metal deposition–dissolution behaviour has been investigated in FSA-based ILs.168,169 Polarisation and impedance tests revealed that addition of water at the ppm level (∼500 ppm) provides positive effects such as smooth morphology and low interfacial resistance as well as contributing to the formation of an ideal SEI.169 Another study revealed that the interfacial resistance of a Na/Na symmetric cell with a Na[FSA]–[C3C1pyrr][FSA] electrolyte containing 2000 ppm water is lower than that for one containing 20 ppm water after 20 cycles of Na metal deposition–dissolution.168 Such observations suggest that water can act as an effective additive in practical Na secondary batteries with FSA-based ILs. DCA-based ILs also improve Na metal deposition–dissolution behaviour.247
2.5 Handling of IL electrolytes
ILs sometimes require special handling. One of the most important considerations for IL electrolytes is controlling water content. Even hydrophobic ILs that form a two-phase state with water can contain water up to a few wt%.170 For example, water-saturated [C2C1im][TFSA] and [C3C1pyrr][FSA] ILs (two-phase conditions) contain 19940 and 18000 ppm water, respectively.221,248 The removal of water from hydrophobic ILs is much easier than that from hydrophilic ILs, with evacuation at elevated temperatures reducing the water content to below 30 ppm. Some BF4−-based ILs are hydrophilic, and removal of water by simple evacuation to the level required for non-aqueous batteries is very troublesome, even at elevated temperatures. The use of molecular sieves can be effective in such cases, but contamination by other components is inevitable as ILs are difficult to separate by distillation.249 Purging dry gas through ILs at elevated temperature has also been reported to be effective for water removal.250,251
There are now many commercially available ILs that need special attention in terms of impurity control. ILs are often contaminated by alkali metal (or silver) and/or halide ion impurities during synthesis. These ions have high polarities and can be removed by washing with water if the ILs are hydrophobic. Passing ILs through activated alumina or activated carbon can sometimes work effectively for purification, but, again, it has to be performed carefully because of possible contamination.252,253 Thus, pre-treatment of the starting materials used for the synthesis is more effective for obtaining high-grade ILs than post-synthesis purification of the ILs obtained.254,255 Another important process for battery testing using IL electrolytes is impregnation of the IL into the separator and electrode materials. Highly viscous ILs are sometimes resistant to soaking into porous materials. In such cases, treating such porous materials in ILs under vacuum at elevated temperatures (e.g., ∼80 °C) sometimes works for impregnation of ILs and derives the true performance of the IL electrolytes to improve the results of battery tests.164,180,248
2.6 Current limitation of IL electrolytes
Cost is undoubtedly a serious issue for most applications of ILs. Battery-grade ILs are generally very expensive due to the lack of large-scale production methods and the need for fluorinated anions for high electrochemical stability.94 Kuzmina described economical aspects of ILs in 2016 and mentioned that it is very difficult to truly estimate the industrial price of ILs at the current stage.256 The problem of the estimation based on catalogue prices was also critically pointed out. Within the nine issues to the commercial use of ILs proposed by Joglekar et al.,257 the lack of scale-up studies is quite serious for practical applications. This makes cost estimation of ILs very difficult and leads to apathy from the industrial side. Furthermore, battery-grade ILs need to be extremely pure, which leads to a further increase in cost. Such pure ILs are not widely synthesized at industrial levels and thus estimation of price is difficult even for lithium ion batteries. Recovery and recycling of ILs are widely mentioned for synthetic applications, but they are not very hopeful for electrolyte applications. In general, quaternary ammonium cations are more expensive than ternary ammonium cations, and a fluorine-containing anion is more expensive than the one without fluorine. Alkylammonium and alkylphosphonium are more cost-effective than dialkylimidazolium.258 One of the most important steps for the application of ILs, including electrolyte applications, is collecting data for true estimation of each purpose. In contrast to TFSA salts, FSA salts can be synthesized without the costly electrofluorination and therefore could potentially achieve a lower price through further improvement in the synthetic process259,260 and mass production109,261 in the near future.As mentioned in the previous section, high viscosity of ILs, especially at low temperature, potentially causes problems in handling such as impregnation onto porous separators and electrodes. Purification including dehydration requires many efforts for ILs. Some ILs are not always environmentally benign, and harmful decomposition products must be taken into account.262 Solubility of inorganic salts into some ILs is very low, which intrinsically makes their use as electrolytes difficult. From the viewpoint of their high thermal stability, disposal of ILs after their use in batteries would be energy-consuming compared to conventional organic electrolytes.
3. Electrode materials investigated in IL electrolytes
3.1 Positive electrode materials
The prospective advantages of Na secondary batteries are their economic feasibility and their ability to act as sustainable energy sources for large-scale applications such as ESSs and electric vehicles. Thus, research into electrode materials for Na secondary batteries should comply with the principle of the utilisation of earth-abundant elements.49,263,264 Positive electrode materials have been widely studied as a means to maximise the performance of Na secondary batteries with respect to power and energy densities, efficiency, cycleability, and safety.Various polyanionic compounds, layered transition-metal oxides, and organic compounds have been reported as positive electrode materials for Na secondary batteries.14,15,264–266 Several compounds exhibit excellent performance and some unique electrochemical results in IL electrolytes, including improved cycleability, high rate capability, and enhanced electrochemical activity at elevated operating temperatures.103,164,180,266–268 These discoveries were enabled by exploiting the electrochemical and thermal stabilities of IL electrolytes.
Fig. 9 illustrates the relationship between theoretical capacity and working potential for positive electrode materials applied in IL electrolytes. The gravimetric theoretical capacities (mA h g−1) and working potentials (V vs. Na+/Na) are presented in Fig. 9. Most of the studies on positive electrode materials utilising IL electrolytes have been carried out using polyanionic compounds and O3- and P2-type layered transition-metal oxides. The “O3” and “P2” nomenclature developed by Delmas et al.269 is used to indicate the Na–O coordination (O for octahedral and P for prismatic) and repetition stacking period in the unit cell (2 or 3). These materials are highly reversible based on Na-intercalation with limited volumetric change. The structural properties of selected representative polyanionic compounds (Na2FeP2O7 and Na3V2(PO4)3) and O3-type and P2-type layered transition-metal oxides are shown in Fig. S1 (ESI†). A comprehensive list of positive electrode materials tested in IL electrolytes is provided in Table S4 (ESI†) along with their structural and electrochemical characteristics. Table 2 shows their detailed electrochemical results in designated IL electrolytes including mass loading, operating temperature, cut-off voltage, and current density. In this section, the electrochemical properties of polyanionic compounds and layered oxide materials tested with IL electrolytes in Table 2, including their structural properties, are reviewed.
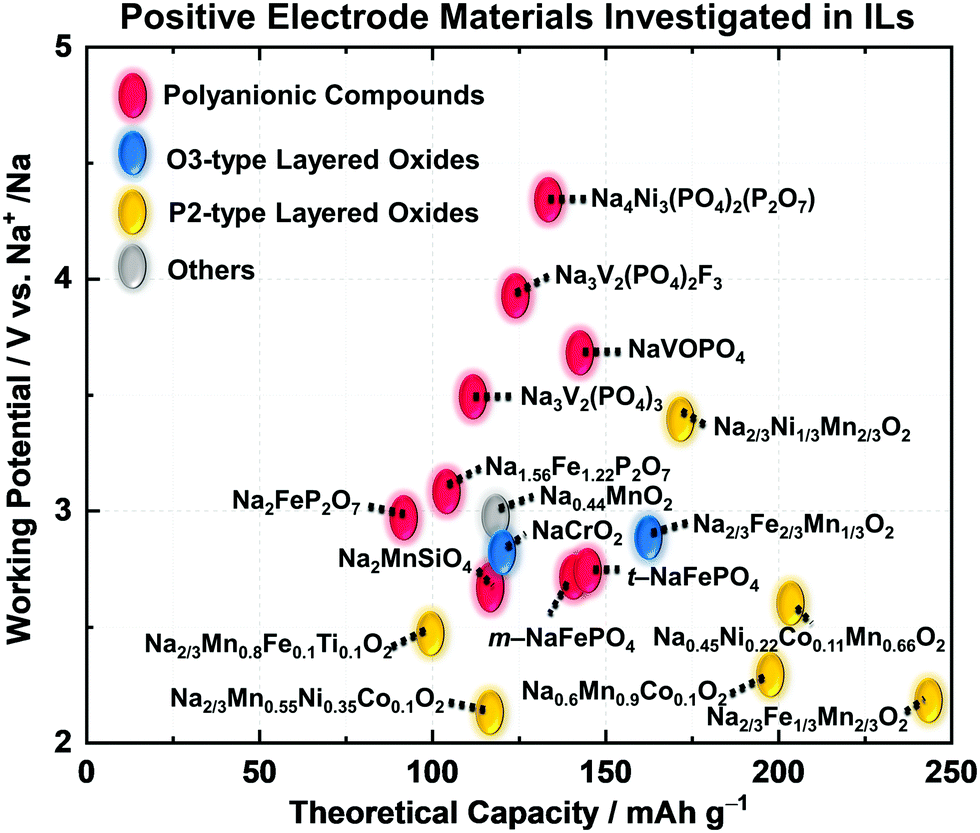 | ||
| Fig. 9 Positive electrode materials for sodium secondary batteries investigated in IL electrolytes. The relationship between theoretical capacity and working potential. | ||
| Cell configuration | Electrolyte | Mass loading/mg cm−2 | Temp./°C | Discharge capacity/mA h g−1 @ mA g−1 | Rate capability/mA h g−1 @ mA g−1 | Cycleability | Ref. |
|---|---|---|---|---|---|---|---|
| a AC = activated carbon. b HC = hard carbon. c CNF = carbon nanofiber. d See Fig. 3 for abbreviation of ionic species in ILs. | |||||||
| Na/Na2FeP2O7 | 56 mol% Na[FSA]–K[FSA] | — | 90 | 91 @ 10 | 59 @ 2000 | 91% after 1000 cycles | 295 |
| Na/Na2FeP2O7 | 30 mol% Na[FSA]–[C2C1im][FSA] | — | 25 | 89 @ 1 | 17 @ 2000 | 97% after 300 cycles | 180 |
| Na/Na2FeP2O7 | 30 mol% Na[FSA]–[C2C1im][FSA] | — | 90 | 93 @ 10 | 83 @ 2000 | 93% after 1500 cycles | 180 |
| Na/Na1.56Fe1.22P2O7 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 2 | 25 | 90 @ 11.8 | 47 @ 1180 | 98.5% after 300 cycles | 298 |
| Na/Na1.56Fe1.22P2O7 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 2 | 90 | 0 | 64 @ 4720 | 71% after 3000 cycle | 298 |
| Na/Na4Ni3(PO4)2(P2O7) | 10 mol% Na[TFSA]–[C4C1pyrr][ FSA] | 2.8 | 20 | 63 @ 10 | — | — | 268 |
| Na3V2(PO4)3/Na3V2(PO4)3 | 0.4 mol dm−3 Na[BF4]–[C2C1im][BF4] | — | 25 | 63 @ 0.1 | ∼15 @ 2.0 | — | 87 |
| Na3V2(PO4)3/Na3V2(PO4)3 | 0.4 mol dm−3 Na[BF4]–[C2C1im][BF4] | — | 80 | 83 @ 0.1 | ∼25 @ 2.0 | — | 87 |
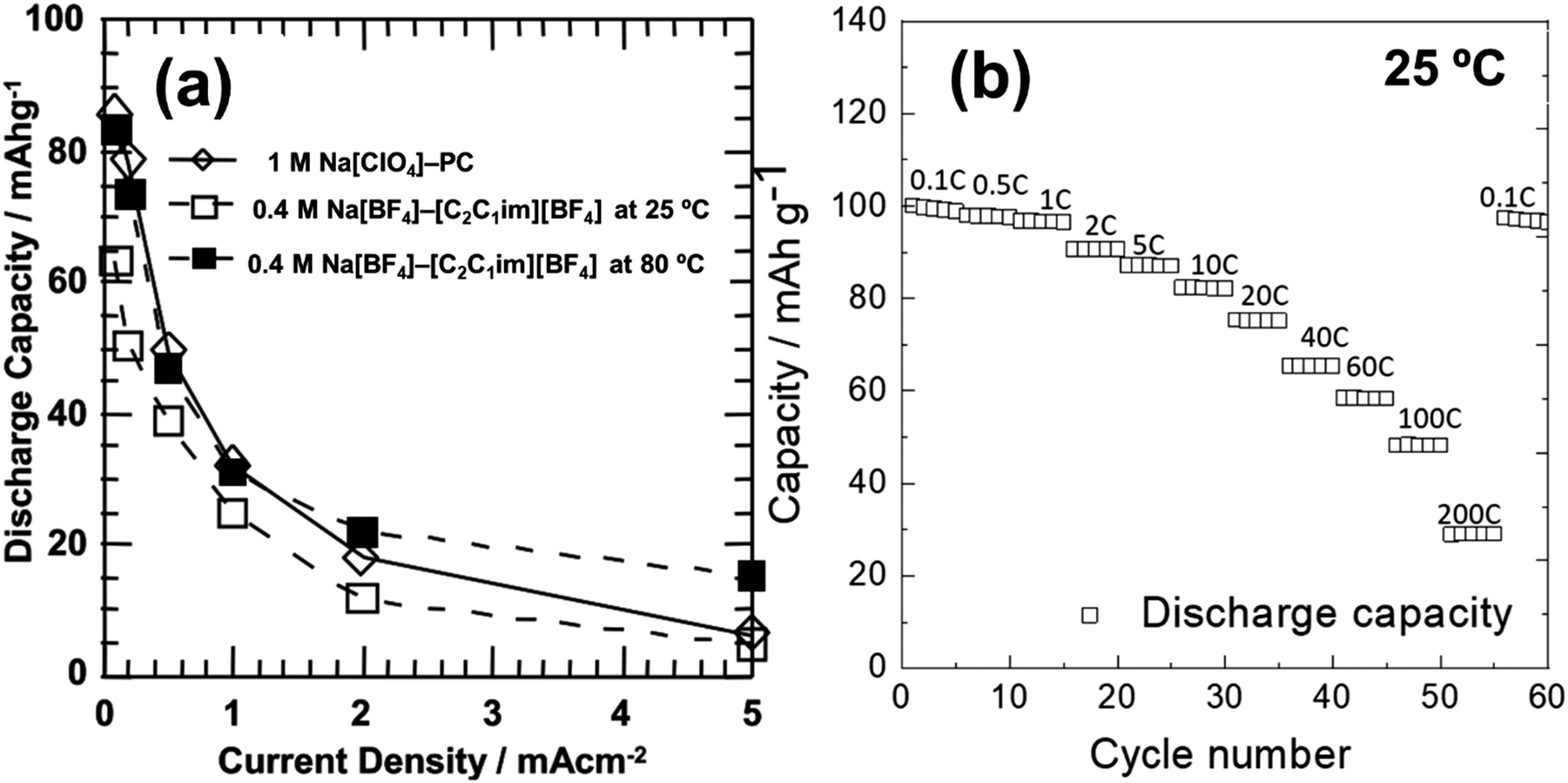
| ACa/Na3V2(PO4)3 | 0.3 mol dm−3 Na[TFSA]–[C4Hpyrr][TFSA] | 1.6 | 40 | 70 @ 118 | ∼25 @ 1180 | 93 mA h g−1 after 100 cycles | 128 |
| HCb//Na3V2(PO4)3 | 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] | — | 25 | 85 @ 11.7 | 60 @ 234 | 75% after | 408 |
| Na/Na3V2(PO4)3@C | 20 mol% Na[FSA]–[C2C1im][FSA] | 3 | -20 | 58 @ 11.7 | 82.7 @ 11700 |
— | 164 |
| Na/Na3V2(PO4)3@C | 20 mol% Na[FSA]–[C2C1im][FSA] | 3 | 25 | 100 @ 11.7 | 41 @ 2340 | 99.6% after 200 cycles | 164 |
| Na/Na3V2(PO4)3@C | 50 mol% Na[FSA]–[C2C1im][FSA] | 3 | 90 | 104 @ 11.7 | 82.7 @ 11700 |
89.2% after 5000 cycles | 164 |
| Na/Na3V2(PO4)3@CNFc | 40 mol% Na[FSA]–[C2C1im][FSA] | 8.5 | 25 | 105 @ 11.7 | 51 @ 1170 | 94 mA h g−1 after 200 cycles | 283 |
| Na/Na3V2(PO4)3@CNFc | 40 mol% Na[FSA]–[C2C1im][FSA] | 8.5 | 90 | 107 @ 11.7 | 77 @ 11700 |
104 mA h g−1 after 200 cycles | 283 |
| Na/Na3V2(PO4)3@C | 0.25 mol dm−3 Na[PF6]–[C4C1im][TFSA] | — | 25 | 107 @ 50 | — | 104 mA h g−1 after 50 cycles | 286 |
| Na/t-NaFePO4 | 0.5 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | — | 50 | 125 @ 7.8 | 75 @ 155 | 83% after 100 cycles | 84 |
| Na/t-NaFePO4 | 1 mol dm−3 Na[BF4]–[C4C1pyrr][TFSA] | 2.5 | 75 | 152 @ 7.8 | 91 @ 155 | 90% after 100 cycles | 119 |
| Na/t-NaFePO4 | 42 mol% Na[FSA]–[N2(20201)3][FSA] | — | 50 | 97 @ 15.5 | 66 @ 155 | 71% after 100 cycles | 289 |
| Na/t-NaFePO4 | 45 mol% Na[FSA]–[P1i4i4i4][FSA] | — | 50 | 113 @ 15.5 | 88 @ 155 | 12% after 100 cycles | 289 |
| Na/t-NaFePO4 | 55 mol% Na[FSA]–[P111i4][FSA] | — | 50 | 107 @ 15.5 | 84 @ 155 | 95% after 100 cycles | 289 |
| Na/m-NaFePO4 | 30 mol% Na[FSA]–[C2C1im][FSA] | 3 | 90 | 107 @ 15.5 | 62 @ 155 | 67% after 100 cycles | 103 |
| Na/NaVOPO4-AB-8 h | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 2 | 25 | 60 @ 5 | — | — | 303 |
| Na/NaVOPO4-AB-8 h | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 2 | 90 | 101 @ 5 | 77 @ 725 | 76% after 300 cycles | 303 |
| Na/NaCrO2 | 56 mol% Na[FSA]–K[FSA] | — | 80 | 77 @ 15 | 58 @ 150 | 89% after 100 cycles | 316 |
| Na/NaCrO2 | 10 mol% Na[TFSA]–Cs[TFSA] | — | 150 | 94 @ 10 | 66.4 @ 100 | 83.4 mA h g−1 after 10 cycles | 237 |
| Na/NaCrO2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 25 | 92 @ 20 | — | — | 245 |
| Na/NaCrO2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 80 | 106 @ 20 | 62 @ 500 | — | 245 |
| HCb/NaCrO2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 13.5 (HC 5.2) | 25 | 27 A h @ 2.7 A | 26 A h @ 10.8 A | — | 411 |
| HCb/NaCrO2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 13.5 (HC 5.2) | 60 | 28 A h @ 2.7 A | 27 A h @ 10.8 A | 87% after 500 cycels | 411 |
| HCb/NaCrO2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 13.5 (HC 5.2) | 90 | 28 A h @ 2.7 A | 27 A h @ 10.8 A | — | 411 |
| Na/NaMnO2 | 10 mol% Na[TFSA]–[C4C1pyrr][TFSA] | 2–4 | 25 | 130 @ 9.7 | — | 81% after 10 cycles | 133 |
| Na/Na0.44MnO2 | [Na(G5)1][TFSA] | — | 60 | 110 @ 12.7 | — | 95% after 30 cycles | 149 |
| Na/Na0.44MnO2 | 1 mol dm−3 Na[ClO4]–[C4C1pyrr][TFSA] | — | 50 | 109 @ 6.4 | 82 @ 127 | 80% after 100 cycles | 127 |
| HCb//Na0.44MnO2 | 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] | 1.5 | 25 | 120 @ 30 | 70 @ 1000 | 97% after 100 cycles | 86 |
| Na/P2-Na2/3Fe1/3Mn2/3O2 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 90 | 227 @ 20 | 65 @ 500 | 64% after 200 cycles | 318 |
| Na/P2-Na2/3Fe2/3Mn1/3O2 | 2.3 mol dm−3 Na[FSA]–[P111i4][FSA] | — | 50 | 130 @ 17.4 | 110 @ 34.8 | — | 135 |
| Na/O3-Na2/3Fe2/3Mn1/3O2 | 2.3 mol dm−3 Na[FSA]–[P111i4][FSA] | — | 50 | 167 @ 17.4 | 130 @ 34.8 | 57% after 100 cycles | 135 |
| Na/P2-Na2/3Ni1/3Mn2/3O2 | 1 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | 3 | 60 | 220 @ 10 | — | 200 mA h g−1 after 14 cycles | 319 |
| Na/Na0.6Mn0.9Co0.1O2 | 0.5 mol dm−3 Na[FSA]–DEME–[TFSA] | — | 25 | 205 @ 20 | 68 @ 400 | 70% after 350 cycles | 131 |
| Na/Na0.6Mn0.9Co0.1O2 | 0.5 mol dm−3 Na[FSA]–MOEMPIP–[TFSA] | — | 25 | 117 @ 20 | 20 @ 400 | 80% after 350 cycles | 131 |
| Na/P2-NaNa0.6Ni0.22Fe0.11Mn0.66O2 | 0.2 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | 1 | 25 | 150 @ 10 | — | — | 409 |
| NaxSB-C/P2-NaNa0.6Ni0.22Fe0.11Mn0.66O2 | 0.2 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | 1 (NaxSB-C 1.9) | 25 | 120 @ 10 | — | — | 409 |
| ACa/Na0.67Mn0.89Mg0.11O2 | 0.3 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | 2.1 | 40 | 57 @ 100 | — | 59 mA h g−1 after 100 cycles | 128 |
| Na2.55V6O16/Na0.6Co0.1Mn0.9O2 | 0.5 mol dm−3 Na[FSA]–[C4C1pyrr][TFSA] | 1.75 (Na2.55V6O16 0.65) | 25 | 125 @ 50 | 60 @ 600 | 76% after 100 cycles | 410 |
| Na/P2-Na0.45Ni0.22Co0.11Mn0.66O2 | 0.4 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] | 2.5 | 20 | 210 @ 23 | — | 80% after 100 cycles | 267 |
| Na/P2-Na2/3Mn0.8Fe0.1Ti0.1O2 | 2.3 mol dm−3 Na[FSA]–[P111i4][FSA] | — | 50 | 176 @ 17.6 | 125 @ 88 | 92% after 50 cycles | 125 |
| Na/P2-Na2/3Mn0.8Fe0.1Ti0.1O2 | 45 mol% Na[FSA][P1i4i4i4][FSA] | — | 50 | 101 @ 17.6 | 60 @ 88 | 28% after 50 cycles | 125 |
| Na/P2-Na2/3Mn0.8Fe0.1Ti0.1O2 | 55 mol% Na[FSA][N2(2O2O1)3][FSA] | — | 50 | 153 @ 17.6 | 30 @ 88 | 58% after 50 cycles | 125 |
| Na/calix[4]quinone | 0.3 mol dm−3 Na[TFSA]–[C3C1pyrr][TFSA] | 0.58 | 25 | 406 @ 20 | — | 99.7 after 300 cycles | 380 |
| Na/5,7,12,14-pentacenetetrone | 0.3 mol dm−3 Na[TFSA]–[C3C1pyrr][TFSA] | 0.58 | 25 | 245 @ 30 | — | — | 380 |
NASICON-type Na3V2(PO4)3 crystallises in the rhombohedral space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c281,282 and is the polyanionic material that has been most extensively studied using IL electrolytes owing to its open structure for facile Na+ transport, high thermal stability, and high energy density. Na3V2(PO4)3 can provide two redox reactions based on V4+/V3+ and V3+/V2+. The V4+/V3+ reaction is based on a two-electron reaction (for two V atoms) with a theoretical capacity of 117 mA h g−1 showing an average potential of 3.4 V vs. Na+/Na (eqn (6)). The V3+/V2+ reaction is based on a one-electron reaction (for one V atom) with a theoretical capacity of 55 mA h g−1 showing an average potential of 1.6 V vs. Na+/Na (eqn (7)).87,164,283,284 This double-redox system facilitates the fabrication of symmetric Na3V2(PO4)3 cells, and is often used for testing new electrolytes.
