
A versatile single-ion electrolyte with a Grotthuss-like Li conduction mechanism for dendrite-free Li metal batteries†

Abstract
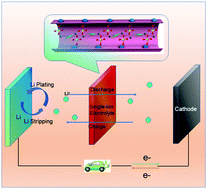
Batteries with Li metal anodes have the desirable feature of high energy density; however, the notorious problem of Li dendrite formation has impeded their practical applications. Herein, we present a versatile single-ion electrolyte, which is achieved by a different strategy of coordinating the anions in the electrolyte on the open metal sites of a metal organic framework. Further investigations of the activation energy and theoretical quantum mechanical calculations suggest that Li ion transport inside the pores of Cu-MOF-74 is via a Grotthuss-like mechanism where the charge is transported by coordinated hopping of Li ions between the perchlorate groups. This single-ion electrolyte is versatile and has wide applications. When the single-ion electrolyte is used for Li‖Li symmetric cells and Li‖LiFePO4 full cells, Li dendrites are suppressed. As a result, an ultralong cycle life is achieved for both cells. In addition, when the single-ion electrolyte is assembled into Li‖LiMn2O4 batteries, the dissolution of Mn2+ into the electrolyte is suppressed even at elevated temperatures, and a long cycle life with improved capacity retention is achieved for Li‖LiMn2O4 batteries. Finally, when the single-ion electrolyte is applied to Li–O2 batteries, an improved cycle life with reduced overpotential is also achieved.
- This article is part of the themed collection: 2019 Energy and Environmental Science HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