c281,282 and is the polyanionic material that has been most extensively studied using IL electrolytes owing to its open structure for facile Na+ transport, high thermal stability, and high energy density. Na3V2(PO4)3 can provide two redox reactions based on V4+/V3+ and V3+/V2+. The V4+/V3+ reaction is based on a two-electron reaction (for two V atoms) with a theoretical capacity of 117 mA h g−1 showing an average potential of 3.4 V vs. Na+/Na (eqn (6)). The V3+/V2+ reaction is based on a one-electron reaction (for one V atom) with a theoretical capacity of 55 mA h g−1 showing an average potential of 1.6 V vs. Na+/Na (eqn (7)).87,164,283,284 This double-redox system facilitates the fabrication of symmetric Na3V2(PO4)3 cells, and is often used for testing new electrolytes.
| Na3V2(PO4)3 ⇄ NaV2(PO4)3 + 2Na+ + 2e− | (6) |
| Na4V2(PO4)3 ⇄ Na3V2(PO4)3 + Na+ + e− | (7) |
 | ||
| Fig. 10 Electrochemical behaviour of NASICON-type Na3V2(PO4)3 in ILs. (a) Rate performance of Na3V2(PO4)3 symmetric cells with a 0.4 mol dm−3 Na[BF4]–[C2C1im][BF4] IL and 1 mol dm−3 Na[ClO4]-PC organic solvent electrolytes. Reproduced with permission.87 Copyright 2010, The Electrochemical Society. (b) Rate performance of CNF-incorporated Na3V2(PO4)3 symmetric cells with a 40 mol% Na[FSA]–[C2C1im][FSA] IL electrolyte. Reproduced with permission.283 Copyright 2019, American Chemical Society. | ||
A more recent study on Na3V2(PO4)3 revealed that Na3V2(PO4)3 combined with carbon nanofibers exhibits good rate performance in IL electrolytes at 25 °C.283 In this study, a symmetric cell comprising a Na3V2(PO4)3/carbon nanofiber positive electrode with a 40 mol% Na[FSA]–[C2C1im][FSA] IL electrolyte exhibited a high rate capability of 48.3 mA h g−1 at 100C (1C = 117 mA g−1) and 25 °C, as shown in Fig. 10b.283 Carbon nanofibers efficiently improve the electronic conductivity of Na3V2(PO4)3. Owing to the high thermal, chemical, and electrochemical stabilities of Na3V2(PO4)3, it has become the standard material for assessing new IL electrolytes. For example, the protic IL Na[TFSA]–[C4Hpyrr][TFSA] (0.3 mol dm−3) has been studied as an electrolyte for Na secondary batteries.128 This protic IL electrolyte exhibits more stable cycle performance than that with a 0.3 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] aprotic IL. A gelled 0.8 mol kg−1 Na[FSA]–[C3C1pyr][FSA]-PVdF-co-HFP (PVdF-co-HFP = polyvinylidene fluoride-co-hexafluoropropylene) IL gel electrolyte showed good cycle performance with 92% capacity retention and 99.9% coulombic efficiency over 150 cycles.285 Another study on Na3V2(PO4)3 with Na[PF6]–[C4C1im][TFSA] showed improved cycling performance over that achieved with an organic solvent electrolyte.286 Cycleability results will be considered in more detail later in Section 4.3.
Olivine-type LiFePO4 is one of the most well studied and commercialised materials for LIBs because of its cost-effective, facile synthesis and high theoretical capacity of 170 mA h g−1.273,287 In the same manner, Fe-based phosphates (NaFePO4) have been investigated as potential positive electrode materials using IL electrolytes. Olivine NaFePO4, which has the highest theoretical capacity of 155 mA h g−1 among the Fe-based phosphates, has two polymorphs, i.e., triphylite and maricite phases, both of which crystallise in the orthorhombic space group Pnma.288 However, triphylite-phase NaFePO4 is not thermally stable and can only be prepared by electrochemical/chemical extraction of Li from LiFePO4 and substitution with Na.288Fig. 11 shows the electrochemical behaviours of triphylite-NaFePO4 and several layered metal oxides in ILs. Triphylite NaFePO4 was assessed using [C4C1pyrr][TFSA] IL electrolytes containing Na[BF4], Na[ClO4], Na[PF6], or Na[TFSA] and a conventional 1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v) organic electrolyte at 25, 50, and 75 °C.84,119 At 25 °C, 1 mol dm−3 Na[ClO4]-EC/DEC exhibited the highest rate performance and cycleability, but at the elevated temperature of 50 °C, the cycleability became poorer. A significant improvement in reversible capacity, rate performance (Fig. 11a), and cycleability were observed in 1 mol dm−3 Na[BF4]–[C4C1pyrr][TFSA]. The discharge capacity was further improved to 152 mA h g−1 (at 0.05C) at 75 °C in 1 mol dm−3 Na[BF4]–[C4C1pyrr][TFSA] electrolyte.84,119
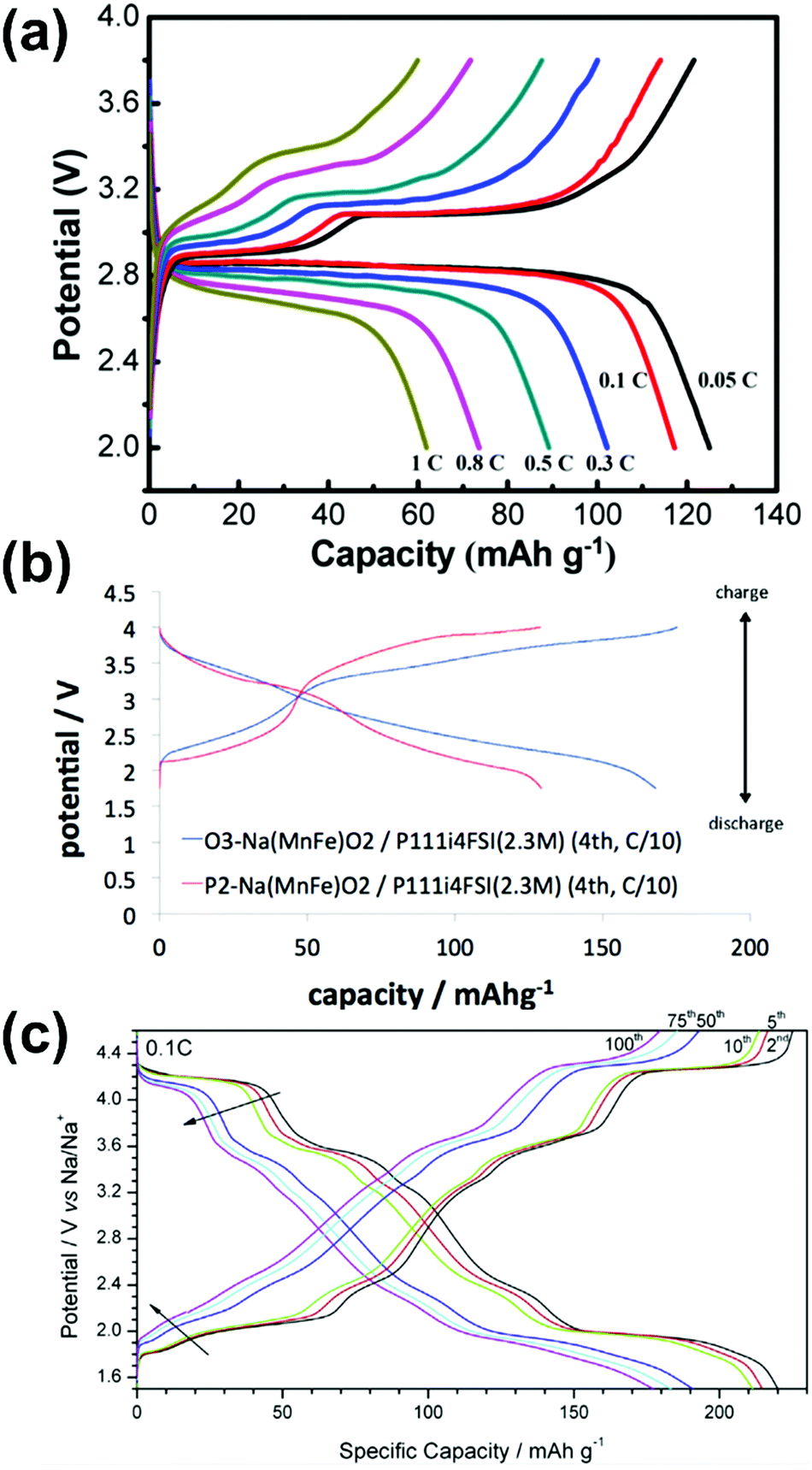 | ||
| Fig. 11 Electrochemical behaviours of triphylite-NaFePO4 and layered metal oxides in ILs. (a) Charge–discharge curves for a Na/1 mol dm−3 Na[BF4]–[C4C1im][TFSA]/NaFePO4 cell at various current rates and 50 °C. Reproduced with permission.119 Copyright 2014, American Chemical Society. (b) Charge–discharge curves for Na/2.3 mol dm−3 Na[FSA]–[P111i4][FSA]/O3- and P2-type Na2/3Fe2/3Mn1/3O2 cells. Reproduced with permission.135 Copyright 2017, Elsevier. (c) Charge–discharge curves for a Na/Na0.45Ni0.22Co0.11Mn0.66O2/10 mol% Na[TFSA]–[C4C1pyrr][FSA] cell. Reproduced with permission.267 Copyright 2014, Elsevier. | ||
Recently, cycle performance improvement was reported for a 1 mol dm−3 Na[FSA]–[P111i4][FSA] IL electrolyte with a capacity retention of 95% over 100 cycles (50 °C, C/2).289 In this study, it was suggested that electrolyte conductivity is not the main factor for determining cell performance and is not significantly related to capacity. Above all, the SEI determines the cycle stability, as is discussed in more detail in the section on negative electrodes below. XPS measurements confirmed the existence of IL-derived decomposition products like NaOH, Na2S, and NaF on the Na metal surface, which provided stable cycle performance in a half-cell.289
Maricite-NaFePO4 can be prepared by the conventional solid-state method. It was previously believed that maricite-NaFePO4 was electrochemically inactive.288,290 However, nano-sized maricite-NaFePO4 transforms into an amorphous FePO4 phase during the first charge (desodiation) and shows unexpected charge–discharge performance.291 Another study on maricite NaFePO4 using ILs revealed that ball-milled maricite-NaFePO4 shows reversible (de)sodiation properties, as confirmed by X-ray diffraction (XRD) and X-ray absorption near edge structure (XANES) spectroscopy.102,103 Further details will be presented in Section 4.1.
Pyrophosphate compounds, such as Na2FeP2O7, Na1.56Fe1.22P2O7, and Na4Fe(PO4)2P2O7, offer stable three-dimensional diffusion paths for Na ions composed of corner-sharing FeO6 units.292 Na2FeP2O7 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .293,294 Its theoretical capacity estimated from the one-electron reaction based on the Fe2+/Fe3+ redox couple reaction is 97 mA h g−1 (eqn (8)).294
.293,294 Its theoretical capacity estimated from the one-electron reaction based on the Fe2+/Fe3+ redox couple reaction is 97 mA h g−1 (eqn (8)).294
| Na2FeP2O7 ⇄ NaFeP2O7 + Na+ + e− | (8) |
| Na1.56Fe1.22P2O7 ⇄ Na0.34Fe1.22P2O7 + Na+ + e− | (9) |
Na2MnSiO4 is a low-cost and environmentally benign material composed of only earth-abundant elements. In a recent study, carbon-coated Na2MnSiO4 was prepared by a sol–gel method and assessed in a 20 mol% Na[FSA]–[C3C1pyrr][FSA] IL.300 This material has theoretical capacities of 278 mA h g−1 and 125 mA h g−1 based on a two-electron reaction and one-electron reaction, respectively.300,301 However, the obtained reversible capacities of 70, 94, and 125 mA h g−1 at 25, 50, and 90 °C, respectively, suggested that the redox activity based on only the one-electron reaction, which corresponds to 0.9 equivalents of Na being reversibly (de)sodiated based on the Mn3+/Mn2+ redox couple reaction.
Vanadium-based phosphate (NaVOPO4) crystallises in the monoclinic space group P21/c. NaVOPO4 has a high operating potential of 3.6 V vs. Na+/Na with a large theoretical capacity of 145 mA h g−1 based on the V5+/V4+ redox couple reaction.302 The charge–discharge behaviour of NaVOPO4 was investigated using 20 mol% Na[FSA]–[C3C1pyrr][FSA] IL electrolytes at 25 and 90 °C. Reversible capacities of 60 and 101 mA h g−1 were obtained at 25 and 90 °C, respectively.303
The structures of O3- and P2-type layered transition-metal oxides are shown in Fig. S1c and d (ESI†), respectively. Despite the success of layered transition-metal oxide compounds as positive electrode materials in LIBs, the number of studies on their use in IL electrolytes for Na secondary batteries is still limited. In terms of cost and resource sustainability, Ni and Co layered transition-metal oxides are not particularly attractive materials for Na secondary batteries. Thus, the layered transition-metal oxides for Na secondary batteries investigated in IL electrolytes mostly contain Mn, Fe, and Cr elements instead of Co and Ni.
The first report on reversible Na insertion and extraction concerned O3-type NaCrO2.314 The theoretical capacity of this material is 125 mA h g−1 for the one-electron reaction based on the Cr4+/Cr3+ redox couple.315 O3-type NaCrO2 exhibits numerous two-phase regions in its charge–discharge plateaus up to 3.5 V. The upper cut-off voltage was 3.6 V because further desodiation causes an irreversible phase transition. The material was assessed with IL electrolytes comprising 56 mol% Na[FSA]–K[FSA] at 80 °C. The Na/Na[FSA]–K[FSA]/NaCrO2 cell exhibited a specific discharge capacity of 77.3 mA h g−1.316 This capacity was improved to 101.4 mA h g−1 at 150 °C using 10 mol% Na[TFSA]–Cs[TFSA].237 Furthermore, the use of NaCrO2 in a wide temperature range (−20 to 90 °C) with 20 mol% Na[FSA]–[C3C1pyrr][FSA] IL electrolyte has been reported (see a later Section 4.1 for the temperature dependence of the performance).166
Recently, binary and ternary mixed transition metal oxides have been investigated using IL electrolytes. O3 and P2-type NaFe1-xMnxO2 consisted of earth-abundant elements and was expected to deliver a high reversible capacity. O3- and P2-type Na2/3Fe2/3Mn1/3O2 was assessed in a 0.5 mol dm−3 Na[PF6]-EC/DMC organic solvent electrolyte. The P2-type Na2/3Fe2/3Mn1/3O2 and O3-type Na2/3Fe2/3Mn1/3O2 delivered the first discharge capacities of 151 and 157 mA h g−1, respectively, over the voltage range 4.2–1.5 V at C/10. However, the capacity of both P2- and O3-Na2/3Fe2/3Mn1/3O2 faded within 10 cycles.317 Interestingly, the capacity and cycle performance were improved in measurements using 2.3 mol dm−3 Na[FSA]–[P111i4][FSA]135 only for O3-type Na2/3Fe2/3Mn1/3O2, which exhibited excellent performance using the IL electrolyte at 50 °C. The capacity of O3-type Na2/3Fe2/3Mn1/3O2 was improved to 184 mA h g−1 over the voltage range of 4.0–1.75 V, while P2-type Na2/3Fe2/3Mn1/3O2 provided a capacity of 130 mA h g−1 because a narrower voltage range was applied (Fig. 11b). The rate capability and cycle performance were improved for both P2-type and O3-type Na2/3Fe2/3Mn1/3O2 upon using IL electrolytes.135
A material with a different Fe/Mn ratio, P2-Na2/3Fe1/3Mn2/3O2, was investigated in a Na[FSA]–[C3C1pyrr][FSA] IL electrolyte, and showed a high discharge capacity of 227 mA h g−1 at a current density of 20 mA g−1 over the voltage range 2.0–4.3 V. With a wider cut-off voltage of 1.5–4.2 V, P2-Na2/3Fe1/3Mn2/3O2 exhibited a very high discharge capacity of 317 mA h g−1 in the IL, but this capacity exceeds its theoretical capacity of 261 mA h g−1, indicating unfavourable side reactions.318
A recent report on P2-Na2/3Ni1/3Mn2/3O2 revealed stable cycling involving an O redox reaction in a 1 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] IL electrolyte. The discharge capacity of 220 mA h g−1 fell to 200 mA h g−1 after 14 cycles in the IL electrolyte at 60 °C, whereas it decreased to 160 mA h g−1 after 14 cycles in 1 mol dm−3 Na[PF6]-EC/DMC at 25 °C.319
The ternary transition metal oxide P2-Na0.45Ni0.22Co0.11Mn0.66O2 provides a good example of the superiority of IL electrolytes (Fig. 11c). Compared to the case in an organic electrolyte, higher reversibility of the P2–O2 phase transition was observed for P2-Na0.45Ni0.22Co0.11Mn0.66O2 using 10 mol% Na[TFSA]–[C4C1pyrr][FSA] above 4.2 V, which is attributable to the wide electrochemical window of the IL. The cell exhibited a capacity of approximately 220 mA h g−1 at 12 mA g−1 and 20 °C. This is because the electrochemical stability of the IL electrolyte is higher than that of the organic electrolyte. Furthermore, the low solubility of Mn in the IL electrolyte at potentials below 2.2 V helps to maintain its good cycleability.267 A discussion of the cycleability of the materials is presented in Section 4.3.
Na0.44MnO2 is an attractive material for Na secondary batteries in terms of its low cost and the earth-abundance of its constituent elements. It crystallises in the orthorhombic space group Pbam and has wide tunnels for Na+ diffusion.320 The initial charge desodiates 0.26 equivalents of Na+ from Na0.44MnO2 (eqn (10)). After the initial charge, the charge–discharge reaction is based on (de)sodiation with 0.46 equivalents of Na+ (eqn (11)). The theoretical capacity is calculated to be 127 mA h g−1 based on the Mn4+/Mn3+ redox couple reaction.
| Na0.44MnO2 → Na0.18MnO2 + 0.26Na+ + 0.26e− | (10) |
| Na0.18MnO2 + 0.46Na+ + 0.46e− ⇄ Na0.64MnO2 | (11) |
3.2 Negative electrode materials
In this section, the performances of negative electrode materials in IL electrolytes will be discussed. Table 3 shows electrochemical results of negative electrode materials in IL electrolytes. The various types of negative electrodes are classified based on their sodiation/desodiation behaviour. They are divided into carbon materials, titanium-based compounds, alloy materials, and conversion-based materials. Each section begins with a brief description of the material characteristics and electrochemical behaviour in organic electrolytes and their performances in ILs. Finally, based on the insight accumulated from the studies reported, future prospective investigations are suggested.| Cell configuration | Electrolytes | Mass loading/mg cm−2 | Temp./°C | 1st cycle discharge capacity @ current (col. eff.)/mA h g−1 @ mA g−1 | Rate capability/mA h g−1 (current) | Cycleability | Ref. |
|---|---|---|---|---|---|---|---|
| a See Fig. 3 for abbreviation of ionic species in ILs. b mAh cm−2. | |||||||
| Na/3-D CF | Na[FSA]–[C3C1pyrr][FSA] | — | 25 | 235 @ 30 (40%) | 115 (1000 mA g−1) | 85% (250 cycles) | 397 |
| 60 | — | — | 80% (250 cycles) | ||||
| Na/semi-graphitic HC | 0.5 M Na[TFSA]–[C3C1pyrr][TFSA] + 2% VEC | — | 30 | 178 @ 74.2 (91%) | 44 (1860 mA g−1) | 100% (55 cycles) | 328 |
| Na/HC | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 0 | 48 @ 20 (—) | — | — | 329 |
| 25 | 193 @ 20 (—) | 44 (200 mA g−1) | >100% (500 cycles) | ||||
| 90 | 277 @ 20 (78.5%) | 230 (1000 mA g−1) | 84% (500 cycles) | ||||
| Na/anatase TiO2 | 10 mol% Na[TFSA]–[C3C1pyrr][FSA] | 1.4–1.6 | 25 | 159 @ 33.5 (43%) | 47 (3350 mA g−1) | 98% (80 cycles) | 336 |
| Na/Nb doped rutile TiO2 | Na[FSA]–[C3C1pyrr][FSA] | — | 30 | 110 @ 50 (—) | — | — | 341 |
| Na[FSA]–[C3C1pyrr][FSA] | 60 | 180 @ 50 (—) | — | 97% (350 cycles) | |||
| Na[FSA]-PC | 30 | 120 @ 50 (—) | — | — | |||
| Na[FSA]-PC | 60 | 200 @ 50 (—) | — | 91% (350 cycles) | |||
| Na/TiO2–Fe2O3 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 90 | 386 @ 10 (52%) | 91 (2000 mA g−1) | 80% (300 cycles) | 337 |
| Na/FeTiO3-C | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 0.8–1.3 | 25 | 120 @ 10 (—) | — | — | 338 |
| 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 90 | 390 @ 10 (46.8%) | 256 (2000 mA g−1) | 113% (2000 cycles) | |||
| Na[PF6]-EC/DMC | 25 | 150 @ 10 (—) | — | — | |||
| Na/TiO2-C | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 1–2 | 25 | 143 @ 10 (—) | — | — | 228 |
| 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 90 | 340 @ 10 (45%) | 93 (2000 mA g−1) | 79% (1000 cycles) | |||
| Na[PF6]-EC/DMC | 25 | 114 @ 10 (—) | — | — | |||
| Na/Sn film | 56 mol% Na[FSA]–K[FSA] | — | 90 | 729 @ 0.619b (92.3%) | — | 66% (30 cycles) | 427 |
| Na/Sn–Fe film | 56 mol% Na[FSA]–K[FSA] | 0.76 | 90 | 443 @ 84.7 (71%) | — | 43% (100 cycles) | 355 |
| Na/Sn–Cu film | 56 mol% Na[FSA]–K[FSA] | 0.88–1.07 | 90 | 100 @ 84.7 (62.5%) | 70 (847 mA g−1) | 100% (1000 cycles) | 353 |
| Na/Sn–Ni film | 56 mol% Na[FSA]–K[FSA] | 0.67–0.85 | 90 | 605 @ 84.7 (86.4%) | — | 56% (100 cycles) | 354 |
| Na/P-AB | 0.25 M Na[FSA]–[C3C1pyrr][FSA] | 0.47–0.56 | — | 1595 @ 125 (72.3%) | — | 93% (80 cycles) | 80 |
| Na[PF6]-PC | — | 1768 @ 125 (76.3%) | — | 26% (80 cycles) | |||
| Na[PF6]-EC/DEC | — | 1755 @ 125 (77.3%) | — | 30% (80 cycles) | |||
| Na[PF6]-EC/PC/3DEC | — | 1654 @ 125 (75.8%) | — | — | |||
| Na/black P | Na[TFSA]–[C(10201)C1][FSA] | — | 30 | 800 @ 50 (—) | — | 38% (100 cycles) | 362 |
| Na[TFSI]–[C2C1im][FSA] | 30 | 1072 @ 50 (—) | — | — | |||
| Na[TFSA]-PC | 30 | 1500 @ 50 (—) | — | 7% (40 cycles) | |||
| Na/CuP2-C | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 25 | 316 @ 100 (73.7%) | 50 (8000 mA g−1) | — | 369 |
| 90 | 595 @ 100 (70.2%) | 368 (8000 mA g−1) | 71% (200 cycles) | ||||
| Na/V4P7-5P | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | 1 | 25 | 560 @ 100 (88%) | 155 (2000 mA g−1) | 54% (100 cycles) | 371 |
| Na[PF6]-EC/DEC | 90 | 738 @ 100 (86%) | 360 (8000 mA g−1) | 48% (100 cycles) | |||
| 25 | 605 @ 100 (88%) | — | SCD | ||||
| Na/V4P7 | 20 mol% Na[FSA]–[C3C1pyrr][FSA] | — | 25 | 270 @ 100 (—) | — | — | 371 |
| 90 | 470 @ 100 (—) | — | — | ||||
| Na/InP | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 476 @ 50 (78.7%) | — | 105% (100 cycles) | 368 |
| Na/CuP2 | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 758 @ 50 (72.7%) | — | SCD | 368 |
| Na/GeP | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 798 @ 50 (87.5%) | — | SCD | 368 |
| Na/GeP5 | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 963 @ 50 (85.6%) | — | SCD | 368 |
| Na/SiP | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 522 @ 50 (71.5%) | — | SCD | 368 |
| Na/LaP | Na[FSA]–[C3C1pyrr][FSA] | 0.8–1 | 30 | 12 @ 50 (7.4%) | — | — | 368 |
| Na/Sn4P3 | Na[FSA]–[C3C1pyrr][FSA] | 1.2 | 0 | 220 @ 100 (55.9%) | — | 396 | |
| 1.6 | 30 | 670 @ 50 (83.5%) | 520 (1000 mA g−1) | 112% (200 cycles) | 395 | ||
| 1.2 | 60 | 976 @ 100 (84.6%) | 250 (3000 mA g−1) | — | 396 | ||
| 1.2 | 90 | 1057 @ 100 (82.5%) | 230 (3000 mA g−1) | — | 396 | ||
| Na/SnP3 | Na[FSA]–[C3C1pyrr][FSA] | 1.6 | 30 | 1100 @ 50 (88.4%) | — | SCD | 395 |
| Na/Sb2S3–graphene | Na[ClO4]-PC | 1–1.2 | 25 | 710 @ 50 (52%) | 340 (1500 mA g−1) | 48% (100 cycles) | 373 |
| Na[ClO4]-PC/FEC | 25 | 710 @ 50 (60%) | 285 (1500 mA g−1) | 83% (100 cycles) | |||
| Na[ClO4]-EC/DEC | 25 | 560 @ 50 (48%) | 55 (1500 mA g−1) | 40% (100 cycles) | |||
| Na[ClO4]-EC/DEC/FEC | 25 | 580 @ 50 (55%) | 150 (1500 mA g−1) | 69% (100 cycles) | |||
| Na[FSA]–[C3C1pyrr][FSA] | 25 | 660 @ 50 (65%) | 240 (1500 mA g−1) | 96% (100 cycles) | |||
| Na[ClO4]-PC/FEC | 60 | 610 @ 50 (50%) | 120 (1500 mA g−1) | 43% (100 cycles) | |||
| Na[FSA]–[C3C1pyrr][FSA] | 60 | 760 @ 50 (75%) | 420 (1500 mA g−1) | 95% (100 cycles) | |||
| Na/SnO2–graphene | Na[ClO4]-PC/FEC | 1–1.2 | 25 | 505 @ 20 (—) | 197 (1000 mA g−1) | 73% (100 cycles) | 374 |
| Na[ClO4]-EC/PC | 25 | 500 @ 20 (—) | 310 (1000 mA g−1) | 71% (100 cycles) | |||
| Na[ClO4]-EC/PC/FEC | 25 | 505 @ 20 (—) | 237 (1000 mA g−1) | 88% (100 cycles) | |||
| Na[FSA]–[C3C1pyrr][FSA] | 25 | 410 @ 20 (—) | 148 (1000 mA g−1) | 99% (100 cycles) | |||
| Na[ClO4]-EC/PC/FEC | 60 | 505 @ 20 (—) | — | 59% (100 cycles) | |||
| Na[FSA]–[C3C1pyrr][FSA] | 60 | 600 @ 20 (—) | 330 (1000 mA g−1) | 96% (100 cycles) | |||
| Na/Na2DBQ-OMC | Na[ClO4]-EC/PC | 0.3–0.6 | 22 | — | — | 38% (50 cycles) | 384 |
| Na[FSA]-EC/PC | 22 | — | — | — | |||
| Na[FSA]–[C3C1pyrr][FSA] | 22 | 145 @ 25 (43%) | — | — | |||
| Na[FSA]–[C3C1pyrr][FSA] | 60 | 277 @ 25 (—) | 175 (300 mA g−1) | 88% (300 cycles) | |||
To date, hard carbon (HC) has been the most extensively studied carbonaceous negative electrode material for Na secondary batteries. HC prepared by the pyrolysis of sugar demonstrated a high storage capacity of 300 mA h g−1 with excellent capacity retention and rate capability.327 The ester-based solvent EC/PC provided lower overpotential and charge transfer resistance than EC/PC with fluoroethylene carbonate (FEC) as an additive. Overall, this study proposed that the performance is highly dependent on the electrolyte. The importance of electrolytes is readily understood by such studies. Therefore, many researchers have recently investigated ILs with carbon materials.
Fig. 12 shows the electrochemical behaviours of negative electrode materials in ILs. The electrochemical properties of semi-graphitic HC prepared by pyrolysing olive pits were investigated with 0.5 mol dm−3 Na[TFSA]–[C4C1pyrr][TFSA] with addition of 2% vinyl ethylene carbonate (VEC) and 0.3 mol dm−3 Na[TFSA]–[C4Hpyrr][TFSA] at 30 °C.328 This carbon material showed good cycleability with Na[TFSA]–[C4C1pyrr][TFSA] but the capacity faded quickly in the case of 0.3 mol dm−3 Na[TFSA]–[C4Hpyrr][TFSA] owing to the instability of the [C4Hpyrr]+ cation below 1 V with the Na metal as a counter electrode. The electrochemical performance of commercial HC powders was investigated with a 20 mol% Na[FSA]–[C3C1pyrr][FSA] IL in the wide temperature range −10 to 90 °C, as shown in Fig. 12a.329 This HC showed a high capacity of 277 mA h g−1 at 20 mA g−1 and remarkable rate capability at 90 °C (Fig. 12b) owing to the high ionic conductivity and low charge transfer resistance of the IL. It also showed a capacity of 193 mA h g−1 at 20 mA g−1 and 25 °C, which is important for room temperature applications. Although only a limited number of studies conducted with ILs is available, the above-mentioned studies demonstrate the high capacity, excellent rate capability at elevated temperature, and superior cycleability of carbon materials achievable in ILs.
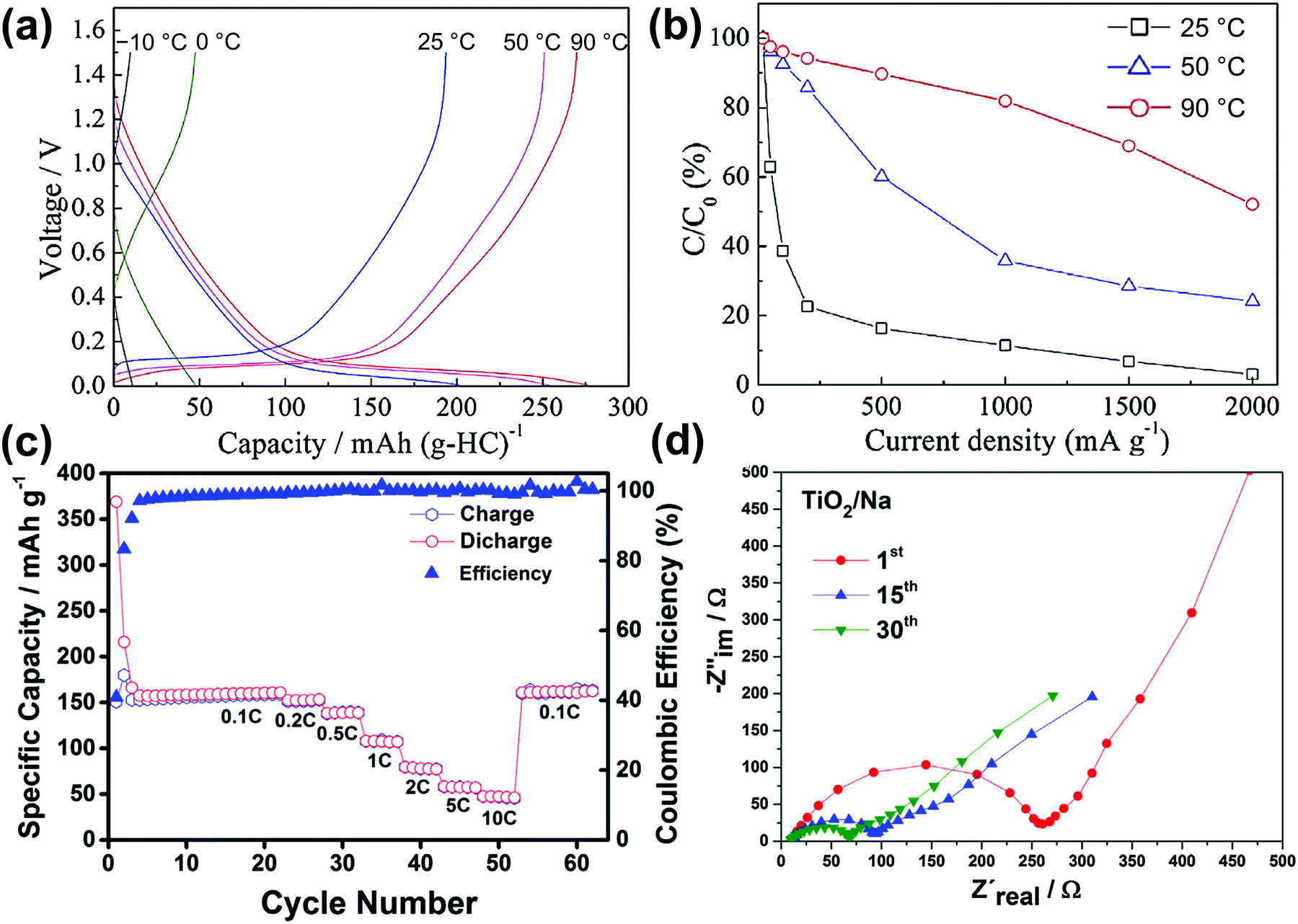 | ||
| Fig. 12 Electrochemical behaviours of hard carbon (HC) and Ti-based negative electrode materials in ILs. (a) Galvanostatic charge–discharge profiles of HC at −10 to 90 °C at a current density of 20 mA g−1 in 20 mol% Na[FSA]–[C3C1pyrr][FSA]. (b) Rate capabilities of the HC electrode at 25, 50, and 90 °C, corresponding to (a). Reproduced with permission.329 Copyright 2015, Elsevier. (c) Rate capability of anatase-TiO2-based electrodes in 10 mol% Na[TFSA]–[C3C1pyrr][FSA]. After two formation cycles at 0.01C (0.1 to 2.0 V), various C rates (1C = 335 mA g−1) were applied to the electrode. (d) Impedance spectra for the 1st, 15th, and 30th cycles recorded for a fully charged (i.e., desodiated) TiO2/Na cell. Reproduced with permission.336 Copyright 2016, Elsevier. | ||
:1, v/v).331 The hexagonal crystal structure of NiTiO3, which features zigzag open tunnels and mesoporosity, enabled this high rate performance.
A NASICON-type NaTi2(PO4)3 (NTP) was investigated in 1 mol dm−3 Na2SO4 aqueous electrolytes.332 Owing to the high Na+ conductivity of the NASICON structure and the nanoparticle conformation of NTP, high rate capability (103 mA h g−1 at 2C) was attained. Another study on an NTP/graphene composite prepared by the sol-gel method revealed the reversible insertion/extraction of Na+ involving the Ti4+/Ti3+ redox couple.333
The charge–discharge mechanism of anatase-TiO2 was found to be rather interesting. It proceeds with the insertion of Na+ into the TiO2 lattice, distorting the structure and subsequently forming Na0.69TiO2, Ti metal (Ti0), and O2 by reduction.334 Although the formation of Ti metal is unfavourable according to the Gibbs energy calculation,335 the larger size of Na+ causing strain in the lattice might be responsible for triggering the disproportionation reaction. The aforementioned studies highlight the attention-grabbing properties of Ti-based compounds. Accordingly, the following studies highlight the performance of Ti-based compounds with IL electrolytes.
The charge–discharge behaviour of commercial anatase-TiO2 in 10 mol% Na[TFSA]–[C3C1pyrr][FSA] has been reported.336 Even at 25 °C, TiO2 showed good rate capability by providing 47 mA h g−1 at 2C (1C = 335 mA g−1) (Fig. 12c). The electrochemical impedance spectroscopy (EIS) results, obtained at the fully desodiated state of the TiO2/Na cell, are shown in Fig. 12d. The semicircle gradually decreases in size from the 1st to the 30th cycle and reaches a stable value of approximately 70 Ω, suggesting a stabilised electrode/electrolyte interface. TiO2/C prepared by heat treatment of citric acid with commercial anatase-TiO2 exhibited remarkable capacity retention of 79% after 1000 cycles and excellent rate performance at 90 °C in 20 mol% Na[FSA]–[C3C1pyrr][FSA].228 To increase the reversible capacity, Fe2O3 was incorporated into TiO2 by heat treatment of commercial TiO2 and Fe(NO3)3. A high reversible capacity of 386 mA h g−1 at 10 mA g−1 and 80% capacity retention after 300 cycles at 200 mA g−1 were achieved at 90 °C using 20 mol% Na[FSA]–[C3C1pyrr][FSA].337 An FeTiO3/C electrode prepared by a two-step heat treatment method from the same precursor as that used above and sucrose as the carbon source showed no discernible capacity fading over 2000 cycles at 90 °C, which is the best cycleability for negative electrodes in ILs currently reported.338
In addition to fabricating composite materials, doping with aliovalent ions has been demonstrated to be an effective technique for improving the electrical conductivities of materials, which has a positive effect on the performance of batteries.339,340 For example, Nb-doped rutile-TiO2 (Ti0.94Nb0.06O2) prepared by a sol–gel method was investigated as a negative electrode material for Na secondary batteries using 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] and 1 mol dm−3 Na[FSA]-PC.341 A high capacity retention of 97% was observed with Na[FSA]–[C3C1pyrr][FSA] at 60 °C. Flammability tests demonstrated no ignition of Na[FSA]–[C3C1pyrr][FSA] even at 300 °C, whereas explosive combustion of Na[FSA]-PC was observed at 150 °C, demonstrating the benefit of ILs in terms of safety as well as performance.
:1, v/v) using Si negative electrodes revealed the high stability of the SEI formed, which was attributed to the volume change suppression and negligible mass change during delithiation observed by electrochemical quartz crystal microbalance analysis.347 Although there are many studies of Si with Li, limited studies have been performed with Na. Theoretically, Na forms NaSi at the maximum capacity (∼954 mA h g−1) but in practical cases, a reversible capacity of ∼300 mA h g−1 (Na0.29Si) has been recorded so far.348 Numerous materials have been investigated as candidates for alloy materials in Na secondary batteries. These have been summarised in several review articles.349–351 Most of these studies adopted organic electrolytes, and there is a very limited number of reports using ILs. This section addresses the reports on alloy-based materials with ILs.
Fig. 13 shows the charge–discharge behaviours of alloy and phosphide-based negative electrode materials for Na secondary batteries. Sn exhibits high capacity (∼847 mA h g−1 for Na15Sn4), low cost (∼20.8 $ kg−1),352 and non-toxicity. Several intermetallic Sn–M compounds (M = Fe, Ni, Cu) with 56 mol% Na[FSA]–K[FSA] IL at 90 °C have been investigated for Na secondary batteries.353–355 The intermetallic compounds were synthesised by electrodeposition followed by annealing for different periods of time. The annealing time was optimised with respect to capacity and cycleability. Longer annealing times caused the evolution of inactive intermetallic compounds, which results in increased buffering at the cost of capacity. The charge–discharge curves for an Sn–Cu film annealed for 4 h show four plateaus, even after 1000 cycles at 90 °C, which highlights the excellent cycleability of this system (Fig. 13a). This result was attributed to the buffering effect of the inactive component Cu3Sn and the randomly dispersed less electrochemically active component Cu6Sn5.
 | ||
| Fig. 13 Electrochemical behaviours of alloy- and conversion-based negative electrode materials in ILs. (a) Charge–discharge curves for the 1st, 10th, 100th, and 1000th cycles of an annealed Sn-Cu film at a current density of 84.7 mA (g-Sn) −1 and 90 °C in 56 mol% Na[FSA]–K[FSA]. Reproduced with permission.353 Copyright 2014, Elsevier. (b) Rate capabilities of a CuP2/C composite in 20 mol% Na[FSA]–[C3C1pyrr][FSA] at 25 and 90 °C. Reproduced with permission.369 Copyright 2018, Wiley. | ||
Phosphorus is another alloyable material, providing a large theoretical capacity in Na secondary batteries (∼2596 mA h g−1) owing to the formation of Na3P. Red P with carbon composites has been extensively investigated in the past decade for Na secondary batteries with organic electrolytes356–358 but there is only one report concerning its use in an IL.80 The red P/carbon composite exhibited stable cycleability in 0.25 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] but the capacity faded quickly in an organic electrolyte. The reason for this behaviour is discussed below in the SEI layer section. Black P, being an extraordinary conductor of electricity and thermodynamically stable under ambient conditions, is widely applied in optoelectronics, electronics,359 and batteries.360,361 Black P was synthesised by heating red phosphorus at 800 °C under high pressure and the effect of electrolyte additives (FEC and VC) in achieving good SEIs was investigated. In another study, black P prepared by the ball-milling of red P showed the best cycleability at 30 °C in ILs among the three different electrolytes: 1 mol dm−3 Na[TFSA]–[C(10201)C1pyrr][FSA], 1 mol dm−3 Na[TFSA]–[C2C1im][FSA], and 1 mol dm−3 Na[TFSA]-PC.362 These results indicate the potential of ILs with alloy-based negative electrodes for better cycleability and safer performance.
| MaXb + (b × c)Na ⇌ aM + bNacX | (12) |
Metal phosphides are emerging as a separate class of negative electrodes for Na secondary batteries. In addition to showing high capacities, they exhibit low overpotential as conversion-based materials due to the low electronegativity of P, as mentioned above. The sodiation of metal phosphides usually results in the formation of Na3P and separation of metal nanoparticles, which improves electronic conduction. However, the large volume change causes electrode pulverisation, which causes severe capacity deterioration. Similar to the situation with alloy-based materials, carbon composites, nanostructuring, and morphology and electrolyte optimisation are some of the strategies used to improve performance. Although transition-metal phosphides have been highly investigated in organic electrolytes,365–367 there are limited reports using ILs. A variety of binary phosphides (GeP5, GeP, SiP, LaP, InP, and CuP2) were evaluated in 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 30 °C.368 The best cycleability was observed for InP, which was attributed to the provision of electronic conductivity and maintenance of interparticle contact by the adjustment of NaIn morphology in response to volume expansion by Na3P.
A CuP2/C carbon composite was investigated using 20 mol% Na[FSA]–[C3C1pyrr][FSA] at 25 and 90 °C.369 Owing to the high ionic conductivity of the IL and enhanced electrode kinetics at 90 °C, good rate capability was achieved (Fig. 13b). Earlier studies with organic electrolytes revealed the reversible formation of CuP2370 but in this case, the product remained amorphous after the first sodiation. The authors proposed that the charge–discharge mechanism can be changed by changing the electrolyte and operating conditions. An early transition-metal phosphide, V4P7, investigated recently,371 shows a peculiar insertion-based mechanism as revealed by ex situ XRD. Moderate capacities of 270 and 470 mA h g−1 at 25 and 90 °C, respectively, were observed using 20 mol% Na[FSA]–[C3C1pyrr][FSA] as the electrolyte. Moreover, high rate capability and good cycleability were also attained at 90 °C. In the same study, a V4P7 P composite (V4P7/5P) was also investigated in 20 mol% Na[FSA]–[C3C1pyrr][FSA] and 1 mol dm−3 Na[PF6]-EC/DEC (1:1, v/v). Stable cycleability over 100 cycles was observed for the IL, whereas the capacity faded after six cycles for the organic electrolyte. This observation was supported by EIS and SEM observations. After 10 cycles, a significant decrease in impedance and a similar particle size compared to the pristine state were observed in the IL case, whereas large impedance and enlargement of particle size were observed for the organic electrolyte.
As discussed above, the metal and non-metal can be varied to tune the properties. Although sulfides and oxides have larger overpotentials than phosphides, they also show high capacities and rate capabilities. Usually, the reaction proceeds with the formation of Na2X (X = S, O) and separation or alloying of the metal. Recently Sn- and Sb-based materials have become popular owing to their alloying properties and relative abundance. Sb2O3 prepared by electrostatic spray deposition provided a reversible capacity of 331 mA h g−1 with stable cycleability and good rate capability using 1 mol dm−3 Na[PF6]-EC/DEC/PC (4:4:2 by vol) with 5% FEC.372 The sodiation product NaSb(OH)6 was observed by ex situ XRD and transmission electron microscopy (TEM) and appeared to be amorphous, which is a common phenomenon in conversion-based materials. The sulfide analogue Sb2S3 was investigated using 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] IL and various organic electrolytes at 25 and 60 °C.373 High first cycle discharge capacities of 710 and 660 mA h g−1 were delivered with 1 mol dm−3 Na[ClO4]-PC/FEC and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 25 °C, respectively.
Fig. 14a shows SEM images of the pristine Sb2S3 and those after charge–discharge in 1 mol dm−3 Na[ClO4]-PC/FEC (Fig. 14b) and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] (Fig. 14c). It can be clearly seen that after charging and discharging in 1 mol dm−3 Na[ClO4]-PC/FEC, the particles are covered by thick SEI layers, whereas in Fig. 14c, the morphology is retained, demonstrating formation of thin and robust SEI layers. In Fig. 14d–f, the capacity, rate capability, and cycleability are compared at 25 °C and 60 °C for 1 mol dm−3 Na[ClO4]-PC/FEC and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA]. It can be observed that at 60 °C, the 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] clearly outperforms the 1 mol dm−3 Na[ClO4]-PC/FEC in all performance aspects. The large difference in the rate performance at 60 °C is attributed to the elevated side reaction in 1 mol dm−3 Na[ClO4]-PC/FEC. Conversely, 1 mol dm−3 Na[ClO4]-PC/FEC delivered higher capacity and better rate performance than 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 25 °C due to the high viscosity in the IL system, which restricts Na ion diffusion.
 | ||
| Fig. 14 Comparison of the electrochemical performances of Sb2S3 in IL and organic electrolytes. SEM micrographs of (a) the as-prepared Sb2S3/graphene electrode and the electrode after the first charge–discharge cycle in (b) 1 mol dm−3 Na[ClO4]-PC/FEC and (c) 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] electrolytes at 25 °C. Effects of temperature on (d) reversible capacity at 50 mA g−1, (e) capacity retention at 1500 mA g−1 (compared to values at 50 mA g−1), and (f) capacity retention after 100 cycles for Sb2S3/graphene electrodes measured in 1 mol dm−3 Na[ClO4]-PC/FEC and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA]. Reproduced with permission.373 Copyright 2017, American Chemical Society. | ||
A SnO2/graphene composite prepared by supercritical-CO2-assisted synthesis was analysed in similar organic and IL electrolytes.374 High rate capability with the incorporation of EC in PC at 25 °C and degradation of rate with the addition of FEC were demonstrated. At the elevated temperature of 60 °C, high reversible capacities of 600 and 330 mA h g−1 at 20 and 1000 mA g−1, respectively, were obtained with 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA].
3.3 Summary and future investigations
Prussian blue analogues have not yet been tested using IL electrolytes. For example, Na4Fe(CN)6, which is made from naturally abundant resources, could be an interesting material that simultaneously achieves good capacity and moderate working potential.377
Finally, organic compounds are very attractive materials for Na secondary batteries in terms of sustainability and diversity.378 One of the most important advantages of organic compounds is their chemical designability and tunable functionality. Thus, various redox-active organic electrodes are currently known. Nevertheless, organic electrode materials for Na secondary batteries have not been well investigated in IL electrolytes. A study on the organic electrode material poly((3,4)-ethylenedioxythiophene)biopolymer in 20 mol% Na[TFSA]–[C4C1pyrr][TFSA] and Na[FSA]–[C2C1im][FSA] IL electrolytes indicated their stable electrochemical performance.379 In another recent study, calix[4]quinone and 5,7,12,14-pentacenetetrone organic-based positive electrode materials were investigated in a 0.3 mol dm−3 Na[TFSA]–[C3C1pyrr][TFSA] IL electrolyte.380 A Na/calix[4]quinone half-cell exhibited the very high revisable capacity of 406 mA h g−1 at 20 mA g−1 and excellent cycleability with a stable capacity retention of 99.7% after 300 cycles. A Na/5,7,12,14-pentacenetetrone half-cell with a 0.3 mol dm−3 Na[TFSA]–[C3C1pyrr][TFSA] IL electrolyte exhibited similarly good performance with a capacity of 245 (30 mA g−1). The authors insisted that these improvements were achieved using the IL electrolyte because it overcame problems associated with the poor dissolution of quinone compounds.380 Thus, IL electrolytes may be useful to overcome problems associated with organic compounds such as suppressing the dissolution of active materials that typically occurs in aprotic solvents and ameliorating the low intrinsic electronic conductivity by elevated temperature operation.378,381–383
There are many candidate materials for Ti-based compounds. For example, Na titanates, especially Na2Ti3O7387,388 and Na2Ti6O13,389 show lower redox potentials than other Ti-based compounds and are highly investigated as negative electrode materials in organic electrolyte systems. Furthermore, the SEI layer should be studied in detail along with ILs. In the study on red P mentioned in this review,80 it included a large amount of binder (∼20 wt%) to facilitate good cycleability, but the amount and type of binder also influence the decomposition of the electrolyte. Therefore, an investigation into the SEI layer should be performed without (or with a minimal amount of) binder. In addition, the surface morphology of the SEI layer should be inspected by characterisation tools like atomic force microscopy (AFM), TEM, and cross-sectional focused ion beam SEM. Recently, there have been reports on volume change suppression of Si-based negative electrodes for LIBs in ILs.390–393 Similar results were reported for V4P7/5P in Na secondary batteries,371 but an understanding of the key mechanism behind the phenomenon is lacking.
4. Benefits of using IL electrolytes
As previously stated, organic electrolytes such as EC, PC, DEC, DMC, and 1,2-dimethoxyethane (DME) coupled with the Na salts Na[ClO4] and Na[PF6] are routinely used for Na secondary battery studies and to achieve good performance. However, the safety issues surrounding secondary batteries are mostly concerned with the volatility and flammability of organic electrolytes. As Na secondary batteries are mainly intended as large-scale batteries for ESSs and electric vehicles, the safety problems become more significant as the battery size increases. In addition to the safety benefits, recent studies have revealed that ILs bring excellent cycle performance and superior rate capability. This is because IL electrolytes can form more stable SEIs that enable more facile Na diffusion and can also form a stable passivation layer on Al current collectors, preventing Al corrosion.394 Furthermore, ILs enable operation at elevated temperatures, which can facilitate Na diffusion in the electrode and electrolyte. In this section, some recent electrochemical discoveries and interesting accomplishments obtained by utilising IL electrolytes are presented.4.1 Thermal and electrochemical stability
The use of ILs extends the temperature limits and cut-off voltage range upon electrochemical measurements. This flexibility in testing conditions is advantageous not only for conducting more comprehensive evaluations of well-developed materials but also for identifying the optimal working environments for new materials.Fig. 15 shows the temperature effects on the charge–discharge performances of some positive electrode materials. In this context, Na3V2(PO4)3 and NaCrO2 were evaluated in the wide temperature range −30 to 90 °C using 20 mol% Na[FSA]–[C2C1im][FSA] and Na[FSA]–[C3C1pyrr][FSA] IL electrolytes, respectively, as shown in Fig. 12a and b.164,166 The Na3V2(PO4)3 cell provided stable reversible performance across the whole temperature range and achieved capacities of 100.8, 78.1, and 57.6 mA h g−1 at 25, −10, and −20 °C, respectively (Fig. 15a).164 Charge–discharge testing of the NaCrO2 cell was performed between −20 and 90 °C, and it also exhibited very stable reversible capacities (Fig. 15b). With a decrease in temperature, the polarisation of the cell increases and the reversible capacity decreases as a consequence of the increase in internal cell resistance, which is due to the reduced ionic conductivity of the electrolyte, retarded Na diffusion in the electrode, and higher charge transfer resistance. Nonetheless, IL electrolytes could support a comprehensive operation temperature range for Na secondary batteries (Fig. 5).
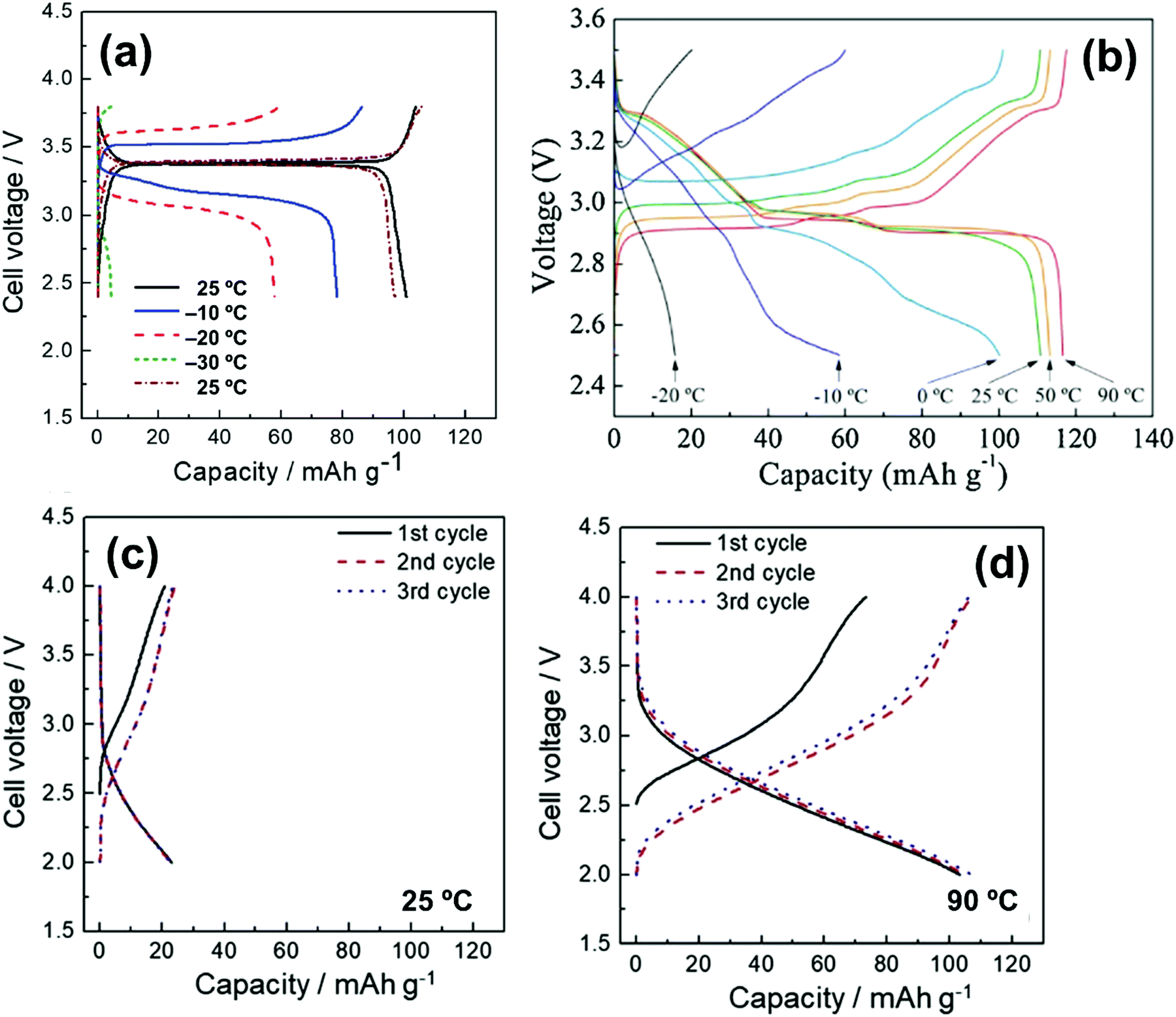 | ||
| Fig. 15 Temperature effects on the charge–discharge behaviours of selected positive electrode materials in ILs. Charge–discharge curves of (a) a Na/20 mol% Na[FSA]–[C2C1im][FSA]/carbon coated Na3V2(PO4)3 cell at −30 to 25 °C. Reproduced with permission.164 Copyright 2018, Wiley-VCH; (b) Na/20 mol% Na[FSA]–[C3C1pyrr][FSA]–NaCrO2 cell at −20 to 90 °C. Reproduced with permission.166 Copyright 2015, The Electrochemical Society of Japan. Charge–discharge curves for a Na/30 mol% Na[FSA]–[C2C1im][FSA]–maricite NaFePO4 cell at (c) 25 °C and (d) 90 °C. Reproduced with permission.103 Copyright 2018, Elsevier. | ||
Research on maricite-NaFePO4 has yielded interesting electrochemical results. As noted earlier, maricite-NaFePO4 is generally considered to be an electrochemically inactive material because of its structural arrangement, which features FeO6–FeO6 (edge-sharing) and PO4–FeO6 (corner-sharing) units that hinder effective diffusion of Na+.288 The elevation of operating temperature possible using IL electrolytes enhanced Na+ diffusion in this structural frame, which effectively allows maricite-NaFePO4 to be electrochemically active. Sodiation/desodiation into/from maricite-NaFePO4 were realised upon elevating the operation temperature.102,103 The charge–discharge of maricite-NaFePO4 in a Na[FSA]–[C2C1im][FSA] IL electrolyte demonstrated a drastic increase in reversible capacity from only 23 mA h g−1 at 25 °C (Fig. 15c) to 107 mA h g−1 at 90 °C (Fig. 15d). The ex situ XRD patterns of desodiated maricite-NaFePO4 continuously shifted to higher 2θ angles, indicating a decrease in d-spacing. Fe K-edge XANES spectra demonstrated the reduction of Fe3+ to Fe2+ by sodiation.103
Fig. 16 illustrates the electrochemical behaviours of a Na4Ni3(PO4)2(P2O7)/C electrode in organic and IL electrolytes. The Ni-based polyanionic compound Na4Ni3(PO4)2(P2O7)/C was expected to exhibit a high operating voltage based on its Ni3+/Ni2+ redox activity. Unfortunately, when a 1 mol dm−3 Na[PF6]-EC/DEC organic solvent electrolyte was used, the cell could only be charged to 4.9 V. The first charge capacity was approximately 97 mA h g−1, which involves 2.3 equivalents of Na based on the capacity during the desodiation process.268 However, only 0.9 equivalents of Na were reversibly sodiated into the composite, corresponding to a coulombic efficiency for the first cycle of about 40% (Fig. 16a). This may indicate electrolyte decomposition at such a high potential. Conversely, this material could be charged up to 5.1 V in 10 mol% Na[TFSA]–[C3C1pyrr][FSA] IL and the composite was able to undergo deeper (de)sodiation; 2.7 and 1.3 equivalents of Na were desodiated and sodiated, respectively, during the first cycle (Fig. 16b), suggesting the possible high stability of the IL at high potentials. The ex situ XRD patterns (Fig. 16c) indicate that neither phase change nor irreversible structural reorganisation occurred during the charge–discharge process, confirming the robustness of this material. Although maricite-NaFePO4 and Na4Ni3(PO4)2(P2O7)/C may appear to be less appealing for practical use owing to the limited achievable capacity, these studies highlight the possibility of using IL electrolytes for electrode studies with new materials.
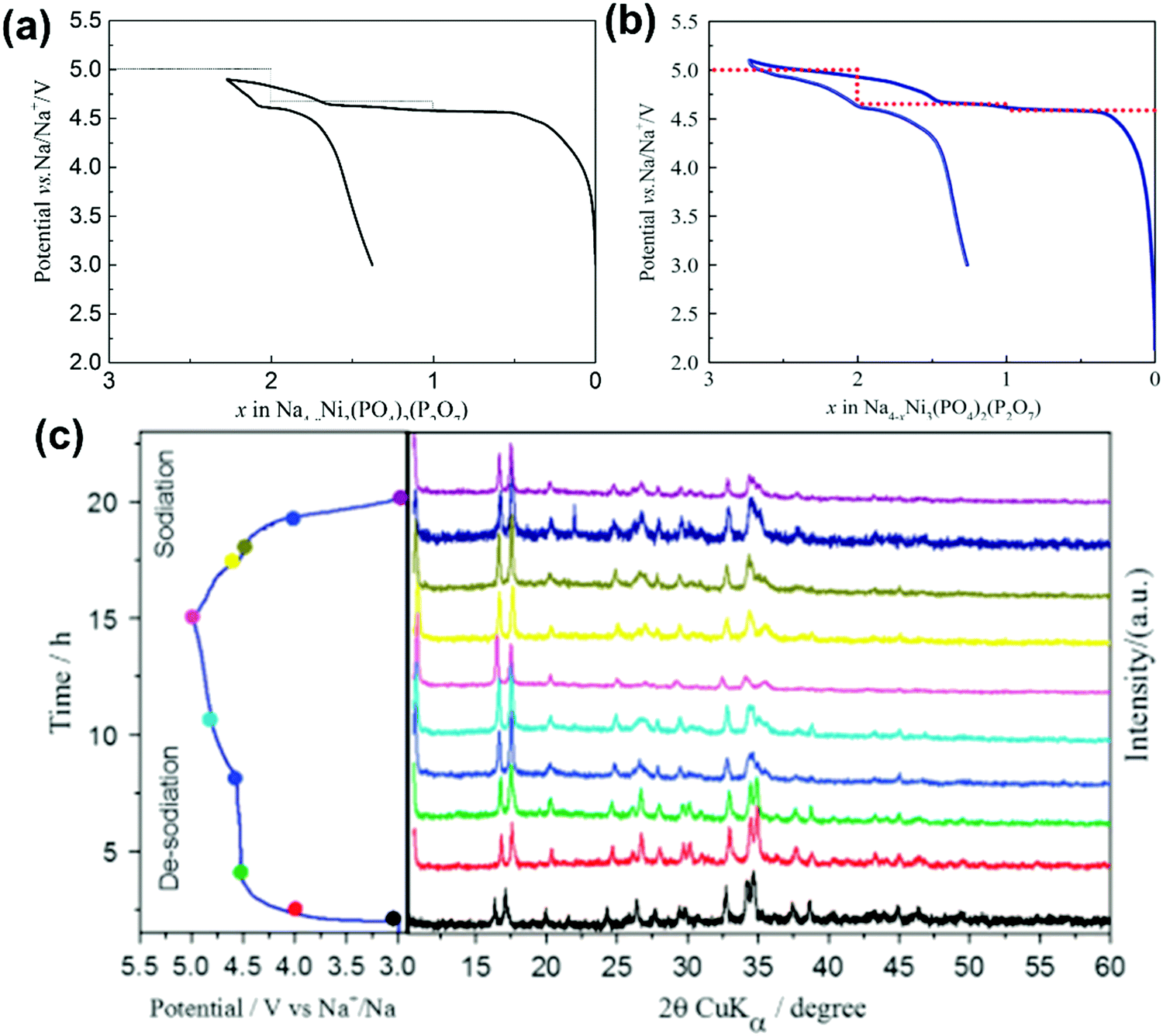 | ||
| Fig. 16 Charge–discharge curves for (a) Na/1 mol dm−3 Na[PF6]-EC/DEC (1:1, w/w)/Na4Ni3(PO4)2(P2O7)/C, cut-off voltages: 3.0–4.9 V, (b) Na/10 mol% Na[TFSA]–[C3C1pyrr][FSA]/Na4Ni3(PO4)2(P2O7)/C, cut-off voltages: 3.0–5.1 V, and (c) ex situ X-ray diffraction patterns for a Na4Ni3(PO4)2(P2O7) electrode during charge–discharge. Reproduced with permission under the Creative Commons Attribution 4.0 International License.268 | ||
4.2 Rate performance and power density
Rate capability and power density are significantly improved by the elevated temperature operation available using ILs owing to the improved interfacial process between electrodes and electrolytes as well as ionic conductivity, as mentioned above. Fig. 17 shows the mass loading and temperature effects on the electrochemical behaviour of Na3V2(PO4)3 in 40 mol% Na[FSA]–[C2C1im][FSA]/carbon.164,283 The synergy of Na3V2(PO4)3 and the IL electrolyte leads to reasonable rate performance at 25 °C. The limited performance in terms of low electronic conductivity at room temperature was easily overcome by carbon coating the electrode material. However, the rate performance dramatically deteriorates at 25 °C as mass loading of the electrode becomes large because the rate performance is very sensitively related to the applied geometric current density (mA cm−2). For example, in the cases of 3.0 mg cm−2 at 10C and 6.0 mg cm−2 at 5C, the capacity retentions are similar (68.3% and 63.2%, respectively, at 25 °C) because the same geometric current density of 3.53 mA cm−2 is applied with doubled mass loading (Fig. 17a). Rate performance is maintained even with a high mass loading of 6 mg cm−2 for Na3V2(PO4)3 at 90 °C (Fig. 17a). The capacity retentions at 90 °C for 3.0 mg cm−2 at 10C and 6.0 mg cm−2 at 5C are 96.1% and 97.3%, respectively.283 Thus, the true rate performance is dependent on geometric current density. Intermediate temperature operation can certainly improve the rate performance, as evidenced in Fig. 17b, which shows that the discharge capacities increase after adjusting the cut-off voltages, giving capacity retentions of 69%, 58%, 36%, and 19% at 500C, 1000C, 1600C, and 2000C, respectively. This level of rate performance could never be achieved by organic solvent electrolytes around room temperature.164 The EIS measurements shown in Fig. 17c reveal that the interfacial resistance, which is indicated by the semicircle (characteristic frequencies of 60–700 Hz), drastically decreases as the operating temperature increases compared to the bulk resistance (Fig. 17c, inset).164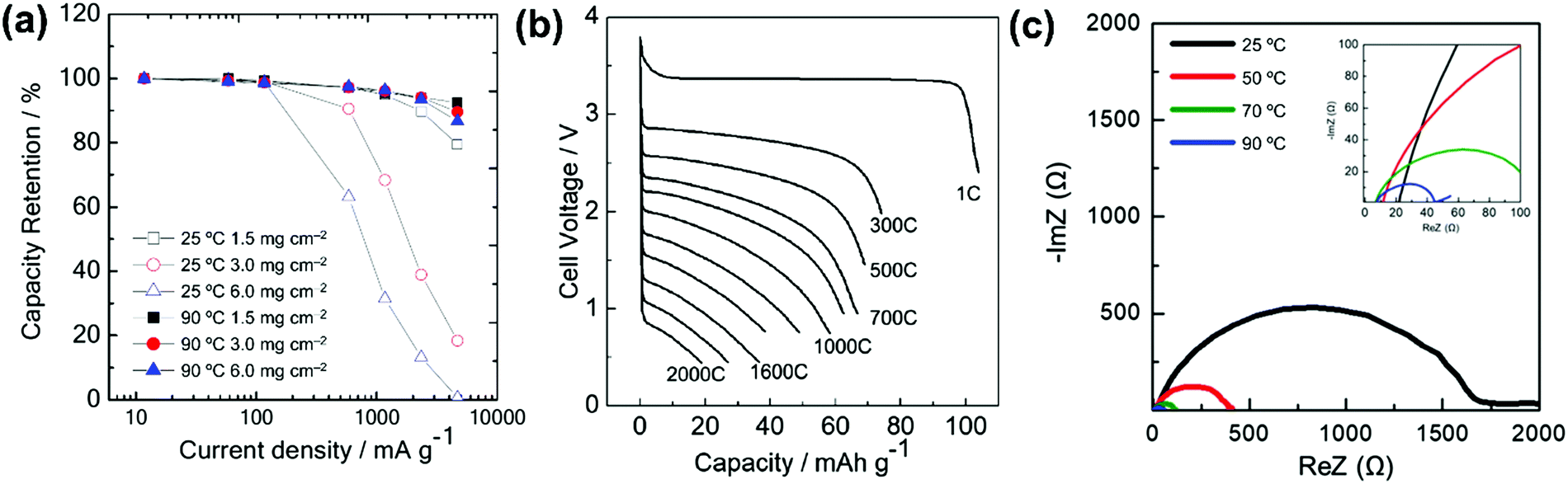 | ||
| Fig. 17 Electrochemical performance of carbon-coated Na3V2(PO4)3 in an IL. (a) Mass loading dependence of rate performance for Na/40 mol% Na[FSA]–[C2C1im][FSA]/carbon-coated Na3V2(PO4)3 cells at 25 and 90 °C. Reproduced with permission.283 Copyright 2019, American Chemical Society. (b) Superfast rate performance test results for a Na/50 mol% Na[FSA]–[C2C1im][FSA]/carbon-coated Na3V2(PO4)3 cell at 90 °C, and (c) electrochemical impedance spectra for Na/50 mol% Na[FSA]–[C2C1im][FSA]/carbon coated Na3V2(PO4)3 cells. Reproduced with permission.164 Copyright 2018, Wiley-VCH. | ||
4.3 Cycle performance and coulombic efficiency
Numerous studies have reported that IL electrolytes exhibit improved cycling performance over a wide temperature range.84,164,180,267,283,286 It is widely acknowledged that IL electrolytes can provide a uniform and robust SEI layer on reactive Na metal (see Section 2.4 for details).Fig. 18 shows the cycle performances of selected electrode materials in IL electrolytes. Cycling tests of a Na/NaFePO4 cell initially confirmed the better cycle performance using a Na[TFSA]–[C4C1pyrr][TFSA] IL (Na[FSA] = 0.1, 0.5, and 1 mol dm−3) electrolyte than that using a 1 mol dm−3 Na[ClO4]-EC/DEC organic solvent electrolyte at 50 °C.84 The capacity retentions after 100 cycles were 62% and 87% with the organic and IL electrolytes, respectively. Furthermore, Na[TFSA]–[C4C1pyrr][TFSA] IL electrolyte containing 0.5 mol dm−3 Na[TFSA] salt gave better cycle performance than that with 0.1 and 1 mol dm−3 Na[TFSA].84 The authors reported that the organic solvent electrolyte at 50 °C presented undesirable electrode–electrolyte interactions and evaporation of the electrolyte, whereas the thermally stable IL electrolyte provided higher Na+ supply as the temperature increased without the above-mentioned drawbacks.84
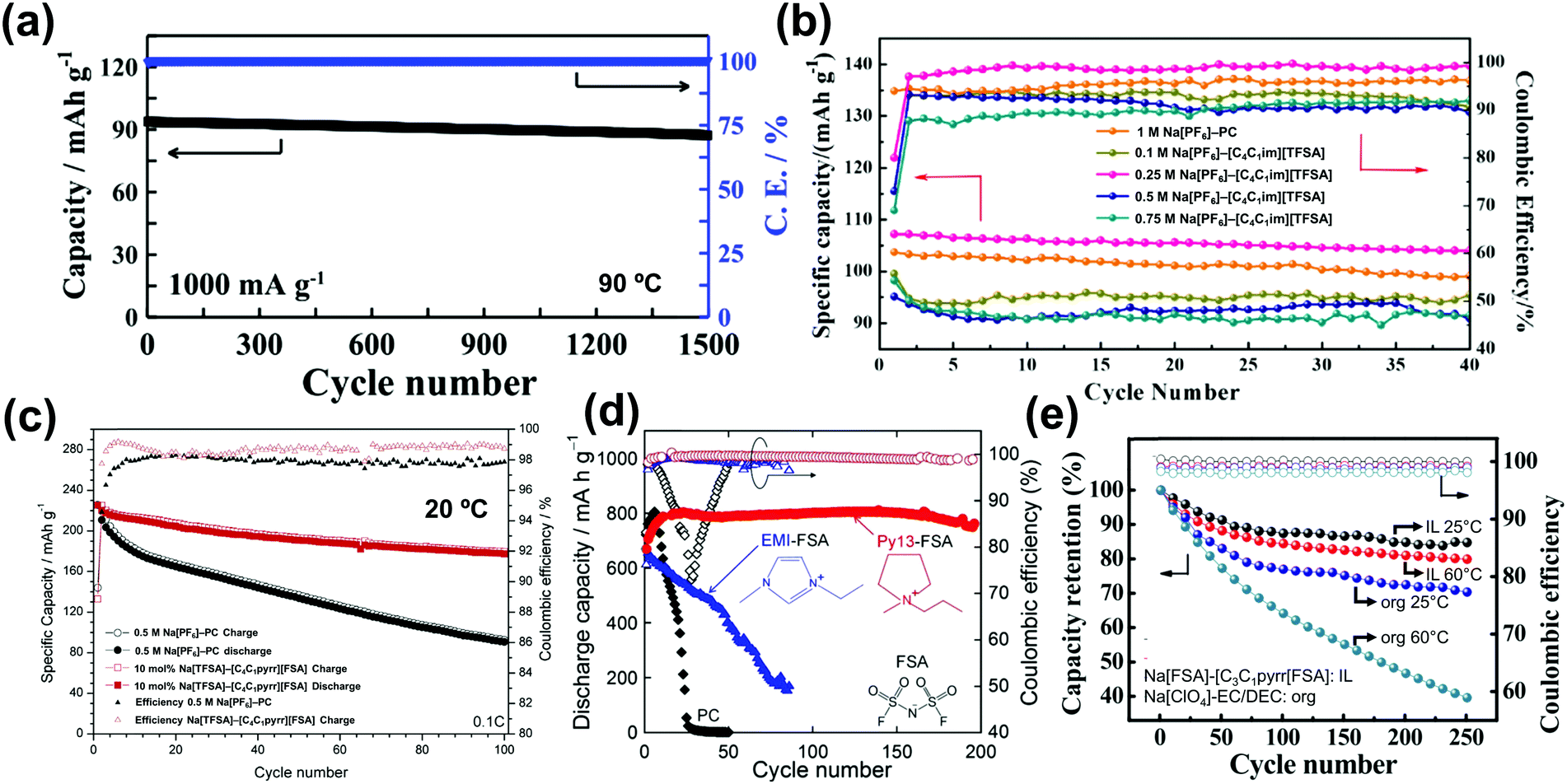 | ||
| Fig. 18 Cycle performances of selected electrode materials in ILs. (a) Cycle performance of a Na/30 mol% Na[FSA]–[C2C1im][FSA]/Na2FeP2O7 cell at 90 °C. Reproduced with permission.180 Copyright 2016, Elsevier. (b) Cycle performances of Na/1 mol dm−3 Na[PF6]-PC/Na3V2(PO4)3 and Na/0.1, 0.25, 0.5, and 0.75 mol dm−3 Na[PF6]–[C2C1im][TFSA]/Na3V2(PO4)3 cells. Reproduced with permission.286 Copyright 2018, Elsevier. (c) Cycle performances of Na/0.5 mol dm−3 Na[PF6]-PC/Na0.45Ni0.22Co0.11Mn0.66O2 and Na/10 mol% Na[TFSA]–[C4C1im][TFSA]/Na0.45Ni0.22Co0.11Mn0.66O2 cells. Reproduced with permission.267 Copyright 2014, Elsevier. (d) Cycling performances and coulombic efficiencies of an Sn4P3 electrode in 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA], 1 mol dm−3 Na[FSA]–[C2C1im][FSA], and 1 mol dm−3 Na[FSA]-PC at 30 °C. Reproduced with permission.395 Copyright 2017, American Chemical Society. (e) Capacity and coulombic efficiency for three-dimensional CF electrodes measured at 100 mA g−1 in 1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v) and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 25 and 60 °C. Reproduced with permission.397 Copyright 2018, Elsevier. | ||
The extended cycle performance of a Na/Na[FSA]–[C2C1im][FSA]/Na2FeP2O7 cell was assessed at 90 °C (Fig. 18a).180 A high capacity retention of 93% was attained for 1500 cycles in this cell with an average coulombic efficiency of 99.9%. Another study using Na3V2(PO4)3 with the same IL electrolyte also revealed very stable cycleability. The capacity retention was 99.9% after 300 cycles at 25 and 90 °C at 1C (117 mA g−1). Furthermore, a faster current cycle test at 20C and an elevated temperature of 90 °C revealed a capacity retention of 89.2%, even after 5000 cycles.164 This extremely good cycle performance was due to the combination of the superior performance of this positive electrode material and stable Na metal deposition–dissolution at the counter electrode in the IL electrolyte at 90 °C (see Fig. 8b for Na metal deposition–dissolution behaviour).246
A study on Na3V2(PO4)3 in Na[PF6]–[C4C1im][TFSA] (Na[PF6] = 0.1, 0.25, 0.5, and 0.75 mol dm−3) IL electrolytes indicated that optimising the salt concentration of the IL electrolyte could improve capacity and cycle performance at the same time.286 As shown in Fig. 18b, 0.25 mol dm−3 Na[PF6]–[C4C1im][TFSA] exhibited a discharge capacity of 107.2 mA h g−1 and retained 104 mA h g−1 after 40 cycles at a current density of 50 mA g−1.286 The discharge capacity and capacity retention with the IL were superior to those with 1 mol dm−3 Na[PF6]-PC. The authors suggested that the IL electrolyte forms a better SEI on Na3V2(PO4)3 than the organic solvent electrolyte and demonstrated that the concentration of Na[PF6] is an important parameter for electrochemical performance.286 It should be noted that the (de)sodiation process can be promoted by supplying a sufficient amount of Na+ while excessive Na+ can negatively affect electrochemical performance owing to the decreased ionic conductivity of the electrolyte.
Another study on Na0.45Ni0.22Co0.11Mn0.66O2 reported that the cycleability with an IL electrolyte was superior to that with an organic solvent electrolyte at 20 °C,267 which clearly indicates that IL electrolytes present more stable cycling behaviour, not only at elevated temperature but also at room temperature, than the organic solvent electrolyte. Cycling of Na0.45Ni0.22Co0.11Mn0.66O2 in 0.5 mol dm−3 Na[PF6]-PC and 10 mol% Na[TFSA][C4C1pyrr][FSA] at 12 mA g−1 also revealed improved cycleability and coulombic efficiency with the IL electrolyte (Fig. 18c).267 The low initial coulombic efficiency was improved after several cycles for both cells. The Na0.45Ni0.22Co0.11Mn0.66O2 cell with the organic solvent electrolyte exhibited a capacity retention of only 40% (approximately 90 mA h g−1) after 100 cycles with an average coulombic efficiency of 97.8%, whereas the cell with the IL electrolyte showed 80% capacity retention (discharge capacity of 177 mA h g−1) after 100 cycles with a coulombic efficiency of 98.7%. The authors claimed that the rapid capacity fading in both electrolytes was due to the irreversibility of the P2–O2 phase transition of Na0.45Ni0.22Co0.11Mn0.66O2 in the organic solvent electrolyte. However, the dissolution of Mn ions in the IL electrolyte is restricted, making the Na0.45Ni0.22Co0.11Mn0.66O2 with IL much more electrochemically reversible and stable.267
With respect to negative electrode materials, Sn4P3 is a special case in which remarkable cycleability (112% for 200 cycles) and high reversible capacity were achieved with 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 30 °C (Fig. 18d).395 The charge–discharge mechanism here was similar to that for InP, and the reason for the better performance in the IL was proposed to be the homogeneity and stability of the SEI layer. Moreover, Sn4P3 exhibits a reversible capacity of 220 mA h g−1 at 0 °C and high rate capability at 90 °C in the IL, which denotes its applicability in a wide temperature range.396 The P-rich phase SnP3 was also investigated using 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] at 30 °C, but severe capacity degradation was observed owing to the insufficient buffering power of SnP3, which is due to its low Sn content.395
The electrochemical performance of three-dimensional CFs prepared by high-speed homogenisation of graphene nanosheets and carbon nanospheres was analysed using 1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v) and 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] IL at 25 and 60 °C (Fig. 18e).397 A capacity retention of 85% was observed after 250 cycles with the IL electrolyte at 25 °C, while only 70% capacity retention was observed with the organic electrolyte at room temperature. At 60 °C, the performance was significantly improved in the IL compared to that in the organic electrolyte. Moreover, the self-discharge in the organic electrolyte (15%) exceeded that in IL (8%) after five days of rest. Robust SEI layer formation owing to the decomposition of FSA− was identified as the main reason for the improved cycleability.
4.4 Solid electrolyte interphase layer
Extensive efforts have been focused on the improvement of negative electrode materials in recent decades, but the investigation of electrode/electrolyte interfacial properties is equally important for ensuring the overall performance and safety of batteries. The SEIs formed by reductive decomposition of conventional ester-based electrolytes are considered not sufficiently stable, leading to severe capacity degradation.398 The success of conventional HC negative electrodes depends on complicated electrolyte formulations and the introduction of SEI forming additives. Thus, for the development of negative electrodes, exploring compatible electrolytes is indispensable.The performance and safety of a battery are highly dependent on SEI layer properties399 which are sensitive to parameters such as the salt concentration of the electrolyte,400 temperature,160,401 and the binder used.402 Investigating the physical and chemical properties of SEI layers is very important for understanding the degradation mechanisms in batteries. However, such a study is often hindered because of the complex structure and nanoscale dimensions of the SEI layer, as well as the limited characterisation tools available. Moreover, owing to the higher reactivity and higher standard redox potential of Na than Li, transferring the knowledge of interfacial properties from LIBs is not always applicable. In addition, the solubility of SEIs in conventional electrolytes for Na secondary batteries aggravates self-discharging properties. Accordingly, self-discharge in Na secondary batteries using conventional 1 mol dm−3 Na[PF6]-EC/DEC has been confirmed, whereas minimal self-discharge has been observed in corresponding LIBs.403 Therefore, it is imperative to investigate the interfacial properties for Na secondary batteries with other types of electrolytes.
Na metal is generally used as the counter electrode in half-cell configurations. However, the electrochemical behaviour of the Na electrode itself is generally not well investigated, causing uncertainty upon interpreting data and identifying the origin of cell degradation.404 The SEI layer formed on Na metal in ester-based EC/DEC and ether-based diglyme and tetraglyme with 1 mol dm−3 Na[PF6] was investigated by XPS, and formation of a thin and stable SEI was observed in the diglyme case,405 whereas a thick and highly resistive film was formed in EC/DEC. In-depth XPS studies identified the formation of a uniform and solvent-impermeable layer consisting of inorganic components in glyme-based electrolytes containing Na2O and NaF, contrasting with the results for the mixed organic (RCH2OCO2Na) and inorganic SEI formed in EC/DEC. It was found that a mixed organic–inorganic SEI is prone to solvent permeation, resulting in dendrite growth.
Recently, the electrochemical properties of Na metal were studied in IL electrolytes (see Section 2.4 for details). Typically, a large interfacial resistance, presumably resulting from the surface film on the Na metal, is observed in the IL electrolytes.86,124,164 Such impedance can hinder the evaluation of the true performance of the target electrode in half-cell configurations. A systematic study on Na/Na symmetric cells using ILs showed that the overall cell resistance dramatically decreases with increasing operating temperature but increases with the Na salt content of the electrolyte.164 The effects of electrolyte on the composition of the SEI layer formed on HC were extensively investigated by Eshetu et al.406 The anion of the electrolyte salt was reported to govern the characteristics of the SEI formed on HC. The organic content decreases in the order Na[PF6] > Na[ClO4] > Na[TFSA] > Na[FTFSA] > Na[FSA]. The effect of electrolyte on the thermal stability of the SEI and corrosion of the Al current collector were also studied by the same group. For a fixed solvent (EC/DMC), the onset temperature of the first exothermic peak (related to SEI cracking) follows the sequence Na[ClO4] < Na[PF6] < Na[TFSA] ≈ Na[FTFSA] < Na[FSA].407 For the corrosion of the Al current collector, the Al dissolution or anion decomposition increases in the order Na[PF6] < Na[ClO4] < Na[TFSA] < Na[FTFSA] < Na[FSA] in EC/DEC. However, Na[FSA] showed stability towards decomposition in the different solvents in the order EC/DEC < EC/DEC + 5% NaPF6 < [C4C1pyrr][FSA]. This result shows that ILs impart the highest thermal stability to the SEI and suppress Al corrosion.
IL-derived SEIs can be distinctly different from those formed in organic electrolytes. Accordingly, the performances and interfacial properties of HC in a 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] IL and a conventional organic electrolyte (1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v)) were compared at 25 °C. The SEIs formed in the HC/HC symmetric cell showed a lower charge transfer resistance in the IL than that in the organic electrolyte. A dense SEI enriched with Na2CO3 and NaCO2R species was confirmed to form in the organic electrolyte by scanning electron microscopy (SEM) and XPS measurements. These species are considered to be the resistive components in SEI layers.397 Similar species were also observed using 1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v) in a three-dimensional carbon framework (CF), whereas polyolefin ((CH2)n) and S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO derived from FSA anions were found in the SEI layer formed in the IL.397
CO derived from FSA anions were found in the SEI layer formed in the IL.397
For alloy-based electrodes, a robust SEI layer is widely acknowledged as a crucial factor for realising good cycleability as these materials undergo significant volume change during sodiation/desodiation. Fig. 19 shows the compositional evolution of SEIs on red phosphorus cycled in organic electrolytes (Fig. 19a) and an IL (Fig. 19b) as revealed by a combination of hard X-ray photoelectron spectroscopy (HAXPES) and time-of-flight secondary ion mass spectroscopy (TOF-SIMS).80 The TOF-SIMS analysis shows that the SEI layer generated in the IL electrolyte is composed of a uniform mixture of organic and inorganic components (with the inner surface primarily covered by inorganic species), whereas mainly inorganic species are observed in the organic electrolyte formed under the same conditions. Moreover, it can be clearly seen that the SEI composition dynamically changes with cycle number in the organic electrolyte, while the composition remains stable in the IL electrolyte. The homogeneous and complete coverage of the SEI layer on red phosphorus was considered to be the reason behind the improved reversible capacity and capacity retention in the IL electrolyte.
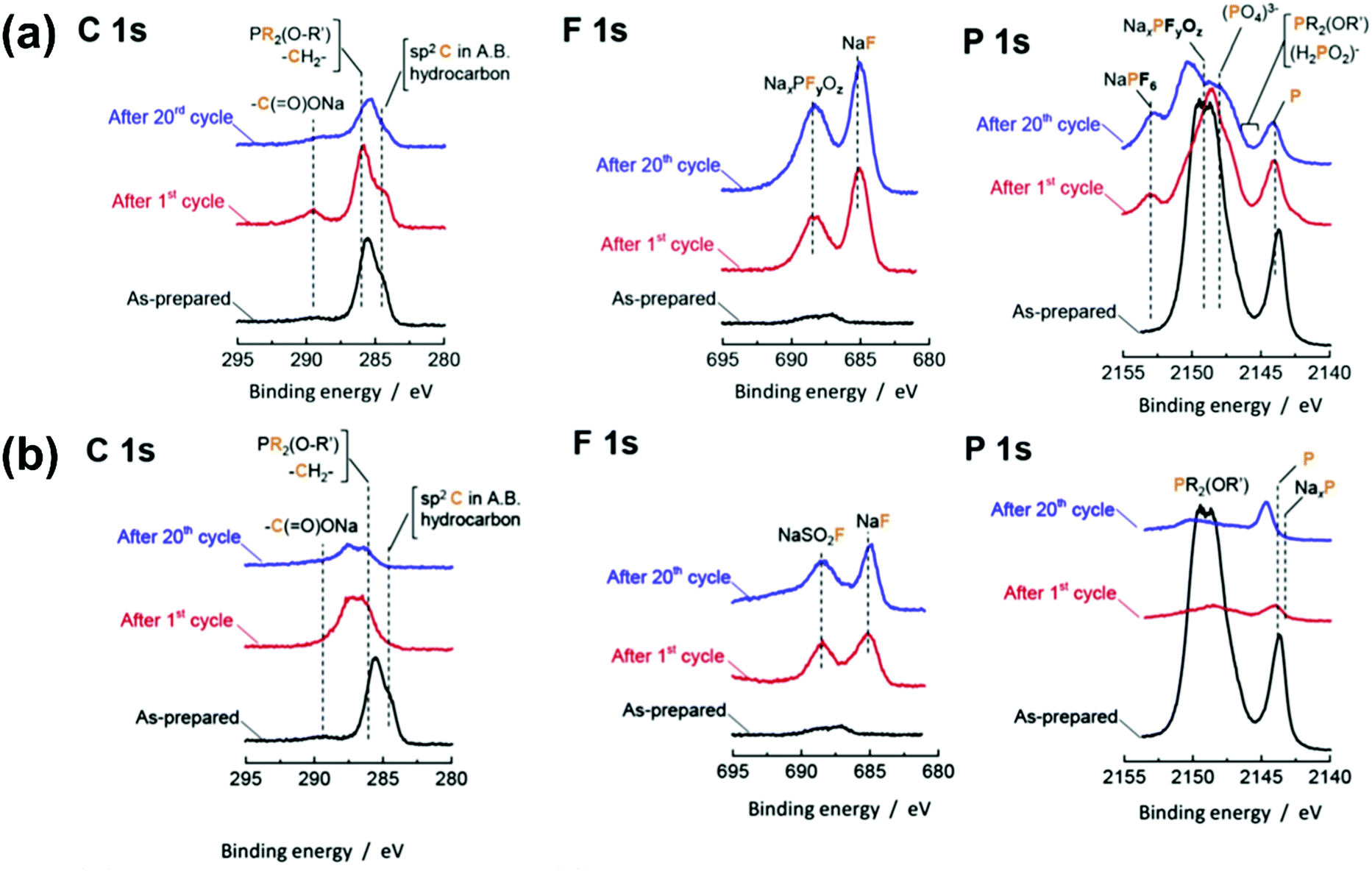 | ||
| Fig. 19 Hard X-ray photoelectron spectra of C1s, F1s, and P1s regions for P/AB/PANa electrodes (as-prepared (black), after first (red), and 20th (blue) cycles) in (a) 1.0 mol dm−3 Na[PF6]-EC/DEC and (b) 0.25 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] electrolytes. Reproduced with permission.80 Copyright 2017, Elsevier. | ||
4.5 Full cell performance
Despite the fact that full cell studies are required to confirm the actual feasibility of Na secondary batteries using IL electrolytes, they are still in their infancy. There are several reports of full cell tests with ILs using coin-cells86,128,380,408–410 and one report for a prismatic cell.411Although full cell tests were carried out using coin-cells, it is worthy of mention that the cycle performance was notably improved using IL electrolytes. A full cell with Na3V2(PO4)3 and HC positive and negative electrodes in 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] achieved the high energy density of 368 W h kg−1 with good capacity retention of 75% after 100 cycles, while the test using a 1 mol dm−3 Na[ClO4]-EC/PC (1:1, v/v) organic electrolyte exhibited a capacity retention of 57% after 100 cycles at room temperature.408 Another study on the combination of Na0.44MnO2 positive and HC negative electrode materials in a 1 mol dm−3 Na[FSA]–[C3C1pyrr][FSA] IL electrolyte achieved a capacity retention of 97% after 100 cycles, which is better than that in a 1 mol dm−3 Na[ClO4]-EC/DEC (1:1, v/v) organic solvent electrolyte, which achieved a capacity retention of 61% after 100 cycles at 25 °C.86 The authors demonstrated that the SEI components generated from the organic solvent electrolyte hinder Na+ transport and desolvation of Na+.86,412 Superior safety was confirmed using a 0.5 mol dm−3 Na[FSA]–[C4C1pyrr][TFSA] IL electrolyte compared to that with a 1 mol dm−3 Na[ClO4]-EC/PC (1:1, w/w) using accelerating rate calorimetry analysis for a full cell with Na0.6Co0.1Mn0.9O2 positive and Na2.55V6O16 negative electrodes. This analysis indicated that this full cell was stable up to 250 °C. In addition, the performance of the full cell with an IL electrolyte was superior to that with an organic electrolyte at room temperature.410
A full cell test with organic electrode materials showed a very interesting electrochemical result. Calix[4]quinone was used as the positive electrode material and disodium terephthalate was used as a negative electrode material. This full cell was prepared in a mass ratio of 1:2.5 (calix[4]quinone:disodium terephthalate) with 0.3 mol dm−3 Na[TFSA]–[C3C1pyrr][TFSA] in a 2032-type coin cell, and stable cycleability was observed compared to that achieved with 0.3 mol dm−3 Na[TFSA]–DME.380 This is because quinone compounds easily dissolve in organic electrolytes but not in ILs.
A large-sized prismatic full cell was fabricated using a NaCrO2 positive electrode and a HC negative electrode with a 20 mol% Na[FSA]–[C3C1pyrr][FSA] electrolyte.411 This prismatic full cell was designed to have a capacity of 27000 mA h with a gravimetric energy density of 75 W h kg−1 and a volumetric energy density of 125 W h dm−3. The discharge capacity was 27300 mA h at 2700 mA at 25 and 60 °C, which is equal to the designed capacity, with high coulombic and energy efficiencies of 99.8% and 93.2%, respectively, at 25 °C and 99.8% and 96.9%, respectively, at 60 °C. Furthermore, excellent cycleability was achieved at 60 °C, with a capacity retention of 87% after 500 cycles at 10000 mA. This large-sized full cell is comparable with prismatic lithium-ion batteries developed in the early 2000s in terms of energy density, power density (rate capability), energy efficiency, and cycleability.413,414
5. Conclusions and perspectives
This article comprehensively reviewed the use of ILs as electrolytes in Na secondary batteries in view of their large-scale applications in devices such as electric vehicles and EESs. Although Na secondary batteries have a long history, their rapid progress over the last decade has revealed the potential of a variety of electrode and electrolyte materials including ILs. Research in this field has certainly advanced by exploiting the knowledge base of Li analogues, but several studies have confirmed different chemistries that are unique to Na secondary batteries.There are a number of advantages for ILs over organic electrolytes. In particular, their non-volatility and wide liquid-phase temperature range are very attractive for large-scale and task-specific applications. Further research is required to identify ideal ILs and develop their formulation in terms of meeting growing demands for suitable potential windows, fast ionic conduction, and workable temperature extremes. Ion and solvation structures, Na salt form and concentration, cell configuration, and operating protocols have to be further optimised to derive optimal performances for both the positive and negative electrodes simultaneously. The merits of FSA-based ILs are remarkable: allowing the formation of robust SEIs, relatively high ionic conductivity, and great thermal stability.
New anionic species with similar or superior functionalities are an interesting target for future studies. Although not fully considered in this review, IL gel polymer electrolytes prepared by incorporating ILs into polymer hosts may be used for constructing safe all-solid-state Na secondary batteries owing to their excellent mechanical strengths and the low probability of leakage.415–418 Moreover, deep eutectic solvents (DESs) and some complex-salt based electrolytes that behave like ILs could be a cheaply obtainable alternative.149,419–421 The mixing of water or organic solvents with ILs to a certain extent is also a method to ameliorate the high viscosity of ILs without sacrificing their safety.168,238 Such ideas should be intensively explored for practical applications. Recent advances using ILs and their analogues as electrode pretreatment agents,422 reaction media for electrode material synthesis, and for the recycling of spent batteries423,424 show the multiple roles of ILs in energy-related fields and that many exciting opportunities are waiting to be discovered.
Electrode materials without expensive minor metals are required for Na secondary batteries because large-scale applications are envisioned. In this sense, Fe-based positive electrode materials are preferable, but Mn- and V-based compounds are also good candidates. Electrode design for IL electrolytes can be different to that for organic electrolytes, as the performance of the electrode material in ILs is better than that in organic electrolytes in some cases and worse in other cases. Various factors can affect such behaviour, but one certainty is the difficulty in evaluating true performance in a half-cell configuration because of the large polarisation at the Na counter electrode. This is an intrinsic problem for research into Na secondary batteries and a sophisticated solution is necessary for reliable and efficient screening of electrode materials.
In addition to the influence of the counter electrode, polarisation caused by the lower ionic conductivity of ILs and thus underestimated capacity and inferior rate capability are also often observed with incautious use of ILs. These issues can be overcome via modifying electrode–electrolyte interfacial properties by operation at elevated temperatures, which is a practically viable feature of thermally stable ILs that allows electrode materials to deliver greatly improved performance as a consequence of better SEI formation at both negative and positive electrodes, favourable ion transport, and stable Na metal deposition–dissolution without dendrite growth. Particularly, relatively stable cycleability over thousands of cycles has readily been attained for various materials in half-cell using ILs at elevated temperatures, confirming that these beneficial effects are universal regardless of electrode chemistry.
The high cost of ILs is another issue for the construction of practical cells. Undoubtedly, ILs show promise for batteries that operate under harsh environmental conditions or that must withstand widely dissimilar temperatures upon charge and discharge. Other specific applications for which the unique advantages of ILs could be used must be explored in more depth in order to make them truly competitive, even though the price of ILs may become more affordable in mass production than that today.
Furthermore, improved academic understanding is still required for the development of Na secondary batteries, including those made with ILs. The bulk properties of electrolytes, interfacial properties between the electrode materials and the IL, and the effects of binders and conductive additives are all valid targets of fundamental research. Thus, further systematic studies should be undertaken along with those concerning commercial opportunities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partly supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) program “Elements Strategy Initiative to Form Core Research Center”.Notes and references
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- R. C. Armstrong, C. Wolfram, K. P. de Jong, R. Gross, N. S. Lewis, B. Boardman, A. J. Ragauskas, K. Ehrhardt-Martinez, G. Crabtree and M. V. Ramana, Nat. Energy, 2016, 1, 15020 CrossRef.
- O. Schmidt, A. Hawkes, A. Gambhir and I. Staffell, Nat. Energy, 2017, 2, 17110 CrossRef.
- D. Andre, S.-J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- B. Nykvist and M. Nilsson, Nat. Clim. Change, 2015, 5, 329 CrossRef.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2014, 7, 19 CrossRef PubMed.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- C. Helbig, A. M. Bradshaw, L. Wietschel, A. Thorenz and A. Tuma, J. Clean. Prod., 2018, 172, 274–286 CrossRef CAS.
- C. Delmas, Adv. Energy. Mater., 2018, 8, 1703137 CrossRef.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy. Mater., 2012, 2, 710–721 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy. Mater., 2018, 8, 1800079 CrossRef.
- H. Kim, J. C. Kim, M. Bianchini, D.-H. Seo, J. Rodriguez-Garcia and G. Ceder, Adv. Energy. Mater., 2018, 8, 1702384 CrossRef.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Chem. Rev., 2017, 117, 4287–4341 CrossRef CAS PubMed.
- R. J. Gummow, G. Vamvounis, M. B. Kannan and Y. He, Adv. Mater., 2018, 30, 1801702 CrossRef PubMed.
- Y. Zhang, S. Liu, Y. Ji, J. Ma and H. Yu, Adv. Mater., 2018, 30, 1706310 CrossRef PubMed.
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CrossRef CAS PubMed.
- R. Cao, K. Mishra, X. Li, J. Qian, M. H. Engelhard, M. E. Bowden, K. S. Han, K. T. Mueller, W. A. Henderson and J.-G. Zhang, Nano Energy, 2016, 30, 825–830 CrossRef CAS.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- Y. Zhao, K. R. Adair and X. Sun, Energy Environ. Sci., 2018, 11, 2673–2695 RSC.
- J. Rouxel, L. Trichet, P. Chevalier, P. Colombet and O. A. Ghaloun, J. Solid State Chem., 1979, 29, 311–321 CrossRef CAS.
- S.-B. Son, T. Gao, S. P. Harvey, K. X. Steirer, A. Stokes, A. Norman, C. Wang, A. Cresce, K. Xu and C. Ban, Nat. Chem., 2018, 10, 532–539 CrossRef CAS PubMed.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324 CrossRef CAS PubMed.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- R. S. Carmichael, Practical Handbook of Physical Properties of Rocks and Minerals, CRC Press, Boca Raton, FL, 1989 Search PubMed.
- J. L. Murray, Bull. Alloy Phase Diagrams, 1983, 4, 407–410 CrossRef.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- M. Okoshi, Y. Yamada, A. Yamada and H. Nakai, J. Electrochem. Soc., 2013, 160, A2160–A2165 CrossRef CAS.
- A. Leblanc-Soreau, M. Danot, L. Trichet and J. Rouxel, Mater. Res. Bull., 1974, 9, 191–197 CrossRef.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734–749 RSC.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- J. Deng, W.-B. Luo, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy. Mater., 2018, 8, 1701428 CrossRef.
- J. Song, B. Xiao, Y. Lin, K. Xu and X. Li, Adv. Energy. Mater., 2018, 8, 1703082 CrossRef.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X.-Z. Liao and Z.-F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- Y. Huang, L. Zhao, L. Li, M. Xie, F. Wu and R. Chen, Adv. Mater., 2019, 31, 1808393 CrossRef PubMed.
- Y. Sun, P. Shi, H. Xiang, X. Liang and Y. Yu, Small, 2019, 15, 1805479 CrossRef PubMed.
- C. Bommier and X. Ji, Small, 2018, 14, 1703576 CrossRef PubMed.
- G. Yan, S. Mariyappan, G. Rousse, Q. Jacquet, M. Deschamps, R. David, B. Mirvaux, J. W. Freeland and J.-M. Tarascon, Nat. Commun., 2019, 10, 585 CrossRef PubMed.
- M. Chen, W. Hua, J. Xiao, D. Cortie, W. Chen, E. Wang, Z. Hu, Q. Gu, X. Wang, S. Indris, S.-L. Chou and S.-X. Dou, Nat. Commun., 2019, 10, 1480 CrossRef PubMed.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS.
- P. Barpanda, G. Oyama, S.-i. Nishimura, S.-C. Chung and A. Yamada, Nat. Commun., 2014, 5, 4358 CrossRef CAS PubMed.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- M. Winter, Z. Phys. Chem., 2009, 223, 1395 CrossRef CAS.
- M. Moshkovich, Y. Gofer and D. Aurbach, J. Electrochem. Soc., 2001, 148, E155–E167 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- F. A. Soto, P. Yan, M. H. Engelhard, A. Marzouk, C. Wang, G. Xu, Z. Chen, K. Amine, J. Liu, V. L. Sprenkle, F. El-Mellouhi, P. B. Balbuena and X. Li, Adv. Mater., 2017, 29, 1606860 CrossRef PubMed.
- R. Dugas, B. Zhang, P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A867–A874 CrossRef CAS.
- H. Yildirim, A. Kinaci, M. K. Y. Chan and J. P. Greeley, ACS Appl. Mater. Interfaces, 2015, 7, 18985–18996 CrossRef CAS PubMed.
- F. A. Soto, A. Marzouk, F. El-Mellouhi and P. B. Balbuena, Chem. Mater., 2018, 30, 3315–3322 CrossRef CAS.
- M.-T. F. Rodrigues, G. Babu, H. Gullapalli, K. Kalaga, F. N. Sayed, K. Kato, J. Joyner and P. M. Ajayan, Nat. Energy, 2017, 2, 17108 CrossRef CAS.
- X. Lin, M. Salari, L. M. R. Arava, P. M. Ajayan and M. W. Grinstaff, Chem. Soc. Rev., 2016, 45, 5848–5887 RSC.
- W. M. Seong, K.-Y. Park, M. H. Lee, S. Moon, K. Oh, H. Park, S. Lee and K. Kang, Energy Environ. Sci., 2018, 11, 970–978 RSC.
- X. Feng, M. Fang, X. He, M. Ouyang, L. Lu, H. Wang and M. Zhang, J. Power Sources, 2014, 255, 294–301 CrossRef CAS.
- N. Williard, W. He, C. Hendricks and M. Pecht, Energies, 2013, 6, 4682–4695 CrossRef.
- D. Linden and T. B. Reddy, Linden's Handbook of Batteries, McGraw-Hill, 4th edn, 2010 Search PubMed.
- C. Zhao, L. Liu, X. Qi, Y. Lu, F. Wu, J. Zhao, Y. Yu, Y.-S. Hu and L. Chen, Adv. Energy. Mater., 2018, 8, 1703012 CrossRef.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS PubMed.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- M.-T. F. Rodrigues, K. Kalaga, H. Gullapalli, G. Babu, A. L. M. Reddy and P. M. Ajayan, Adv. Energy. Mater., 2016, 6, 1600218 CrossRef.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- X. Zheng, C. Bommier, W. Luo, L. Jiang, Y. Hao and Y. Huang, Energy Storage Mater., 2019, 16, 6–23 CrossRef.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS PubMed.
- M. Forsyth, H. Yoon, F. Chen, H. Zhu, D. R. MacFarlane, M. Armand and P. C. Howlett, J. Phys. Chem. C, 2016, 120, 4276–4286 CrossRef CAS.
- M. Dahbi, M. Fukunishi, T. Horiba, N. Yabuuchi, S. Yasuno and S. Komaba, J. Power Sources, 2017, 363, 404–412 CrossRef CAS.
- H. Usui, Y. Domi, S. Ohshima and H. Sakaguchi, Electrochim. Acta, 2017, 246, 280–284 CrossRef CAS.
- L. Otaegui, E. Goikolea, F. Aguesse, M. Armand, T. Rojo and G. Singh, J. Power Sources, 2015, 297, 168–173 CrossRef CAS.
- C. Liu, X. Ma, F. Xu, L. Zheng, H. Zhang, W. Feng, X. Huang, M. Armand, J. Nie, H. Chen and Z. Zhou, Electrochim. Acta, 2014, 149, 370–385 CrossRef CAS.
- N. Wongittharom, T.-C. Lee, C.-H. Wang, Y.-C. Wang and J.-K. Chang, J. Mater. Chem. A, 2014, 2, 5655–5661 RSC.
- H. Yoon, P. C. Howlett, A. S. Best, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2013, 160, A1629–A1637 CrossRef CAS.
- C.-H. Wang, C.-H. Yang and J.-K. Chang, Chem. Commun., 2016, 52, 10890–10893 RSC.
- L. S. Plashnitsa, E. Kobayashi, Y. Noguchi, S. Okada and J.-I. Yamaki, J. Electrochem. Soc., 2010, 157, A536–A543 CrossRef CAS.
- B. R. Long, S. G. Rinaldo, K. G. Gallagher, D. W. Dees, S. E. Trask, B. J. Polzin, A. N. Jansen, D. P. Abraham, I. Bloom, J. Bareño and J. R. Croy, J. Electrochem. Soc., 2016, 163, A2999–A3009 CrossRef CAS.
- J. Betz, G. Bieker, P. Meister, T. Placke, M. Winter and R. Schmuch, Adv. Energy. Mater., 2019, 9, 1803170 CrossRef.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- G. T. Kim, S. S. Jeong, M. Joost, E. Rocca, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2011, 196, 2187–2194 CrossRef CAS.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- Q. Yang, Z. Zhang, X.-G. Sun, Y.-S. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- A. Basile, M. Hilder, F. Makhlooghiazad, C. Pozo-Gonzalo, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Adv. Energy Mater., 2018, 8, 1703491 CrossRef.
- T. Torimoto, T. Tsuda, K.-i. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed.
- T. Tsuda, G. R. Stafford and C. L. Hussey, J. Electrochem. Soc., 2017, 164, H5007–H5017 CrossRef CAS.
- T. Tsuda, C. Y. Chen and C. L. Hussey, Novel analytical techniques for smart ionic liquid materials, Royal Society of Chemistry, Cambridge, 2017, pp. 1–29 Search PubMed.
- R. Hagiwara, K. Matsumoto, J. Hwang and T. Nohira, Chem. Rec., 2019, 19, 758–770 CrossRef CAS PubMed.
- J. Hwang, K. Matsumoto, T. Nohira and R. Hagiwara, Electrochemistry, 2017, 85, 675–679 CrossRef CAS.
- J. Hwang, K. Matsumoto, Y. Orikasa, M. Katayama, Y. Inada, T. Nohira and R. Hagiwara, J. Power Sources, 2018, 377, 80–86 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- R. Hagiwara and Y. Ito, J. Fluorine Chem., 2000, 105, 221–227 CrossRef CAS.
- H. Xue, R. Verma and J. n. M. Shreeve, J. Fluorine Chem., 2006, 127, 159–176 CrossRef CAS.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, John Wiley & Sons Inc., Hoboken, NJ, 2nd edn, 2011 Search PubMed.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. Silva Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- C. D. Hubbard, P. Illner and R. van Eldik, Chem. Soc. Rev., 2011, 40, 272–290 RSC.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- K. Matsumoto and R. Hagiwara, J. Fluorine Chem., 2007, 128, 317–331 CrossRef CAS.
- R. Hagiwara and J. S. Lee, Electrochemistry, 2007, 75, 23–34 CrossRef CAS.
- G. B. Appetecchi, M. Montanino and S. Passerini, Ionic Liquids: Science and Applications, American Chemical Society, 2012, ch. 4, vol. 1117, pp. 67–128 Search PubMed.
- M. Forsyth, G. M. A. Girard, A. Basile, M. Hilder, D. R. MacFarlane, F. Chen and P. C. Howlett, Electrochim. Acta, 2016, 220, 609–617 CrossRef CAS.
- K. Matsumoto, Y. Okamoto, T. Nohira and R. Hagiwara, J. Phys. Chem. C, 2015, 119, 7648–7655 CrossRef CAS.
- D. Monti, E. Jonsson, M. R. Palacin and P. Johansson, J. Power Sources, 2014, 245, 630–636 CrossRef CAS.
- F. Wu, N. Zhu, Y. Bai, L. Liu, H. Zhou and C. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 21381–21386 CrossRef CAS PubMed.
- N. Wongittharom, C.-H. Wang, Y.-C. Wang, C.-H. Yang and J.-K. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 17564–17570 CrossRef CAS PubMed.
- C. R. Pope, M. Kar, D. R. MacFarlane, M. Armand, M. Forsyth and L. A. O'Dell, ChemPhysChem, 2016, 17, 3187–3195 CrossRef CAS.
- T. Hosokawa, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai and K. Nitta, J. Phys. Chem. C, 2016, 120, 9628–9636 CrossRef CAS.
- K. Matsumoto, R. Taniki, T. Nohira and R. Hagiwara, J. Electrochem. Soc., 2015, 162, A1409–A1414 CrossRef CAS.
- M. Hilder, M. Gras, C. R. Pope, M. Kar, D. R. MacFarlane, M. Forsyth and L. A. O’Dell, Phys. Chem. Chem. Phys., 2017, 19, 17461–17468 RSC.
- K. Matsumoto, T. Hosokawa, T. Nohira, R. Hagiwara, A. Fukunaga, K. Numata, E. Itani, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2014, 265, 36–39 CrossRef CAS.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, A. Basile, M. Armand, T. Rojo, M. Kar, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2018, 268, 94–100 CrossRef CAS.
- F. Makhlooghiazad, R. Yunis, D. Mecerreyes, M. Armand, P. C. Howlett and M. Forsyth, Solid State Ionics, 2017, 312, 44–52 CrossRef CAS.
- C.-H. Wang, Y.-W. Yeh, N. Wongittharom, Y.-C. Wang, C.-J. Tseng, S.-W. Lee, W.-S. Chang and J.-K. Chang, J. Power Sources, 2015, 274, 1016–1023 CrossRef CAS.
- T. Vogl, C. Vaalma, D. Buchholz, M. Secchiaroli, R. Marassi, S. Passerini and A. Balducci, J. Mater. Chem. A, 2016, 4, 10472–10478 RSC.
- S. A. M. Noor, N. C. Su, L. T. Khoon, N. S. Mohamed, A. Ahmad, M. Z. A. Yahya, H. Zhu, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2017, 247, 983–993 CrossRef CAS.
- H. Yoon, H. Zhu, A. Hervault, M. Armand, D. R. MacFarlane and M. Forsyth, Phys. Chem. Chem. Phys., 2014, 16, 12350–12355 RSC.
- P. J. Fischer, M. P. Do, R. M. Reich, A. Nagasubramanian, M. Srinivasan and F. E. Kuehn, Phys. Chem. Chem. Phys., 2018, 20, 29412–29422 RSC.
- S. A. Mohd Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef CAS.
- S. Brutti, M. A. Navarra, G. Maresca, S. Panero, J. Manzi, E. Simonetti and G. B. Appetecchi, Electrochim. Acta, 2019, 306, 317–326 CrossRef CAS.
- J. Serra Moreno, G. Maresca, S. Panero, B. Scrosati and G. B. Appetecchi, Electrochem. Commun., 2014, 43, 1–4 CrossRef CAS.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, M. Armand, T. Rojo, D. R. MacFarlane and M. Forsyth, J. Power Sources, 2017, 349, 45–51 CrossRef CAS.
- J. Fuller, R. T. Carlin, H. C. De Long and D. Haworth, J. Chem. Soc., Chem. Commun., 1994, 299–300 RSC.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965–967 RSC.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko and M. Kono, J. Power Sources, 2006, 160, 1308–1313 CrossRef CAS.
- H. Zhang, W. Feng, J. Nie and Z. Zhou, J. Fluorine Chem., 2015, 174, 49–61 CrossRef CAS.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Phys. Chem. Chem. Phys., 2018, 20, 8382–8402 RSC.
- R. Hagiwara, K. Tamaki, K. Kubota, T. Goto and T. Nohira, J. Chem. Eng. Data, 2008, 53, 355–358 CrossRef CAS.
- K. Kubota, T. Nohira and R. Hagiwara, J. Chem. Eng. Data, 2010, 55, 3142–3146 CrossRef CAS.
- K. Kubota, T. Nohira and R. Hagiwara, Electrochim. Acta, 2012, 66, 320–324 CrossRef CAS.
- T. Mandai, K. Dokko and M. Watanabe, Chem. Rec., 2019, 19, 708–722 CrossRef CAS.
- W. A. Henderson, J. Phys. Chem. B, 2006, 110, 13177–13183 CrossRef CAS PubMed.
- W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young and R. Frech, Chem. Mater., 2005, 17, 2284–2289 CrossRef CAS.
- T. Mandai, K. Yoshida, S. Tsuzuki, R. Nozawa, H. Masu, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2015, 119, 1523–1534 CrossRef CAS PubMed.
- S. Terada, T. Mandai, R. Nozawa, K. Yoshida, K. Ueno, S. Tsuzuki, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 11737–11746 RSC.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- H. Sakaebe, H. Matsumoto and K. Tatsumi, Electrochim. Acta, 2007, 53, 1048–1054 CrossRef CAS.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacín, Energy Environ. Sci., 2012, 5, 8572–8583 RSC.
- J. Zhao, L. Zhao, K. Chihara, S. Okada, J.-i. Yamaki, S. Matsumoto, S. Kuze and K. Nakane, J. Power Sources, 2013, 244, 752–757 CrossRef CAS.
- X. Xia, M. N. Obrovac and J. R. Dahn, Electrochem. Solid State Lett., 2011, 14, A130–A133 CrossRef CAS.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A515–A519 CrossRef CAS.
- Y. Lee, H. Lim, S.-O. Kim, H.-S. Kim, K. J. Kim, K.-Y. Lee and W. Choi, J. Mater. Chem. A, 2018, 6, 20383–20392 RSC.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A1048–A1051 CrossRef CAS.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A647–A650 CrossRef CAS.
- J. Zhao, L. Zhao, N. Dimov, S. Okada and T. Nishida, J. Electrochem. Soc., 2013, 160, A3077–A3081 CrossRef CAS.
- M.-T. F. Rodrigues, F. N. Sayed, H. Gullapalli and P. M. Ajayan, J. Power Sources, 2018, 381, 107–115 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170–6178 CrossRef CAS.
- C. T. Moynihan, P. B. Macedo, C. J. Montrose, C. J. Montrose, P. K. Gupta, M. A. DeBolt, J. F. Dill, B. E. Dom, P. W. Drake, A. J. Easteal, P. B. Elterman, R. P. Moeller, H. Sasabe and J. A. Wilder, Ann. N. Y. Acad. Sci., 1976, 279, 15–35 CrossRef CAS.
- J. Hwang, K. Matsumoto and R. Hagiwara, Adv. Sustainable Syst., 2018, 2, 1700171 CrossRef.
- C. Ding, T. Nohira, R. Hagiwara, K. Matsumoto, Y. Okamoto, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2014, 269, 124–128 CrossRef CAS.
- C. Ding, T. Nohira, A. Fukunaga and R. Hagiwaraa, Electrochemistry, 2015, 83, 91–94 CrossRef CAS.
- R. A. Guidotti, F. W. Reinhardt and J. Odinek, J. Power Sources, 2004, 136, 257–262 CrossRef CAS.
- S. A. Ferdousi, M. Hilder, A. Basile, H. Zhu, L. A. O'Dell, D. Saurel, T. Rojo, M. Armand, M. Forsyth and P. C. Howlett, ChemSusChem, 2019, 12, 1700–1711 CrossRef CAS PubMed.
- A. Basile, S. A. Ferdousi, F. Makhlooghiazad, R. Yunis, M. Hilder, M. Forsyth and P. C. Howlett, J. Power Sources, 2018, 379, 344–349 CrossRef CAS.
- R. Seddon Kenneth, A. Stark and M.-J. Torres, Pure Appl. Chem., 2000, 72, 2275 Search PubMed.
- J. M. Slattery, C. Daguenet, P. J. Dyson, T. J. S. Schubert and I. Krossing, Angew. Chem., Int. Ed., 2007, 46, 5384–5388 CrossRef CAS PubMed.
- K. Yuyama, G. Masuda, H. Yoshida and T. Sato, J. Power Sources, 2006, 162, 1401–1408 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962–4967 RSC.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- K. R. Harris, J. Phys. Chem. B, 2010, 114, 9572–9577 CrossRef CAS PubMed.
- K. Kuratani, N. Uemura, H. Senoh, H. T. Takeshita and T. Kiyobayashi, J. Power Sources, 2013, 223, 175–182 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- K. M. Abraham, Z. Jiang and B. Carroll, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS.
- P. G. Bruce and C. A. Vincent, J. Electroanal. Chem., 1987, 225, 1–17 CrossRef CAS.
- C.-Y. Chen, T. Kiko, T. Hosokawa, K. Matsumoto, T. Nohira and R. Hagiwara, J. Power Sources, 2016, 332, 51–59 CrossRef CAS.
- F. Chen, P. Howlett and M. Forsyth, J. Phys. Chem. C, 2018, 122, 105–114 CrossRef CAS.
- T. Carstens, A. Lahiri, N. Borisenko and F. Endres, J. Phys. Chem. C, 2016, 120, 14736–14741 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- A. Noda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2001, 105, 4603–4610 CrossRef CAS.
- H. Tokuda, K. Hayamizu, K. Ishii, M. A. B. H. Susan and M. Watanabe, J. Phys. Chem. B, 2004, 108, 16593–16600 CrossRef CAS.
- R. Richert and C. A. Angell, J. Chem. Phys., 1998, 108, 9016–9026 CrossRef CAS.
- M.-M. Huang, Y. Jiang, P. Sasisanker, G. W. Driver and H. Weingärtner, J. Chem. Eng. Data, 2011, 56, 1494–1499 CrossRef CAS.
- C. Wakai, A. Oleinikova, M. Ott and H. Weingärtner, J. Phys. Chem. B, 2005, 109, 17028–17030 CrossRef CAS PubMed.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef PubMed.
- I. Krossing, J. M. Slattery, C. Daguenet, P. J. Dyson, A. Oleinikova and H. Weingärtner, J. Am. Chem. Soc., 2006, 128, 13427–13434 CrossRef CAS PubMed.
- C. Reichardt, Green Chem., 2005, 7, 339–351 RSC.
- M. Caricato, B. Mennucci and J. Tomasi, Mol. Phys., 2006, 104, 875–887 CrossRef CAS.
- K. A. Fletcher, I. A. Storey, A. E. Hendricks, S. Pandey and S. Pandey, Green Chem., 2001, 3, 210–215 RSC.
- S. N. Baker, G. A. Baker, M. A. Kane and F. V. Bright, J. Phys. Chem. B, 2001, 105, 9663–9668 CrossRef CAS.
- H. Jin, G. A. Baker, S. Arzhantsev, J. Dong and M. Maroncelli, J. Phys. Chem. B, 2007, 111, 7291–7302 CrossRef CAS PubMed.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- R. W. Soukup and K. Sone, Bull. Chem. Soc. Jpn., 1987, 60, 2286–2288 CrossRef CAS.
- M. J. Muldoon, C. M. Gordon and I. R. Dunkin, J. Chem. Soc., Perkin Trans. 2, 2001, 433–435 RSC.
- S. V. Dzyuba and R. A. Bartsch, Tetrahedron Lett., 2002, 43, 4657–4659 CrossRef CAS.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- J. L. Kaar, A. M. Jesionowski, J. A. Berberich, R. Moulton and A. J. Russell, J. Am. Chem. Soc., 2003, 125, 4125–4131 CrossRef CAS PubMed.
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323–11331 CrossRef CAS PubMed.
- H. Tokuda, S. Tsuzuki, M. A. B. H. Susan, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593–19600 CrossRef CAS PubMed.
- R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 2886–2894 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- T. Yokoyama, R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 3233–3237 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- K. Ueno, H. Tokuda and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 1649–1658 RSC.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- S. P. Ong, O. Andreussi, Y. Wu, N. Marzari and G. Ceder, Chem. Mater., 2011, 23, 2979–2986 CrossRef CAS.
- C. Howlett Patrick, I. Izgorodina Ekaterina, M. Forsyth and R. MacFarlane Douglas, Z. Phys. Chem., 2006, 220, 1483 CrossRef.
- S. Kazemiabnavi, Z. Zhang, K. Thornton and S. Banerjee, J. Phys. Chem. B, 2016, 120, 5691–5702 CrossRef CAS PubMed.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H.-P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS PubMed.
- T. Nishi, T. Iwahashi, H. Yamane, Y. Ouchi, K. Kanai and K. Seki, Chem. Phys. Lett., 2008, 455, 213–217 CrossRef CAS.
- K. Kanai, T. Nishi, T. Iwahashi, Y. Ouchi, K. Seki, Y. Harada and S. Shin, J. Chem. Phys., 2008, 129, 224507 CrossRef CAS PubMed.
- K. Kanai, T. Nishi, T. Iwahashi, Y. Ouchi, K. Seki, Y. Harada and S. Shin, J. Electron Spectrosc. Relat. Phenom., 2009, 174, 110–115 CrossRef CAS.
- K. Matsumoto, R. Hagiwara, R. Yoshida, Y. Ito, Z. Mazej, P. Benkič, B. Žemva, O. Tamada, H. Yoshino and S. Matsubara, Dalton Trans., 2004, 144–149 RSC.
- A. M. O’Mahony, D. S. Silvester, L. Aldous, C. Hardacre and R. G. Compton, J. Chem. Eng. Data, 2008, 53, 2884–2891 CrossRef.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461 Search PubMed.
- V. Borgel, E. Markevich, D. Aurbach, G. Semrau and M. Schmidt, J. Power Sources, 2009, 189, 331–336 CrossRef CAS.
- M. Yamagata, N. Nishigaki, S. Nishishita, Y. Matsui, T. Sugimoto, M. Kikuta, T. Higashizaki, M. Kono and M. Ishikawa, Electrochim. Acta, 2013, 110, 181–190 CrossRef CAS.
- Y. Fukuda, R. Tanaka and M. Ishikawa, Electrochemistry, 2007, 75, 589–591 CrossRef CAS.
- T. Iwahashi, Y. Miwa, W. Zhou, Y. Sakai, M. Yamagata, M. Ishikawa, D. Kim and Y. Ouchi, Electrochem. Commun., 2016, 72, 54–58 CrossRef CAS.
- I. A. Shkrob, T. W. Marin, Y. Zhu and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 19661–19671 CrossRef CAS.
- C. Ding, T. Nohira and R. Hagiwara, J. Mater. Chem. A, 2015, 3, 20767–20771 RSC.
- T. Ma, G.-L. Xu, Y. Li, L. Wang, X. He, J. Zheng, J. Liu, M. H. Engelhard, P. Zapol, L. A. Curtiss, J. Jorne, K. Amine and Z. Chen, J. Phys. Chem. Lett., 2017, 8, 1072–1077 CrossRef CAS PubMed.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320–325 CrossRef CAS.
- M. Dahbi, F. Ghamouss, F. Tran-Van, D. Lemordant and M. Anouti, J. Power Sources, 2011, 196, 9743–9750 CrossRef CAS.
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, J. Power Sources, 2011, 196, 3623–3632 CrossRef CAS.
- A. Abouimrane, J. Ding and I. J. Davidson, J. Power Sources, 2009, 189, 693–696 CrossRef CAS.
- C. Peng, L. Yang, Z. Zhang, K. Tachibana and Y. Yang, J. Power Sources, 2007, 173, 510–517 CrossRef CAS.
- K. Matsumoto, E. Nishiwaki, T. Hosokawa, S. Tawa, T. Nohira and R. Hagiwara, J. Phys. Chem. C, 2017, 121, 9209–9219 CrossRef CAS.
- A. Hofmann, L. Merklein, M. Schulz and T. Hanemann, Electrochim. Acta, 2014, 116, 388–395 CrossRef CAS.
- T. Nohira, T. Ishibashi and R. Hagiwara, J. Power Sources, 2012, 205, 506–509 CrossRef CAS.
- D. Monti, A. Ponrouch, M. R. Palacín and P. Johansson, J. Power Sources, 2016, 324, 712–721 CrossRef CAS.
- D. S. Tchitchekova, D. Monti, P. Johansson, F. Bardé, A. Randon-Vitanova, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2017, 164, A1384–A1392 CrossRef CAS.
- G. Damas, C. F. N. Marchiori and C. M. Araujo, J. Phys. Chem. C, 2018, 122, 26876–26888 CrossRef CAS.
- T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A1748–A1752 CrossRef CAS.
- A. Rudola, D. Aurbach and P. Balaya, Electrochem. Commun., 2014, 46, 56–59 CrossRef CAS.
- R. Wibowo, L. Aldous, E. I. Rogers, S. E. Ward Jones and R. G. Compton, J. Phys. Chem. C, 2010, 114, 3618–3626 CrossRef CAS.
- A. Basile, F. Makhlooghiazad, R. Yunis, D. R. MacFarlane, M. Forsyth and P. C. Howlett, ChemElectroChem, 2017, 4, 986–991 CrossRef CAS.
- C. Ding, T. Nohira, K. Kuroda, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 238, 296–300 CrossRef CAS.
- K. Matsumoto, C. Y. Chen, T. Kiko, J. Hwang, T. Hosokawa, T. Nohira and R. Hagiwara, ECS Trans., 2016, 75(15), 139–145 CrossRef CAS.
- A. Basile, H. Yoon, D. R. MacFarlane, M. Forsyth and P. C. Howlett, Electrochem. Commun., 2016, 71, 48–51 CrossRef CAS.
- K. Matsumoto, R. Hagiwara and Y. Ito, Electrochemistry, 2016, 84, 626–630 CrossRef CAS.
- M. Gnahm and D. M. Kolb, J. Electroanal. Chem., 2011, 651, 250–252 CrossRef CAS.
- R. Jarosova and G. M. Swain, J. Electrochem. Soc., 2015, 162, H507–H511 CrossRef CAS.
- S. Ren, Y. Hou, W. Wu and W. Liu, J. Chem. Eng. Data, 2010, 55, 5074–5077 CrossRef CAS.
- A. Stark, P. Behrend, O. Braun, A. Müller, J. Ranke, B. Ondruschka and B. Jastorff, Green Chem., 2008, 10, 1152–1161 RSC.
- B. R. Clare, P. M. Bayley, A. S. Best, M. Forsyth and D. R. MacFarlane, Chem. Commun., 2008, 2689–2691 RSC.
- M. J. Muldoon, A. J. McLean, C. M. Gordon and I. R. Dunkin, Chem. Commun., 2001, 2364–2365 RSC.
- W. L. F. Armarego and C. Cha, Purification of Laboratory Chemicals, Butterworth-Heinemann, London, 7th edn, 2012 Search PubMed.
- O. Kuzumina and J. Hallett, Economical Aspects of Ionic Liquid Applications, Application, Purification, and Recovery of Ionic Liquids, Elsevier, Amsterdam, 2016 Search PubMed.
- H. G. Joglekar, I. Rahman and B. D. Kulkarni, Chem. Eng. Technol., 2007, 30, 819–828 CrossRef CAS.
- Z. P. Visak, J. Solution Chem., 2012, 41, 1673–1695 CrossRef CAS.
- R. P. Singh, J. L. Martin and J. C. Poshusta, Synthesis of bis(fluorosulfonyl)imide, US Pat., 8377406, 2013 Search PubMed.
- R. P. Singh and H. Qichao, J. Fluorine Chem., 2019, 226, 109333 CrossRef.
- M. O'Meara, A. Alemany, M. Maase, U. Vagt and I. Malkowsky, Met. Finish., 2009, 107, 38–39 CrossRef.
- R. P. Swatloski, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 361–363 RSC.
- D. Wu, X. Li, B. Xu, N. Twu, L. Liu and G. Ceder, Energy Environ. Sci., 2015, 8, 195–202 RSC.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy. Mater., 2016, 6, 1600943 CrossRef.
- L. G. Chagas, D. Buchholz, L. Wu, B. Vortmann and S. Passerini, J. Power Sources, 2014, 247, 377–383 CrossRef CAS.
- H. Zhang, I. Hasa, D. Buchholz, B. Qin, D. Geiger, S. Jeong, U. Kaiser and S. Passerini, NPG Asia Mater., 2017, 9, e370 CrossRef CAS.
- C. Delmas, C. Fouassier and P. Hagenmuller, Phys. B + C, 1980, 99, 81–85 CrossRef CAS.
- Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223–3242 RSC.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552–6591 CrossRef CAS PubMed.
- M. Dutreilh, C. Chevalier, M. El-Ghozzi, D. Avignant and J. M. Montel, J. Solid State Chem., 1999, 142, 1–5 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- J. M. Clark, S.-i. Nishimura, A. Yamada and M. S. Islam, Angew. Chem., Int. Ed., 2012, 51, 13149–13153 CrossRef CAS PubMed.
- W. Song, X. Ji, Z. Wu, Y. Zhu, Y. Yang, J. Chen, M. Jing, F. Li and C. E. Banks, J. Mater. Chem. A, 2014, 2, 5358–5362 RSC.
- Z. Jian, Y.-S. Hu, X. Ji and W. Chen, Adv. Mater., 2017, 29, 1601925 CrossRef PubMed.
- J. Gaubicher, C. Wurm, G. Goward, C. Masquelier and L. Nazar, Chem. Mater., 2000, 12, 3240–3242 CrossRef CAS.
- P. Barpanda, L. Lander, S.-i. Nishimura and A. Yamada, Adv. Energy. Mater., 2018, 8, 1703055 CrossRef.
- B. Senthilkumar, C. Murugesan, L. Sharma, S. Lochab and P. Barpanda, Small Methods, 2019, 3, 1800253 CrossRef CAS.
- S.-P. Guo, J.-C. Li, Q.-T. Xu, Z. Ma and H.-G. Xue, J. Power Sources, 2017, 361, 285–299 CrossRef CAS.
- I. Zatovsky, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, i12 CrossRef CAS PubMed.
- B. L. Cushing and J. B. Goodenough, J. Solid State Chem., 2001, 162, 176–181 CrossRef CAS.
- J. Hwang, K. Matsumoto and R. Hagiwara, ACS Appl. Energy Mater., 2019, 2, 2818–2827 CrossRef CAS.
- W. Duan, Z. Zhu, H. Li, Z. Hu, K. Zhang, F. Cheng and J. Chen, J. Mater. Chem. A, 2014, 2, 8668–8675 RSC.
- T. C. Mendes, X. Zhang, Y. Wu, P. C. Howlett, M. Forsyth and D. R. Macfarlane, ACS Sustainable Chem. Eng., 2019, 7, 3722–3726 CrossRef CAS.
- F. Wu, N. Zhu, Y. Bai, Y. Li, Z. Wang, Q. Ni, H. Wang and C. Wu, Nano Energy, 2018, 51, 524–532 CrossRef CAS.
- J. Wang and X. Sun, Energy Environ. Sci., 2015, 8, 1110–1138 RSC.
- M. Avdeev, Z. Mohamed, C. D. Ling, J. Lu, M. Tamaru, A. Yamada and P. Barpanda, Inorg. Chem., 2013, 52, 8685–8693 CrossRef CAS PubMed.
- M. Hilder, P. C. Howlett, D. Saurel, H. Anne, M. Casas-Cabanas, M. Armand, T. Rojo, D. R. MacFarlane and M. Forsyth, J. Power Sources, 2018, 406, 70–80 CrossRef CAS.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 RSC.
- J. Kim, D.-H. Seo, H. Kim, I. Park, J.-K. Yoo, S.-K. Jung, Y.-U. Park, W. A. Goddard Iii and K. Kang, Energy Environ. Sci., 2015, 8, 540–545 RSC.
- Y. Niu, Y. Zhang and M. Xu, J. Mater. Chem. A, 2019, 7, 15006 RSC.
- P. Barpanda, G. Liu, C. D. Ling, M. Tamaru, M. Avdeev, S.-C. Chung, Y. Yamada and A. Yamada, Chem. Mater., 2013, 25, 3480–3487 CrossRef CAS.
- T. Honma, T. Togashi, N. Ito and T. Komatsu, J. Ceram. Soc. Jpn., 2012, 120, 344–346 CrossRef CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira, R. Hagiwara, Y. Orikasa and Y. Uchimoto, J. Power Sources, 2014, 246, 783–787 CrossRef CAS.
- T. Honma, N. Ito, T. Togashi, A. Sato and T. Komatsu, J. Power Sources, 2013, 227, 31–34 CrossRef CAS.
- K.-H. Ha, S. H. Woo, D. Mok, N.-S. Choi, Y. Park, S. M. Oh, Y. Kim, J. Kim, J. Lee, L. F. Nazar and K. T. Lee, Adv. Energy. Mater., 2013, 3, 770–776 CrossRef CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira and R. Hagiwara, J. Electrochem. Soc., 2015, 162, A176–A180 CrossRef CAS.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy. Mater., 2013, 3, 156–160 CrossRef CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira and R. Hagiwara, Electrochem. Commun., 2014, 45, 63–66 CrossRef CAS.
- M. Law, V. Ramar and P. Balaya, J. Power Sources, 2017, 359, 277–284 CrossRef CAS.
- J. Song, M. Xu, L. Wang and J. B. Goodenough, Chem. Commun., 2013, 49, 5280–5282 RSC.
- C.-Y. Chen, K. Matsumoto, T. Nohira and R. Hagiwara, J. Electrochem. Soc., 2015, 162, A2093–A2098 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- T. Ohzuku and A. Ueda, Solid State Ionics, 1994, 69, 201–211 CrossRef CAS.
- K. Takada, N. Aotani, K. Iwamoto and S. Kondo, Solid State Ionics, 1995, 79, 284–287 CrossRef CAS.
- P. He, H. Yu, D. Li and H. Zhou, J. Mater. Chem., 2012, 22, 3680–3695 RSC.
- W. Liu, P. Oh, X. Liu, M.-J. Lee, W. Cho, S. Chae, Y. Kim and J. Cho, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef CAS PubMed.
- S. Okada, Y. Takahashi, T. Kiyabu, T. Doi, J.-I. Yamaki and T. Nishida, ECS Meet. Abstr., 2006, MA2006-02, 201 Search PubMed.
- I. Hasa, D. Buchholz, S. Passerini and J. Hassoun, ACS Appl. Mater. Interfaces, 2015, 7, 5206–5212 CrossRef CAS PubMed.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC.
- K. Kubota and S. Komaba, J. Electrochem. Soc., 2015, 162, A2538–A2550 CrossRef CAS.
- J. J. Braconnier, C. Delmas and P. Hagenmuller, Mater. Res. Bull., 1982, 17, 993–1000 CrossRef CAS.
- S. Komaba, C. Takei, T. Nakayama, A. Ogata and N. Yabuuchi, Electrochem. Commun., 2010, 12, 355–358 CrossRef CAS.
- A. Fukunaga, T. Nohira, Y. Kozawa, R. Hagiwara, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2012, 209, 52–56 CrossRef CAS.
- E. Gonzalo, M. H. Han, J. M. López del Amo, B. Acebedo, M. Casas-Cabanas and T. Rojo, J. Mater. Chem. A, 2014, 2, 18523–18530 RSC.
- C. Ding, T. Nohira and R. Hagiwara, Electrochim. Acta, 2017, 231, 412–416 CrossRef CAS.
- T. Risthaus, D. Zhou, X. Cao, X. He, B. Qiu, J. Wang, L. Zhang, Z. Liu, E. Paillard, G. Schumacher, M. Winter and J. Li, J. Power Sources, 2018, 395, 16–24 CrossRef CAS.
- F. Sauvage, L. Laffont, J. M. Tarascon and E. Baudrin, Inorg. Chem., 2007, 46, 3289–3294 CrossRef CAS PubMed.
- S. Terada, H. Susa, S. Tsuzuki, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2018, 122, 16589–16599 CrossRef CAS.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- P. Ge and M. Fouletier, Solid State Ionics, 1988, 28-30, 1172–1175 CrossRef.
- H. Moriwake, A. Kuwabara, C. A. J. Fisher and Y. Ikuhara, RSC Adv., 2017, 7, 36550–36554 RSC.
- K. Nobuhara, H. Nakayama, M. Nose, S. Nakanishi and H. Iba, J. Power Sources, 2013, 243, 585–587 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- A. Ponrouch, A. R. Goñi and M. R. Palacín, Electrochem. Commun., 2013, 27, 85–88 CrossRef CAS.
- M. Arnaiz, P. Huang, J. Ajuria, T. Rojo, E. Goikolea and A. Balducci, Batteries Supercaps, 2018, 1, 204–208 CrossRef CAS.
- C. Ding, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai and K. Nitta, Electrochim. Acta, 2015, 176, 344–349 CrossRef CAS.
- C. Wu, Y. Ramaswamy, A. Soeparto and H. Zreiqat, J. Biomed. Mater. Res. A, 2008, 86, 402–410 CrossRef PubMed.
- R. S. Kalubarme, A. I. Inamdar, D. S. Bhange, H. Im, S. W. Gosavi and C.-J. Park, J. Mater. Chem. A, 2016, 4, 17419–17430 RSC.
- T.-F. Hung, W.-H. Lan, Y.-W. Yeh, W.-S. Chang, C.-C. Yang and J.-C. Lin, ACS Sustainable Chem. Eng., 2016, 4, 7074–7079 CrossRef CAS.
- D. Luo, P. Lei, Y. Huang, G. Tian and X. Xiang, J. Electroanal. Chem., 2019, 838, 66–72 CrossRef CAS.
- L. Wu, D. Bresser, D. Buchholz, G. A. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy. Mater., 2015, 5, 1401142 CrossRef.
- F. Klein, B. Jache, A. Bhide and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 15876–15887 RSC.
- L. Wu, A. Moretti, D. Buchholz, S. Passerini and D. Bresser, Electrochim. Acta, 2016, 203, 109–116 CrossRef CAS.
- C. S. Ding, T. Nohira and R. Hagiwara, Sustainable Energy Fuels, 2017, 1, 371–376 RSC.
- C. Ding, T. Nohira and R. Hagiwara, J. Power Sources, 2018, 388, 19–24 CrossRef CAS.
- T. Song, S. Ye, H. Liu and Y.-G. Wang, J. Alloys Compd., 2018, 767, 820–828 CrossRef CAS.
- C. Ortiz-Cervantes, P. I. Román-Román, J. Vazquez-Chavez, M. Hernández-Rodríguez and D. Solis-Ibarra, Angew. Chem., Int. Ed., 2018, 57, 13882–13886 CrossRef CAS PubMed.
- H. Usui, Y. Domi, M. Shimizu, A. Imoto, K. Yamaguchi and H. Sakaguchi, J. Power Sources, 2016, 329, 428–431 CrossRef CAS.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. Liu, L. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- X. Han, Y. Liu, Z. Jia, Y.-C. Chen, J. Wan, N. Weadock, K. J. Gaskell, T. Li and L. Hu, Nano Lett., 2014, 14, 139–147 CrossRef CAS PubMed.
- T. Ramireddy, T. Xing, M. M. Rahman, Y. Chen, Q. Dutercq, D. Gunzelmann and A. M. Glushenkov, J. Mater. Chem. A, 2015, 3, 5572–5584 RSC.
- M. N. Obrovac and L. Christensen, Electrochem. Solid State Lett., 2004, 7, A93–A96 CrossRef CAS.
- C.-H. Lim, T.-Y. Huang, P.-S. Shao, J.-H. Chien, Y.-T. Weng, H.-F. Huang, B. J. Hwang and N.-L. Wu, Electrochim. Acta, 2016, 211, 265–272 CrossRef CAS.
- D. M. Piper, T. Evans, K. Leung, T. Watkins, J. Olson, S. C. Kim, S. S. Han, V. Bhat, K. H. Oh, D. A. Buttry and S.-H. Lee, Nat. Commun., 2015, 6, 6230 CrossRef CAS PubMed.
- Y. Xu, E. Swaans, S. Basak, H. W. Zandbergen, D. M. Borsa and F. M. Mulder, Adv. Energy. Mater., 2016, 6, 1501436 CrossRef.
- L. Wang, J. Światowska, S. Dai, M. Cao, Z. Zhong, Y. Shen and M. Wang, Mater. Today Energy, 2019, 11, 46–60 CrossRef.
- H. Tan, D. Chen, X. Rui and Y. Yu, Adv. Funct. Mater., 2019, 29, 1808745 CrossRef.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef PubMed.
- U. S. G. S. Mineral Commodity Summaries 2019, 2019, 192.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, Electrochim. Acta, 2014, 135, 60–67 CrossRef CAS.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai and K. Nitta, Electrochim. Acta, 2016, 193, 275–283 CrossRef CAS.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai and K. Nitta, Electrochim. Acta, 2016, 211, 234–244 CrossRef CAS.
- Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Manivannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372 CrossRef CAS PubMed.
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X.-F. Yu, H. Ushiyama, K. Yamashita, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- K. Y. Masahiro Shimizu, T. Sakata, T. Nokami, T. Itoh and H. Sakaguchi, Int. J. Electrochem. Sci., 2015, 10, 10145–10156 Search PubMed.
- H. Li, P. Balaya and J. Maier, J. Electrochem. Soc., 2004, 151, A1878–A1885 CrossRef CAS.
- Y. Oumellal, A. Rougier, G. A. Nazri, J. M. Tarascon and L. Aymard, Nat. Mater., 2008, 7, 916 CrossRef CAS PubMed.
- K.-H. Nam, K.-J. Jeon and C.-M. Park, Energy Storage Mater., 2019, 17, 78–87 CrossRef.
- Z. Huang, H. Hou, C. Wang, S. Li, Y. Zhang and X. Ji, Chem. Mater., 2017, 29, 7313–7322 CrossRef CAS.
- K. Zhang, M. Park, J. Zhang, G.-H. Lee, J. Shin and Y.-M. Kang, Nano Res., 2017, 10, 4337–4350 CrossRef CAS.
- H. Usui, Y. Domi, R. Yamagami, K. Fujiwara, H. Nishida and H. Sakaguchi, ACS Appl. Energy Mater., 2018, 1, 306–311 CrossRef CAS.
- S. Kaushik, J. Hwang, K. Matsumoto, Y. Sato and R. Hagiwara, ChemElectroChem, 2018, 5, 1340–1344 CrossRef CAS.
- S.-O. Kim and A. Manthiram, Chem. Commun., 2016, 52, 4337–4340 RSC.
- S. Kaushik, K. Matsumoto, Y. Sato and R. Hagiwara, Electrochem. Commun., 2019, 102, 46–51 CrossRef CAS.
- M. Hu, Y. Jiang, W. Sun, H. Wang, C. Jin and M. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 19449–19455 CrossRef CAS PubMed.
- C.-Y. Li, J. Patra, C.-H. Yang, C.-M. Tseng, S. B. Majumder, Q.-F. Dong and J.-K. Chang, ACS Sustainable Chem. Eng., 2017, 5, 8269–8276 CrossRef CAS.
- H.-C. Chen, J. Patra, S.-W. Lee, C.-J. Tseng, T.-Y. Wu, M.-H. Lin and J.-K. Chang, J. Mater. Chem. A, 2017, 5, 13776–13784 RSC.
- Y. Kawabe, N. Yabuuchi, M. Kajiyama, N. Fukuhara, T. Inamasu, R. Okuyama, I. Nakai and S. Komaba, Electrochem. Commun., 2011, 13, 1225–1228 CrossRef CAS.
- X. Lin, X. Hou, X. Wu, S. Wang, M. Gao and Y. Yang, RSC Adv., 2014, 4, 40985–40993 RSC.
- J. Qian, M. Zhou, Y. Cao, X. Ai and H. Yang, Adv. Energy. Mater., 2012, 2, 410–414 CrossRef CAS.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- N. Casado, M. Hilder, C. Pozo-Gonzalo, M. Forsyth and D. Mecerreyes, ChemSusChem, 2017, 10, 1783–1791 CrossRef CAS PubMed.
- X. Wang, Z. Shang, A. Yang, Q. Zhang, F. Cheng, D. Jia and J. Chen, Chem, 2019, 5, 364–375 CrossRef CAS PubMed.
- H.-G. Wang and X.-B. Zhang, Chem. – Eur. J., 2018, 24, 18235–18245 CrossRef CAS PubMed.
- Y. Xu, M. Zhou and Y. Lei, Mater. Today, 2018, 21, 60–78 CrossRef CAS.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan, S. Yu, Z. Sun, Y. Liu and Z. Lu, Nat. Commun., 2016, 7, 13318 CrossRef CAS PubMed.
- B. E. Gurkan, Z. Qiang, Y.-M. Chen, Y. Zhu and B. D. Vogt, J. Electrochem. Soc., 2017, 164, H5093–H5099 CrossRef CAS.
- Y. Park, D.-S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- F. Wan, X.-L. Wu, J.-Z. Guo, J.-Y. Li, J.-P. Zhang, L. Niu and R.-S. Wang, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- A. Rudola, K. Saravanan, C. W. Mason and P. Balaya, J. Mater. Chem. A, 2013, 1, 2653–2662 RSC.
- P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- Y. Zhang, H. Hou, X. Yang, J. Chen, M. Jing, Z. Wu, X. Jia and X. Ji, J. Power Sources, 2016, 305, 200–208 CrossRef CAS.
- C.-Y. Chen, A. Sawamura, T. Tsuda, S. Uchida, M. Ishikawa and S. Kuwabata, ACS Appl. Mater. Interfaces, 2017, 9, 35511–35515 CrossRef CAS PubMed.
- E. Markevich, G. Salitra, A. Rosenman, Y. Talyosef, D. Aurbach and A. Garsuch, RSC Adv., 2014, 4, 48572–48575 RSC.
- T. Sugimoto, Y. Atsumi, M. Kono, M. Kikuta, E. Ishiko, M. Yamagata and M. Ishikawa, J. Power Sources, 2010, 195, 6153–6156 CrossRef CAS.
- Y. Domi, H. Usui, K. Yamaguchi, S. Yodoya and H. Sakaguchi, ACS Appl. Mater. Interfaces, 2019, 11, 2950–2960 CrossRef CAS PubMed.
- R.-S. Kühnel, M. Lübke, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2012, 214, 178–184 CrossRef.
- H. Usui, Y. Domi, K. Fujiwara, M. Shimizu, T. Yamamoto, T. Nohira, R. Hagiwara and H. Sakaguchi, ACS Energy Lett., 2017, 2, 1139–1143 CrossRef.
- H. Usui, Y. Domi, H. Nishida, K. Yamaguchi, R. Yamagami and H. Sakaguchi, ChemistrySelect, 2018, 3, 8462–8467 CrossRef CAS.
- X.-F. Luo, A. S. Helal, C.-T. Hsieh, J. Li and J.-K. Chang, Nano Energy, 2018, 49, 515–522 CrossRef CAS.
- J. Zhang, D.-W. Wang, W. Lv, S. Zhang, Q. Liang, D. Zheng, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 370–376 RSC.
- M. B. Pinson and M. Z. Bazant, J. Electrochem. Soc., 2013, 160, A243–A250 CrossRef CAS.
- M. Wang, L. Huai, G. Hu, S. Yang, F. Ren, S. Wang, Z. Zhang, Z. Chen, Z. Peng, C. Shen and D. Wang, J. Phys. Chem. C, 2018, 122, 9825–9834 CrossRef CAS.
- M. Haruta, T. Okubo, Y. Masuo, S. Yoshida, A. Tomita, T. Takenaka, T. Doi and M. Inaba, Electrochim. Acta, 2017, 224, 186–193 CrossRef CAS.
- T. Jaumann, J. Balach, M. Klose, S. Oswald, U. Langklotz, A. Michaelis, J. Eckert and L. Giebeler, Phys. Chem. Chem. Phys., 2015, 17, 24956–24967 RSC.
- R. Mogensen, D. Brandell and R. Younesi, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS.
- C. Bommier, D. Leonard, Z. Jian, W. F. Stickle, P. A. Greaney and X. Ji, Adv. Mater. Interfaces, 2016, 3, 1600449 CrossRef.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Nano Energy, 2019, 55, 327–340 CrossRef CAS.
- G. G. Eshetu, S. Grugeon, H. Kim, S. Jeong, L. Wu, G. Gachot, S. Laruelle, M. Armand and S. Passerini, ChemSusChem, 2016, 9, 462–471 CrossRef CAS PubMed.
- C. V. Manohar, T. C. Mendes, M. Kar, D. Wang, C. Xiao, M. Forsyth, S. Mitra and D. R. MacFarlane, Chem. Commun., 2018, 54, 3500–3503 RSC.
- I. Hasa, S. Passerini and J. Hassoun, J. Power Sources, 2016, 303, 203–207 CrossRef CAS.
- M. P. Do, P. J. Fischer, A. Nagasubramanian, J. Geder, F. E. Kühn and M. Srinivasan, J. Electrochem. Soc., 2019, 166, A944–A952 CrossRef CAS.
- A. Fukunaga, T. Nohira, R. Hagiwara, K. Numata, E. Itani, S. Sakai and K. Nitta, J. Appl. Electrochem., 2016, 46, 487–496 CrossRef CAS.
- K. C. Klavetter, S. Garcia, N. Dahal, J. L. Snider, J. Pedro de Souza, T. H. Cell, M. A. Cassara, A. Heller, S. M. Humphrey and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 14209–14221 RSC.
- T. Tanaka, K. Ohta and N. Arai, J. Power Sources, 2001, 97–98, 2–6 CrossRef CAS.
- M. Majima, S. Ujiie, E. Yagasaki, K. Koyama and S. Inazawa, J. Power Sources, 2001, 101, 53–59 CrossRef CAS.
- A. Boschin and P. Johansson, Electrochim. Acta, 2016, 211, 1006–1015 CrossRef CAS.
- V. K. Singh, Shalu, S. K. Chaurasia and R. K. Singh, RSC Adv., 2016, 6, 40199–40210 RSC.
- Y.-S. Ye, J. Rick and B.-J. Hwang, J. Mater. Chem. A, 2013, 1, 2719–2743 RSC.
- Q. Chen, Y. Tang, T. Huang, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 5286–5289 CrossRef CAS PubMed.
- A. Boisset, S. Menne, J. Jacquemin, A. Balducci and M. Anouti, Phys. Chem. Chem. Phys., 2013, 15, 20054–20063 RSC.
- D. Ruiz-Martínez, A. Kovacs and R. Gómez, Energy Environ. Sci., 2017, 10, 1936–1941 RSC.
- M. Angell, C.-J. Pan, Y. Rong, C. Yuan, M.-C. Lin, B.-J. Hwang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 834–839 CrossRef CAS PubMed.
- A. Basile, A. I. Bhatt and A. P. O’Mullane, Nat. Commun., 2016, 7, 11794 CrossRef PubMed.
- D. Dwibedi, C. D. Ling, R. B. Araujo, S. Chakraborty, S. Duraisamy, N. Munichandraiah, R. Ahuja and P. Barpanda, ACS Appl. Mater. Interfaces, 2016, 8, 6982–6991 CrossRef CAS PubMed.
- M. K. Tran, M.-T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- S. A. Mohd Noor, H. Yoon, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2015, 169, 376–381 CrossRef CAS.
- K. Kubota, T. Nohira, T. Goto and R. Hagiwara, Electrochem. Commun., 2008, 10, 1886–1888 CrossRef CAS.
- T. Yamamoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2013, 237, 98–103 CrossRef CAS.
- I. D. Scott, Y. S. Jung, A. S. Cavanagh, Y. Yan, A. C. Dillon, S. M. George and S.-H. Lee, Nano Lett., 2011, 11, 414–418 CrossRef CAS PubMed.
- X.-L. Wu, L.-Y. Jiang, F.-F. Cao, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS.
- W. Liu, J. Jiang, K. R. Yang, Y. Mi, P. Kumaravadivel, Y. Zhong, Q. Fan, Z. Weng, Z. Wu, J. J. Cha, H. Zhou, V. S. Batista, G. W. Brudvig and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3578–3583 CrossRef CAS PubMed.
- B. Li, S. Li, J. Liu, B. Wang and S. Yang, Nano Lett., 2015, 15, 3073–3079 CrossRef CAS.
- S. Wu, J. Tang, F. Li, X. Liu and H. Zhou, Chem. Commun., 2015, 51, 16860–16863 RSC.
- B. G. Kim, H.-J. Kim, S. Back, K. W. Nam, Y. Jung, Y.-K. Han and J. W. Choi, Sci. Rep., 2014, 4, 4225 CrossRef PubMed.
- G. Li, X. Lu, J. Y. Kim, K. D. Meinhardt, H. J. Chang, N. L. Canfield and V. L. Sprenkle, Nat. Commun., 2016, 7, 10683 CrossRef CAS PubMed.
- K. W. Nam, S. Kim, S. Lee, M. Salama, I. Shterenberg, Y. Gofer, J.-S. Kim, E. Yang, C. S. Park, J.-S. Kim, S.-S. Lee, W.-S. Chang, S.-G. Doo, Y. N. Jo, Y. Jung, D. Aurbach and J. W. Choi, Nano Lett., 2015, 15, 4071–4079 CrossRef CAS PubMed.
- T. Ling, P. Da, X. Zheng, B. Ge, Z. Hu, M. Wu, X.-W. Du, W.-B. Hu, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2018, 4, eaau6261 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- P. Geysens, V. S. Rangasamy, S. Thayumanasundaram, K. Robeyns, L. Van Meervelt, J.-P. Locquet, J. Fransaer and K. Binnemans, J. Phys. Chem. B, 2018, 122, 275–289 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee02041a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |