
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular engineering for efficient photosynthetic biosynthesis of 1-butanol from CO2 in cyanobacteria†
Xufeng
Liu
,
Rui
Miao
,
Pia
Lindberg
and
Peter
Lindblad
 *
*
Microbial Chemistry, Department of Chemistry-Ångström, Uppsala University, Box 523, SE-751 20 Uppsala, Sweden. E-mail: Peter.Lindblad@kemi.uu.se
First published on 16th July 2019
Abstract
Cyanobacteria are photoautotrophic microorganisms which can be engineered to directly convert CO2 and water into biofuels and chemicals via photosynthesis using sunlight as energy. However, the product titers and rates are the main challenges that need to be overcome for industrial applications. Here we present systematic modular engineering of the cyanobacterium Synechocystis PCC 6803, enabling efficient biosynthesis of 1-butanol, an attractive commodity chemical and gasoline substitute. Through introducing and re-casting the 1-butanol biosynthetic pathway at the gene and enzyme levels, optimizing the 5′-regions of expression units for tuning transcription and translation, rewiring the carbon flux and rewriting the photosynthetic central carbon metabolism to enhance the precursor supply, and performing process development, we were able to reach a cumulative 1-butanol titer of 4.8 g L−1 with a maximal rate of 302 mg L−1 day−1 from the engineered Synechocystis. This represents the highest 1-butanol production from CO2 reported so far. Our multi-level modular strategy for high-level production of chemicals and advanced biofuels represents a blue-print for future systematic engineering in photosynthetic microorganisms.
Broader contextIn order to reduce the impact of human activities on climate change, we must address our dependence on fossil resources. Biological systems are able to make molecules identical to petroleum-derived compounds used in fuels and in the chemical industry. This has led to increasing attention on the field of metabolic engineering, where microorganisms are genetically engineered to produce compounds of industrial interest. Most such microbial systems employ heterotrophic organisms fed substrates generated from plant biomass. However, photosynthetic microorganisms could be used to perform direct production of fuels and chemicals from photosynthesis, enabling a more efficient conversion of solar energy and carbon dioxide to desirable products, thus leading to more sustainable processes. Engineered strains of photosynthetic cyanobacteria can make many different compounds on a proof-of-concept level, but few products so far show titers approaching those achieved in heterotrophic organisms. We have systematically engineered the unicellular cyanobacterium Synechocystis sp. PCC 6803 to produce 1-butanol. The resulting titers are among the highest for any heterologous product from cyanobacteria, and we show that long-term productivity is feasible. The employed strategy can be used for other products when engineering photosynthetic microorganisms for direct production of solar chemicals and biofuels. |
Introduction
The increasing energy demand and concerns about CO2 emissions and global climate change caused by reliance on fossil fuels as our primary energy source give rise to an urgent requirement for renewable energy, such as biofuels.1,2 Biofuels, produced from renewable resources or biomass feedstocks by microbial cell factories, are already implemented as drop-in fuels.1,2 In traditional microbial fermentation processes for biofuel production by heterotrophic microorganisms, including Escherichia coli and Saccharomyces cerevisiae, bio-based sugar feedstocks are required as the carbon source. Additionally, biodiesel can be obtained from crude plant oils. However, for both cases, extensive processes and large agricultural land areas are needed.1,2 On the other hand, CO2 can be directly recycled into biofuels using solar energy by engineered photosynthetic microorganisms, such as cyanobacteria.3–11 These so called solar fuels and chemicals represent potentially cheaper and more sustainable alternatives to traditional biofuels, with the capacity to replace fossil fuels. A prerequisite for pursuing this option is to develop cyanobacteria as efficient green microbial cell factories for directly producing biofuels and biochemicals.Systematic metabolic engineering holds great promise to develop microorganisms for optimal production of a wide range of biofuels and biochemicals.12 For heterotrophic model microorganisms, there are by now decades of experience to build upon, where multiple layers of metabolic engineering have been performed in single microorganisms to successfully manipulate the complex cellular metabolism.12 In cyanobacteria, efforts have been made in proof-of-concept studies towards tailoring the metabolism to produce solar chemicals and fuels. Previous studies demonstrated the potential of cyanobacteria for high-level production of chemicals derived from pyruvate, including ethanol (5.5 g L−1),5 isoprene (1.26 g L−1)8 and 2,3-butanediol (2.38 g L−1).13 However, for higher alcohols (e.g. 1-butanol), alkanes, fatty acid esters and other acetyl-CoA-derived products, which act as attractive drop-in biofuels for the transportation sector, the reported titers are significantly lower. Thus, systematic engineering of cyanobacteria metabolism is required to enable efficient production of solar fuels and chemicals.
An effective strategy to launch systematic engineering is to use modular pathway engineering, which allows for efficiently evaluating portions of the overall biosynthetic pathway, then combining these to collectively form an altered and interconnected metabolic network.14–19 In the present study, we implement for the first time a modular engineering strategy in a photosynthetic organism, using the unicellular cyanobacterium Synechocystis PCC 6803 (Synechocystis) as the host. The target is complete biosynthesis of 1-butanol from CO2. This is achieved through four work modules (Fig. 1 and Fig. S1, ESI†): Module 1, introducing and re-casting the 1-butanol biosynthesis by systematic screening of genes and pathways; Module 2, optimizing the 5′-regions of expression units to tune the protein expression levels; Module 3, rewiring the carbon flux by editing the acetate metabolism; and Module 4, rewriting the photosynthetic central carbon metabolism by installing a phosphoketolase (PK) pathway. Modules 1 to 4 were established, optimized and assembled consecutively to improve the 1-butanol production stepwise.
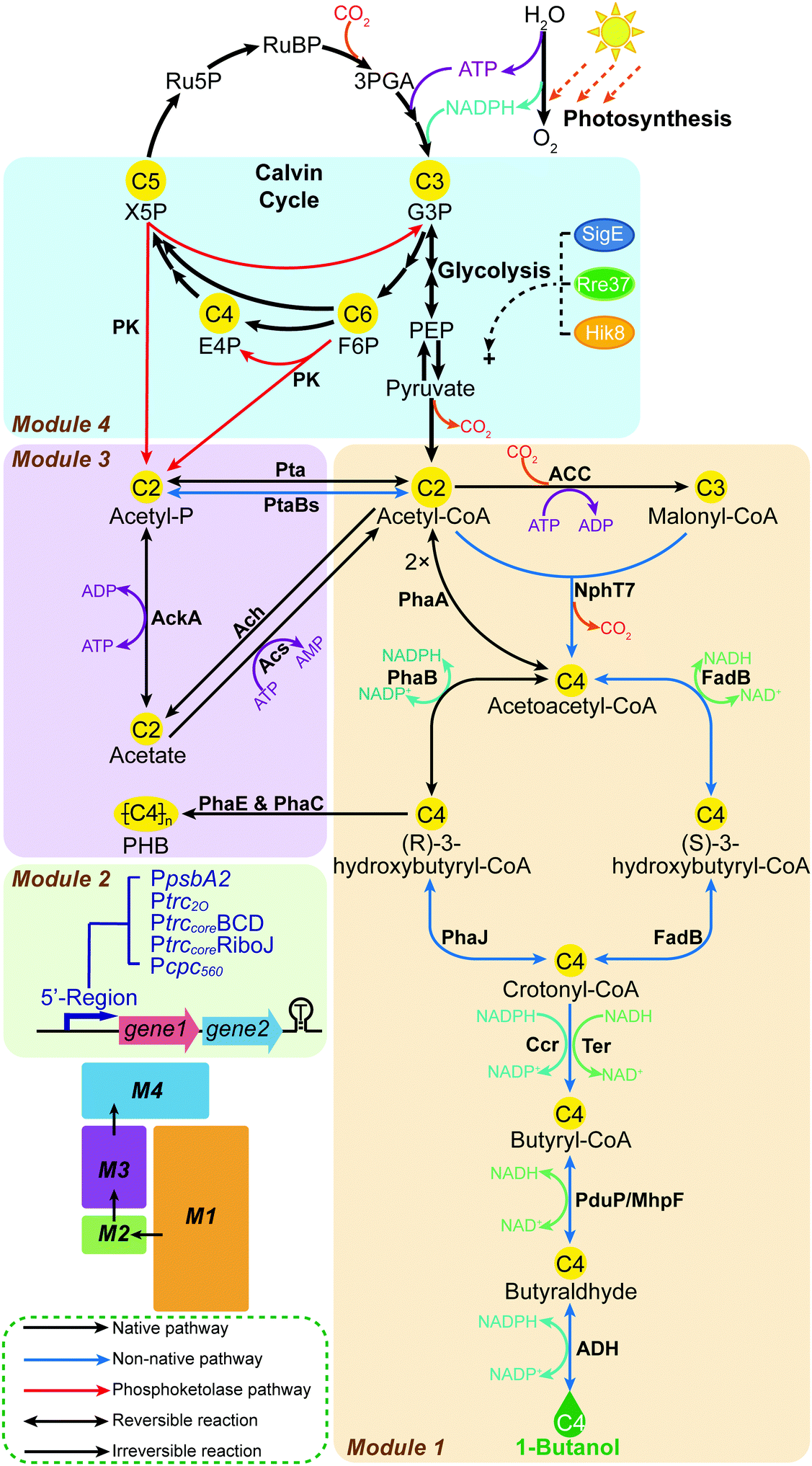 | ||
| Fig. 1 Biosynthetic scheme for complete biosynthesis of 1-butanol from CO2 in Synechocystis PCC 6803, and the main engineering targets implemented in this study. To establish the boundaries, the engineered biosynthesis is visualized in four Modules, Module 1, 2, 3 and 4, respectively. Metabolite abbreviations: Ru5P, ribulose-5-phosphate; RuBP, ribulose-1,5-bisphosphate; 3PGA, 3-phosphoglycerate; G3P, glyceraldehyde-3-phosphate; F6P, fructose-6-phosphate; E4P, erythrose-4-phosphate; X5P, xylulose-5-phosphate; PEP, phosphoenolpyruvate; acetyl-P, acetyl-phosphate; PHB, poly-3-hydroxybutyrate. | ||
Results and discussion
Introducing and re-casting the 1-butanol biosynthetic pathway (Module 1)
In bacterial metabolism, 1-butanol can be produced via four distinct biosynthetic pathways (ESI,† Table S1). In the present study, we firstly introduced and optimized two bacterial 1-butanol biosynthetic pathways, consisting of a redesigned Clostridium-derived clostridial pathway6,20,21 and an artificial reversed β-oxidation pathway which was previously demonstrated in E. coli.22 In both pathways, the biosynthesis of 1-butanol is accomplished by six reactions of reduction and dehydration, using acetyl-CoA as the building blocks and involving multiple acyl-CoA thioester intermediates (Fig. 2a). First, two acetyl-CoA or one acetyl-CoA and one malonyl-CoA initiate the pathways, forming acetoacetyl-CoA catalyzed by acetoacetyl-CoA synthase. In the second step of the pathways, acetoacetyl-CoA is reduced to the corresponding 3-hydroxybutyryl-CoA by 3-hydroxyacyl-CoA dehydrogenase. Then, enoyl-CoA hydratase catalyzes the dehydration of 3-hydroxybutyryl-CoA to crotonyl-CoA in the third step. In the fourth step, crotonyl-CoA hydrogenation is catalyzed by enoyl-CoA reductase to synthesize butyryl-CoA. The reduction from butyryl-CoA to butyraldehyde is then catalyzed by CoA-acylating aldehyde dehydrogenase in the fifth step. Alcohol dehydrogenase (ADH) works as the final step by reducing butyraldehyde into 1-butanol. The two CoA-dependent pathways are basically identical, but they differ in the sequential reduction of acetoacetyl-CoA to form intermediate crotonyl-CoA. While two individual enzymes, PhaB and PhaJ, are required for this conversion in the clostridial pathway, one unique enzyme, FadB, in the reversed β-oxidation pathway can produce crotonyl-CoA via enantioselective catalysis. Since both pathways consist of multiple enzymes, the corresponding genes were divided and cloned into three artificial operons, with the genes encoding the last two steps in one operon, the third and fourth steps of the clostridial pathway or the second to the fourth steps of the reversed β-oxidation pathway in another, and the first two steps of the clostridial pathway or the first step of the reversed β-oxidation pathway in a third operon (ESI,† Fig. S2). For each step, genes from different sources were selected and overexpressed with various combinations to systematically compare their efficiency for 1-butanol production.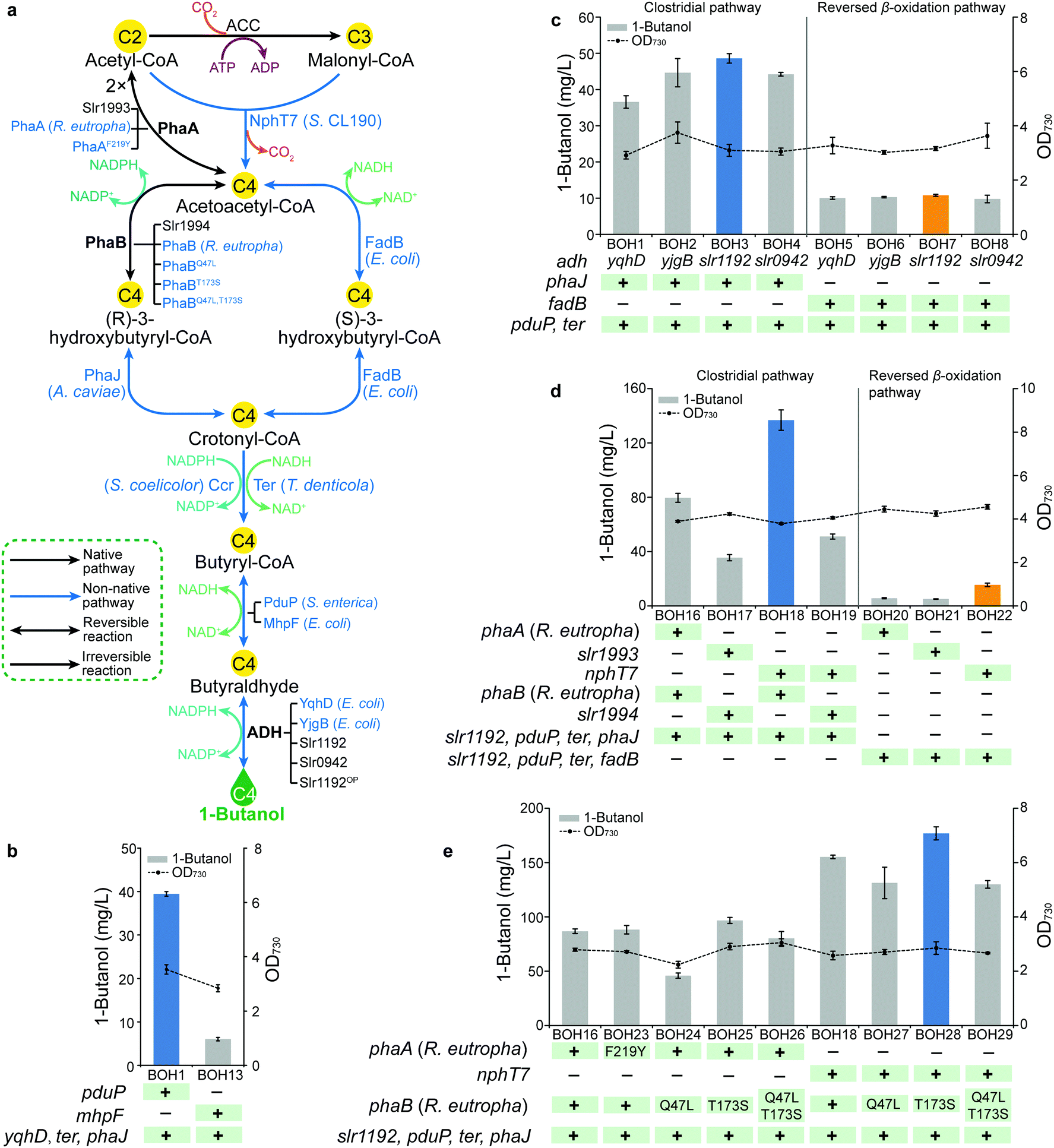 | ||
| Fig. 2 Introducing and re-casting the 1-butanol biosynthesis in Synechocystis PCC 6803 by systematic screening of genes and pathways (Module 1). (a) Biosynthetic scheme of 1-butanol biosynthetic pathways, and the detailed engineering targets implemented in Module 1. Heterologous and endogenous enzymes are shown in blue and black fonts, respectively. (b) 1-Butanol production and cell density of strains BOH1 and BOH13 to compare PduP and MhpF via the last four steps of the clostridial pathway. (c) 1-Butanol production and cell density of strains BOH1–8 to compare four ADHs via the last four steps of the clostridial pathway and last five steps of the reversed β-oxidation pathway. (d) 1-Butanol production and cell density of strains BOH16–22 to compare the clostridial pathway and the reversed β-oxidation pathway. (e) 1-Butanol production and cell density of strains BOH16, BOH18 and BOH23–29 to compare enzymes modified by amino acid substitutions via the clostridial pathway. For (b–e), the time point of the 1-butanol production and cell density is when the maximal in-flask 1-butanol titer is reached. The blue columns indicate the highest 1-butanol production in the clostridial pathway, while the orange columns represent the highest 1-butanol production in the reversed β-oxidation pathway. “ +” and “−” indicate with and without target gene engineering, respectively. All data are presented as mean ± SD of biological triplicates. | ||
In Synechocystis, the intermediates acetoacetyl-CoA and (R)-3-hydroxybutyryl-CoA are endogenously generated for poly-3-hydroxybutyrate (PHB) accumulation23 (Fig. 1). Hence, we started by optimizing the downstream exogenous part of both biosynthetic pathways, i.e. the conversion of (R)-3-hydroxybutyryl-CoA or acetoacetyl-CoA to 1-butanol in the clostridial and the reversed β-oxidation pathways, respectively (Fig. 2a), resulting in 1-butanol producing strains BOH1–15 (ESI,† Fig. S2a and b). For the butyraldehyde forming step, two CoA-acylating aldehyde dehydrogenase-coding genes, S. enterica pduP24,25 and E. coli mhpF,22 were selected. When overexpressed in combination with genes yqhD, ter and phaJ for the clostridial pathway, the pduP-expressing strain BOH1 (39.5 mg L−1) generated an almost 5.5 times higher 1-butanol titer than that of the mhpF-expressing strain BOH13 (6 mg L−1) (Fig. 2b). This effect might be attributed to the fact that PduP is an oxygen-tolerant enzyme6 and therefore exhibits better performance in the presence of oxygen evolved through photosynthesis (Fig. 1).
Subsequently, we moved to optimize the last step, catalyzed by ADH (Fig. 2a). To enhance the photosynthetic 1-butanol production in Synechocystis, the co-factor preference of the selected catalytic enzymes must be taken into account due to the prevalence of reducing force NADPH,26 directly generated from the light reactions of photosynthesis (Fig. 1). Therefore, among strains BOH1–8, four NADPH-specific ADHs, including YqhD and YjgB from E. coli, and the endogenous Slr1192 and Slr0942,27 were investigated in the context of both 1-butanol biosynthetic pathways (Fig. 2c). Overexpression of the Synechocystis ADH Slr1192, which here has been used for the first time for 1-butanol biosynthesis, resulted in the highest 1-butanol production in both pathways (48.6 mg L−1 in strain BOH3 and 10.8 mg L−1 in strain BOH7), even though the E. coli ADH YqhD has been widely used in engineering microbial cell factories for production of higher alcohols,3,4,6,21,22,28 such as 1-butanol and isobutanol.
Similarly, we attempted to replace the NADH-dependent Ter20 with NADPH-dependent Ccr29 to catalyze the conversion of crotonyl-CoA to butyryl-CoA (Fig. 2a). However, no recombinant plasmid harboring the synthetic ccr-phaJ operon could be successfully constructed in the intermediate host E. coli, and no further comparison was made for the clostridial pathway (ESI,† Fig. S2a). Additionally, with genes slr1192, pduP and fadB overexpressed in the reversed β-oxidation pathway, the 1-butanol titer of the ccr-expressing strain BOH11 was similar to that of the ter-expressing strain BOH7 (ESI,† Fig. S3). Consequently, the choice between ccr and ter was not decided, but brought into the next module.
After evaluating the gene candidates for the downstream exogenous part of the two 1-butanol biosynthetic pathways, we turned our attention to optimizing the upstream part starting from central metabolite acetyl-CoA, based on strains BOH3 and BOH7, respectively (Fig. 2a and Fig. S2d–f, ESI†). We found that the clostridial pathway constructed in strains BOH16–19 resulted in significantly higher 1-butanol titers compared with those of the reversed β-oxidation pathway constructed in strains BOH20–22 (Fig. 2d). One reason for the lower production of the reversed β-oxidation pathway may be the co-factor preference in cyanobacteria, which is unfavorable for the NADH-utilizing FadB,22 while NADPH-dependent PhaB21 in the clostridial pathway would be preferred (Fig. 2a). Furthermore, PhaB and PhaJ specifically use the (R)-stereoisomer of 3-hydroxybutyryl-CoA as an intermediate,21 instead of the (S)-isomer used by FadB,22 making it impossible for the reversed β-oxidation pathway to tap into the native carbon flux generated from the endogenous PhaB (Slr1994) (Fig. 2a).
The construction of the initial two steps for the clostridial pathway was inspired by PhaA and PhaB from the PHB production pathway in Synechocystis23 and Ralstonia eutropha30–32 (Fig. 2a), with two different, homologous pairs of phaA and phaB co-overexpressed. Interestingly, strain BOH16 expressing PhaA and PhaB from R. eutropha produced 79.6 mg L−1 1-butanol, which was 1.2× higher than that of strain BOH17 (35.5 mg L−1) overexpressing the endogenous counterparts Slr1993 and Slr1994 (Fig. 2d). For the generation of key intermediate acetoacetyl-CoA, two distinct acetyl-CoA-derived biosynthetic routes were investigated, i.e. the PhaA-mediated reversible condensation of two molecules of acetyl-CoA31 and the irreversible condensation of acetyl-CoA with malonyl-CoA molecules, catalyzed by NphT733 (Fig. 2a). Since NphT7 exhibits no thiolysis activity against acetoacetyl-CoA, and one of its substrates, malonyl-CoA, is generated by the irreversible ATP-activated acetyl-CoA carboxylase (ACC), implementation of this activity should establish a driving force to efficiently channel metabolic flux into 1-butanol biosynthesis.21 As expected, the introduction of NphT7 (strain BOH18) enhanced the formation of 1-butanol to 136 mg L−1, representing a 70% increase compared with that of the PhaA-expressing strain BOH16 (Fig. 2d).
To further increase the 1-butanol production, R. eutropha PhaB variants PhaBQ47L and PhaBT173S, which have been shown to exhibit improved affinity for cofactor NADPH or substrate acetoacetyl-CoA, respectively,34 were evaluated via the NphT7 route in strains BOH27 and BOH28. An 14% increase in the 1-butanol titer (177 mg L−1) was generated by the PhaBT173S-harboring strain BOH28, while introduction of PhaBQ47L (strain BOH27) exerted a negative effect on 1-butanol formation compared with that of strain BOH18 (Fig. 2e). A double mutant PhaBQ47L,T173S was also constructed and overexpressed in strain BOH29. However, the 1-butanol production of strain BOH29 was comparable with that of strain BOH27 expressing PhaBQ47L (Fig. 2e). Furthermore, we tested the 1-butanol biosynthesis via the PhaA route in which a PhaAF219Y variant, possessing more than two-fold improved activity in vitro,35 and the PhaB variants were incorporated (strains BOH23–26). As observed for the NphT7 route, only overexpression of PhaBT173S (strain BOH25) enabled a 11% increase of the 1-butanol titer (96.7 mg L−1) compared with that of strain BOH16 (Fig. 2e). Similar protein levels were observed for the PhaA or PhaB homologs (ESI,† Fig. S4).
In Module 1, we implemented, to our knowledge, the first example of a reversed β-oxidation pathway constructed in cyanobacteria. However, the clostridial pathway performed better and resulted in a 1-butanol production of 177 mg L−1 (strain BOH28). As the final experiment in Module 1, the slr1192 gene codon optimized for Synechocystis (slr1192OP) was generated in order to avoid recombination of overexpressed slr1192 to its native chromosome site, and its effect on the translation level was examined (ESI,† Fig. S2c). When overexpressed with genes pduP, ter and phaJ, the gene slr1192OP in strain BOH31 led to elevated protein expression and a 25% increase of 1-butanol production to 61.4 mg L−1 compared with that of strain BOH3 (ESI,† Fig. S5).
Altogether, a re-cast 1-butanol synthesis pathway using better performing enzymes was obtained in Module 1, with the selection between Ter and Ccr still undetermined (ESI,† Fig. S6). However, the NphT7 route may be limited by the efficiency of the native ACC reaction and malonyl-CoA level. Since PhaA directly uses acetyl-CoA as the source of two-carbon units, PhaA from R. eutropha is still a promising alternative enzyme for further engineering.
Optimizing the 5′-regions of expression units (Module 1 + 2)
In Module 1, the 1-butanol biosynthetic genes were organized as three expression units all driven by the same native promoter PpsbA236 (ESI,† Fig. S2), which may cause a limitation for the protein expression and 1-butanol production. Therefore, in Module 2, all the enzyme components of the optimized 1-butanol biosynthetic pathway were subject to systematical tuning at both transcriptional and translational levels. In the former category, two strong constitutive promoters, Ptrc2O37 and Pcpc560,38 were used to regulate the transcript level of genes, while two genetic insulators, BCD (bicistronic design)39 or RiboJ (self-cleaving ribozyme),40 were introduced to potentially enhance the protein translation in the latter category (Fig. 3). The genetic insulators were coupled with promoter Ptrccore, forming two artificial 5′-regions PtrccoreBCD and PtrccoreRiboJ, respectively. The performance of the 5′-regions, including promoters, was compared with the original PpsbA2 promoter in each expression unit. Stepwise screening of the three expression units via the substitution of promoters or 5′-regions resulted in strains BOH31–57 (Fig. 3). Firstly, while keeping the ter-phaJ operon under the control of the PpsbA2 promoter in strains BOH31 to BOH35, we found that a medium expression level of PduP and Slr1192OP, under the control of Ptrc2O, resulted in the highest 1-butanol titer of 66.3 mg L−1 in strain BOH32, representing a 19% increase compared with that of strain BOH31 harboring the PpsbA2-pduP-slr1192OP unit (Fig. 3a and Fig. S7a, ESI†).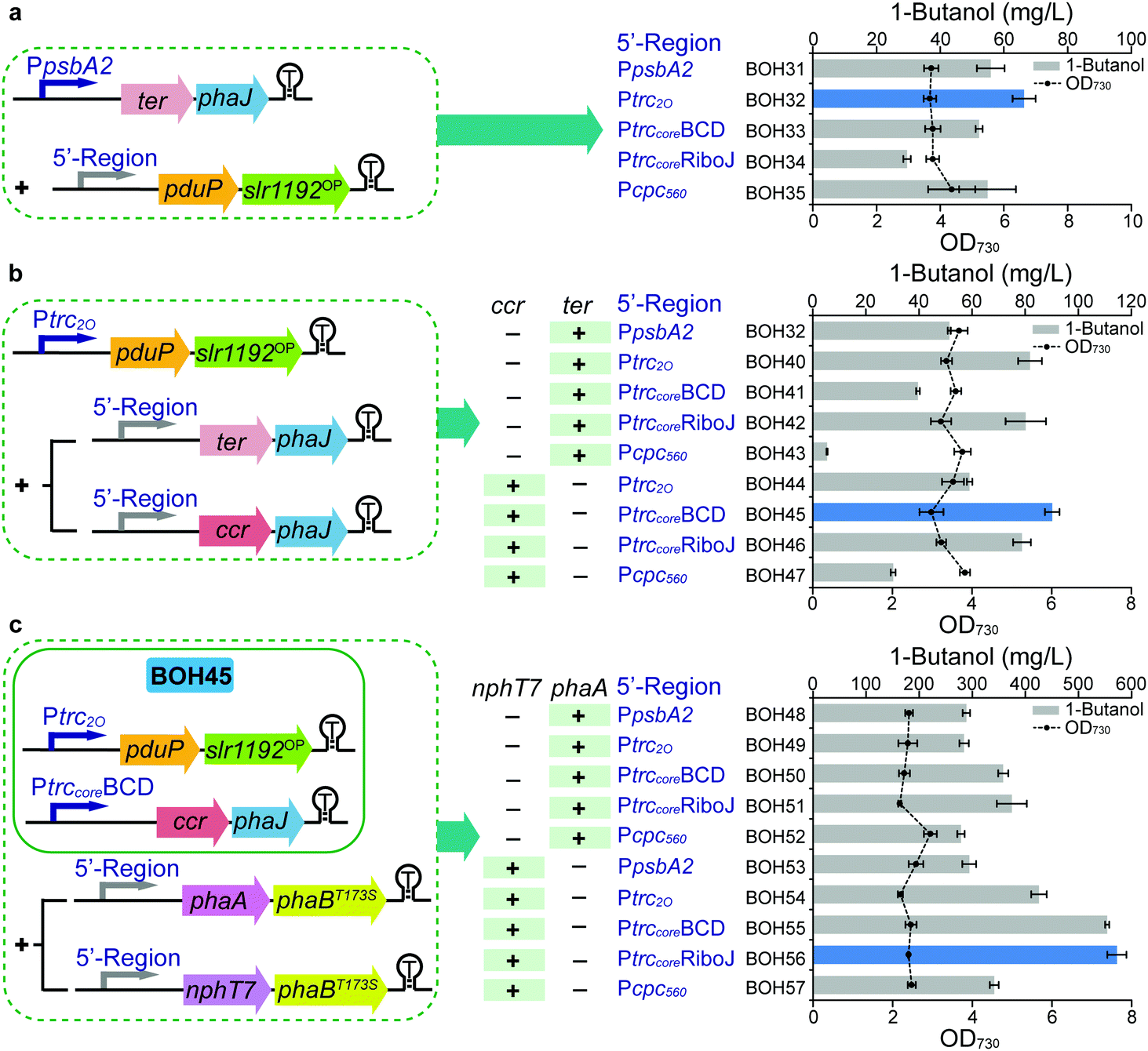 | ||
| Fig. 3 Systematic evaluation and optimization of the 5′-regions of expression units (Module 1 + 2) in Synechocystis PCC 6803. (a) 1-Butanol production and cell density of strains BOH31–BOH35 to screen the 5′-regions of the pduP-slr1192OP operon via the last four steps of the clostridial pathway. (b) 1-Butanol production and cell density of strains BOH32 and BOH40–47 to screen the 5′-regions of the ter-phaJ and ccr-phaJ operons via the last four steps of the clostridial pathway. (c) 1-Butanol production and cell density of strains BOH48–57 to screen the 5′-regions of the nphT7-phaBT173S and phaA-phaBT173S operons via the clostridial pathway. For (a–c), the time point, colored columns, “+”, “−” and other detailed annotations are as described in the Fig. 2(b–e) legend. All data are presented as mean ± SD of biological triplicates. Illustration of the expression units in the strategy designed for 5′-region screening is show on the left of each panel. | ||
Inspired by this, we moved on to the ter/ccr-phaJ operon. In previous studies of the driving forces of the biosynthesis of 1-butanol and other higher alcohols, the implementation of the irreversible activity of NADH-specific Ter is a widely used strategy.6,16,18,20,21,41 Since this step was recognized as a bottleneck of the pathway,20 we speculated that the 1-butanol biosynthesis could be improved by the likewise irreversible reaction catalyzed by NADPH-binding Ccr29 (Fig. 2a), which should be favored by the abundant NADPH content in Synechocystis, as noted above. In Module 1, our attempt to express ccr and phaJ under the PpsbA2 promoter had failed, but by changing the promoters or 5′-regions, four ccr-phaJ operons could be successfully constructed (Fig. 3b). Together with five ter-phaJ operons, the 1-butanol production performance of these operons was compared based on the previous selection of the Ptrc2O-pduP-slr1192OP unit (strains BOH40 to BOH47). Among the tested strains, strain BOH45 harboring the PtrccoreBCD-ccr-phaJ unit had the maximal 1-butanol titer (90.2 mg L−1) (Fig. 3b). Consistent with the titer, the expression levels of Ccr and PhaJ were the highest in this strain (ESI,† Fig. S7b). In the previous screening, only a medium expression level of PduP and Slr1192OP was achieved from the Ptrc2O-pduP-slr1192OP unit. However, after installing the PtrccoreBCD-ccr-phaJ unit in strains BOH36 to BOH39, it was found that the Ptrc2O promoter for the pduP-slr1192OP operon (strain BOH45) still worked best, and did not limit the 1-butanol production (ESI,† Fig. S8). The fact that a medium-strength expression level of PduP is beneficial for 1-butanol formation may be because the natural direction of PduP favors reversed butyryl-CoA formation.24,25
Based on the optimized constructs Ptrc2O-pduP-slr1192OP and PtrccoreBCD-ccr-phaJ in strain BOH45, the performance of the phaA/nphT7-phaBT173S operon under different promoters or 5′-regions was further investigated (Fig. 3c). Among the resultant strains BOH48 to BOH57, strain BOH56 harboring the PtrccoreRiboJ-nphT7-phaBT173S unit had the largest improvement in the 1-butanol titer to 572 mg L−1, a 5.3-fold increase compared with that of strain BOH45 (Fig. 3c), together with the highest protein expression levels of NphT7 and PhaB (ESI,† Fig. S7c). The 1-butanol titer of the NphT7 route in strain BOH56 was 0.5× higher than that of the PhaA route in strain BOH51 (374 mg L−1), demonstrating that NphT7 in fact is not limited by the ACC reaction and malonyl-CoA level, and thus could more efficiently supply acetoacetyl-CoA.
By successively applying transcriptional and translational modification via promoters and 5′-regions in Module 2, 1-butanol production was greatly improved to 572 mg L−1 (strain BOH56), which was 2.3-fold higher compared to that of strain BOH28 in Module 1 (Fig. 3c). Combining Module 1 with Module 2, a further optimized 1-butanol biosynthetic pathway and 5′-regions of the three expression units were thus obtained, including an increase in the ratio of required NADPH/NADH to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI,† Fig. S9). Our systematic evaluation at the transcriptional and translational levels clearly demonstrates the importance of the components and sequences of the 5′-regions in expression units.
:1 (ESI,† Fig. S9). Our systematic evaluation at the transcriptional and translational levels clearly demonstrates the importance of the components and sequences of the 5′-regions in expression units.
Rewiring the carbon flux (Module 1 + 2 + 3)
In the previous modules described above, the phaEC, acs, and pta sites were selected as genomic integration loci for the three expression units (ESI,† Table S5). PHB accumulation, resulting from the irreversible conversion of intermediate (R)-3-hydroxybutyryl-CoA catalyzed by PHB synthase (PhaE and PhaC) in Synechocystis,23 directly competes with 1-butanol biosynthesis (Fig. 1). Hence, phaEC is good choice of knock-out/integration site. In addition, as the building block of 1-butanol, acetyl-CoA can be interconverted to acetate via the “triangular” acetate metabolism, consisting of the Pta (phosphotransacetylase), AckA (acetate kinase), Ach (acetyl-CoA hydrolase) and Acs (acetyl-coenzyme A synthetase) pathways (Fig. 1).42 Since acetyl-CoA acts as the branching point for 1-butanol biosynthesis, acetate metabolism, and other competing metabolic pathways, the availability of acetyl-CoA may affect the carbon flux towards 1-butanol production.Therefore, in Module 3, we manipulated the flow of acetyl-CoA by deleting genes responsible for the acetate metabolism to save this building block for 1-butanol biosynthesis. Furthermore, more possible genomic integration sites for integration of gene expression units were explored. 1-Butanol production levels were compared among strains using four integration loci, acs (strain BOH45), ach (strain BOH58), pta (strain BOH59), and the neutral site slr016843 (strain BOH60), for installing the PtrccoreBCD-ccr-phaJ unit (Fig. 4a). Moreover, the expression unit of Ptrc2O-pduP-slr1192OP was placed at the phaEC site in all tested strains to allow functional 1-butanol production. Among the tested sites, the 1-butanol titer produced by strain BOH58 (96.5 mg L−1) indicated that integration in and deletion of the ach site worked best (Fig. 4a). From western-immunoblotting data, the enzyme expression levels were comparable in all four strains (ESI,† Fig. S10), demonstrating that blocking the conversion of acetyl-CoA to acetate could enhance 1-butanol biosynthesis. The differences of 1-butanol production between strains with different knockouts in the acetate metabolism indicated that one of the key points in 1-butanol biosynthesis is most likely the cytosolic acetyl-CoA level.
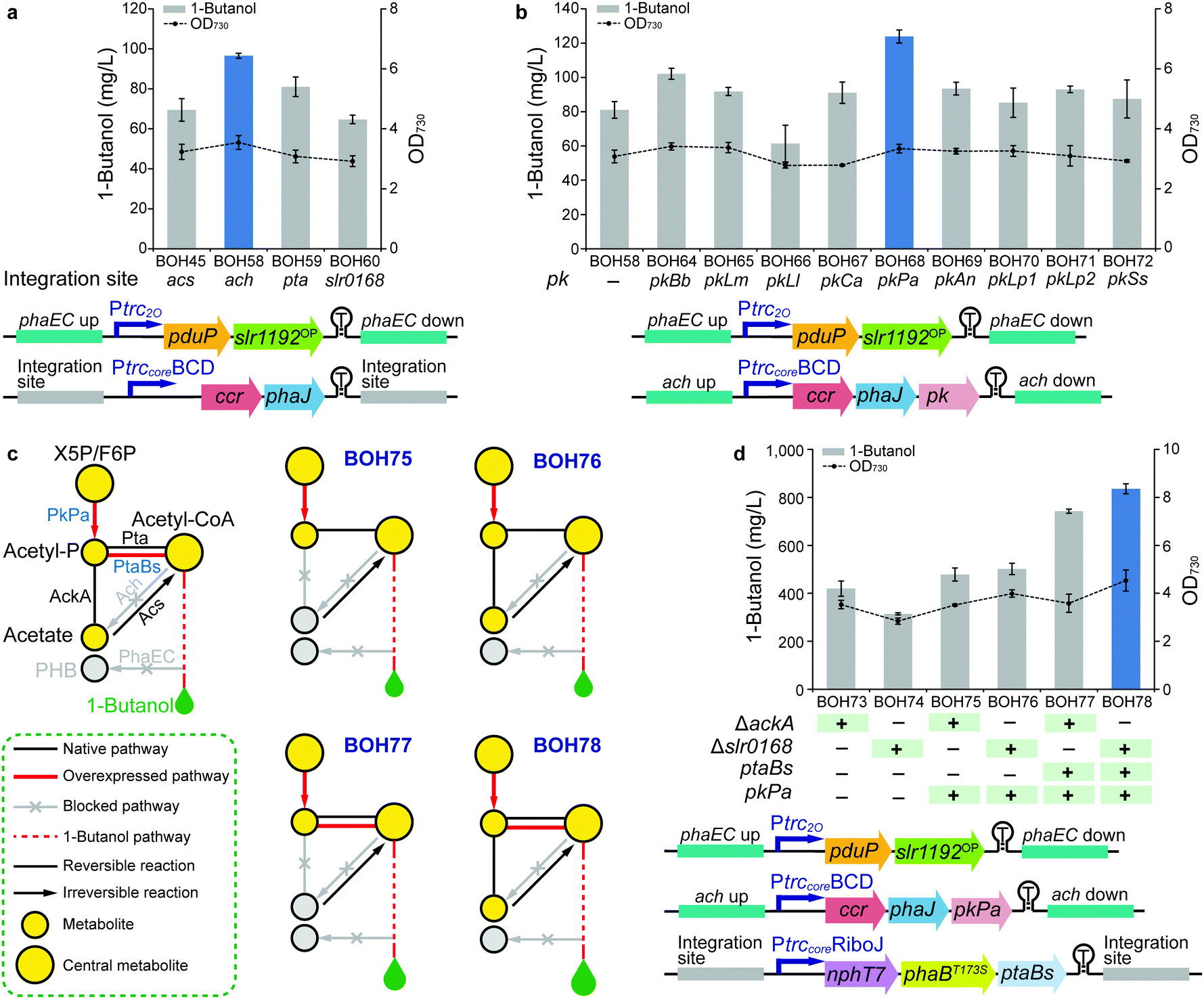 | ||
| Fig. 4 Editing the acetate metabolism and installing the PK pathway in Synechocystis PCC 6803 (Module 1 + 2 + 3 + 4). (a) 1-Butanol production and cell density of strains BOH45 and BOH58–60 to compare four integration sites via the last four steps of the clostridial pathway. (b) 1-Butanol production and cell density of strains BOH58 and BOH64–72 to screen nine PKs via the last four steps of the clostridial pathway. (c) Simplified schematics of four rewiring and rewriting strategies in strains BOH75–78 by assembling Module 3 and 4. The pathway overview model in the left panel describes related metabolites, pathways and enzymes. Heterologous, endogenous and inactivated endogenous enzymes are shown in blue, black and grey fonts, respectively. See Fig. 1 regarding the abbreviations of the metabolites. (d) 1-Butanol production and cell density of strains BOH73–78 to compare six rewiring and rewriting strategies via the clostridial pathway. For (a, b and d), the time point, colored columns, “ +”, “−” and other detailed annotations are as described in the Fig. 2(b–e) legend. All data are presented as mean ± SD of biological triplicates. Genetic architectures of expression units are shown at the bottom of each panel. | ||
Rewriting the central carbon metabolism (Module 1 + 2 + 3 + 4)
The main carbon flux in cyanobacterial cells, generated from the Calvin–Benson–Bassham (CBB) cycle, is channeled through the lower part of the glycolysis pathway, then via pyruvate to acetyl-CoA9 (Fig. 1). The sugar catabolism (e.g. glycolysis pathway) in Synechocystis is subject to regulation at the transcriptional level. Therefore, as the first experiment in Module 4, SigE, Rre37 and Hik8, known as positive transcriptional regulators of the sugar catabolism in Synechocystis,44 were overexpressed to enhance 1-butanol production. However, when compared with strain BOH56, the introduction of the regulators in strains BOH61–63 did not exert a positive effect on 1-butanol production, and resulted in certain negative effects on protein expression levels (ESI,† Fig. S11).Another strategy to increase the carbon flux via acetyl-CoA toward 1-butanol production is to install a phosphoketolase (PK) pathway (Fig. 1). PK catalyzes the cleavage of a 5-carbon xylulose-5-phosphate (X5P) into a 3-carbon glyceraldehyde-3-phosphate (G3P) and a 2-carbon acetyl-P, or a 6-carbon fructose-6-phosphate (F6P) into a 4-carbon erythrose-4-phosphate (E4P) and an acetyl-P.45,46 This pathway enables a direct and irreversible conversion of sugar phosphate in the central carbon metabolism to acetyl-P and further on to acetyl-CoA, which is highly efficient when compared with the original acetyl-CoA synthesis via the glycolysis pathway. It also serves as a carbon-conserving shunt, compared to the carbon-wasting glycolysis pathway, in which one carbon is lost in the form of CO2 with each acetyl-CoA generated from pyruvate45 (Fig. 1). In cyanobacteria, where the carbon flux through the OPP (oxidative pentose phosphate) pathway is low under photoautotrophic conditions, the PK could divert X5P and F6P from the CBB cycle,47 forming an effective shortcut from the CBB cycle to acetyl-P, and thus redirecting flux into 1-butanol biosynthesis (Fig. 1). Overexpression of a PK enzyme was shown to lead to higher production of PHB in one study in Synechocystis.42 PK genes and the corresponding enzymes, which have previously been experimentally verified and characterized in several filamentous fungi, two cyanobacteria and some prokaryotic heterotrophic bacteria (ESI,† Table S3), display significant differences in their substrate specificities and catalytic kinetics towards X5P and F6P. Therefore, nine PKs were chosen as candidates to investigate their distinct efficiencies towards 1-butanol production (ESI,† Table S3). For the strain construction, each individual pk gene was cloned into the ccr-phaJ operon and integrated in the ach site, combined with the two optimized expression units selected in Module 3 (Fig. 4b). Among the nine resultant strains, BOH64-BOH72, the strain BOH68 overexpressing a PK from Pseudomonas aeruginosa (PKPa) had the highest 1-butanol titer, 124 mg L−1, representing a 53% increase compared with that of strain BOH58 without PK overexpression (Fig. 4b). This is the first application of PKPa in metabolic engineering, and it represents a promising option of a PK for engineering biosynthetic pathways utilizing acetyl-CoA as a precursor.
After fixing the most efficient PKPa pathway, we then combined Module 3 and 4 to look for potential bottlenecks in the acetate metabolism for acetyl-CoA formation from acetyl-P that are increased by PK overexpression (Fig. 4c). For the formation of acetyl-CoA, there are two distinct routes, namely the Pta route and the AckA-Acs route. In the Pta-catalyzed route, acetyl-P is directly converted into acetyl-CoA, while the second route consists of two reactions catalyzed by AckA and Acs, in which acetyl-P is first converted into acetate and then forms acetyl-CoA (Fig. 1 and 4c). Improving the carbon flux from acetyl-P to acetyl-CoA via the Pta route seems to be a more effective strategy. Thus, the Pta from Bacillus subtilis (PtaBs), a shorter variant of Pta compared to the Pta found in E. coli and Synechocystis,48 was selected to construct a synthetic PKPa-PtaBs route (Fig. 1 and 4c). Then, the PKPa or PKPa-PtaBs route was combined with the deletion of either ackA or the neutral site slr0168, resulting in strains BOH75–78 (Fig. 4c). Strains BOH73–74, lacking overexpressed PKPa, were constructed as controls. The difference between strain BOH75/77 and strain BOH76/78 was thus the blocked or open metabolic flux through the native AckA-Acs route. We conclude that combining PKPa and PtaBs enabled an enhanced carbon flux to acetyl-CoA, since strains BOH77/78 showed significantly higher 1-butanol production than strains BOH75/76 (Fig. 4d). Interestingly, the slr0168-deletion strain BOH78 generated a 13% increase in the 1-butanol titer (836 mg L−1) compared with that of strain BOH77 harboring the ackA deletion (742 mg L−1), probably because the native AckA-Acs route could provide additional flux from acetyl-P to acetyl-CoA. Hence, the optimal way to convert sugar to acetyl-CoA in Synechocystis is via a combination of the PKPa-PtaBs and PKPa-AckA-Acs routes (Fig. 4c).
From all information collected in the four modules of work described above, we conclude that the combined PKPa-PtaBs and PKPa-AckA-Acs routes are capable of efficiently diverting the carbon flux towards acetyl-CoA and drawing the equilibrium towards 1-butanol, when effectively irreversible steps have been implemented downstream in the 1-butanol biosynthetic pathway, such as the steps catalyzed by ACC, NphT7 and Ccr, and finally secretion of 1-butanol from the cells (ESI,† Fig. S12). The 1-butanol titer of strain BOH78, 836 mg L−1, was at the conclusion of this part of our work the highest 1-butanol titer observed in cyanobacteria (ESI,† Table S1).
Long-term cultivation of BOH78
So far, we established, optimized and assembled four consecutive work modules with the best strain BOH78 producing 836 mg L−1 of 1-butanol (Fig. 5a and Fig. S12, ESI†). Then, the photosynthetic conditions in the sealed cultivation system were optimized to further enhance the 1-butanol titer of strain BOH78. A modified long-term cultivation of Synechocystis was set up using a pH adjustment approach49 (ESI,† Fig. S13). The growth and 1-butanol production under different light intensities and NaHCO3 feeding rates were examined for strain BOH78 and control strain EmptyV5 (ESI,† Fig. S14). The cell growth of strain BOH78 was slightly slower than the control, which maintained a longer steady state than strain BOH78, with no dramatic drop in growth in 30 days. Compared to the control, the effect of the light intensity on the cell growth of strain BOH78 was less significant, and strain BOH78 showed similar growth trends under two different NaHCO3 feeding rates. The 1-butanol titer of strain BOH78 observed in cultures grown under 100 μmol photons m−2 s−1 was not enhanced compared to that in cultures grown under lower light intensities. We observed the highest 1-butanol titer from cells grown under 50 μmol photons m−2 s−1 with NaHCO3 feeding every day. A maximal in-flask 1-butanol titer of 2.13 g L−1 was observed at day 23 with a continuous decrease of the cell density from day 12 (Fig. 5b). This indicates that the cell growth does not correlate with the 1-butanol production levels. The 1-butanol concentration reached is higher than that observed to affect growth when 1-butanol was added externally to cells of Synechocystis.50 However, the response may be different when the 1-butanol is produced internally. We continuously measured the 1-butanol production until day 30, when the cultivation was terminated due to a drop in the cumulative 1-butanol titer for the last two days. A final cumulative titer of 4.8 g L−1 1-butanol at day 28 was obtained from strain BOH78. However, when feeding NaHCO3 only every second day (ESI,† Fig. S14c), the maximal in-flask titer reached 1.86 g L−1 at day 23 with a cumulative titer of 3.0 g L−1 at day 26, demonstrating the importance of efficient removal of the produced 1-butanol.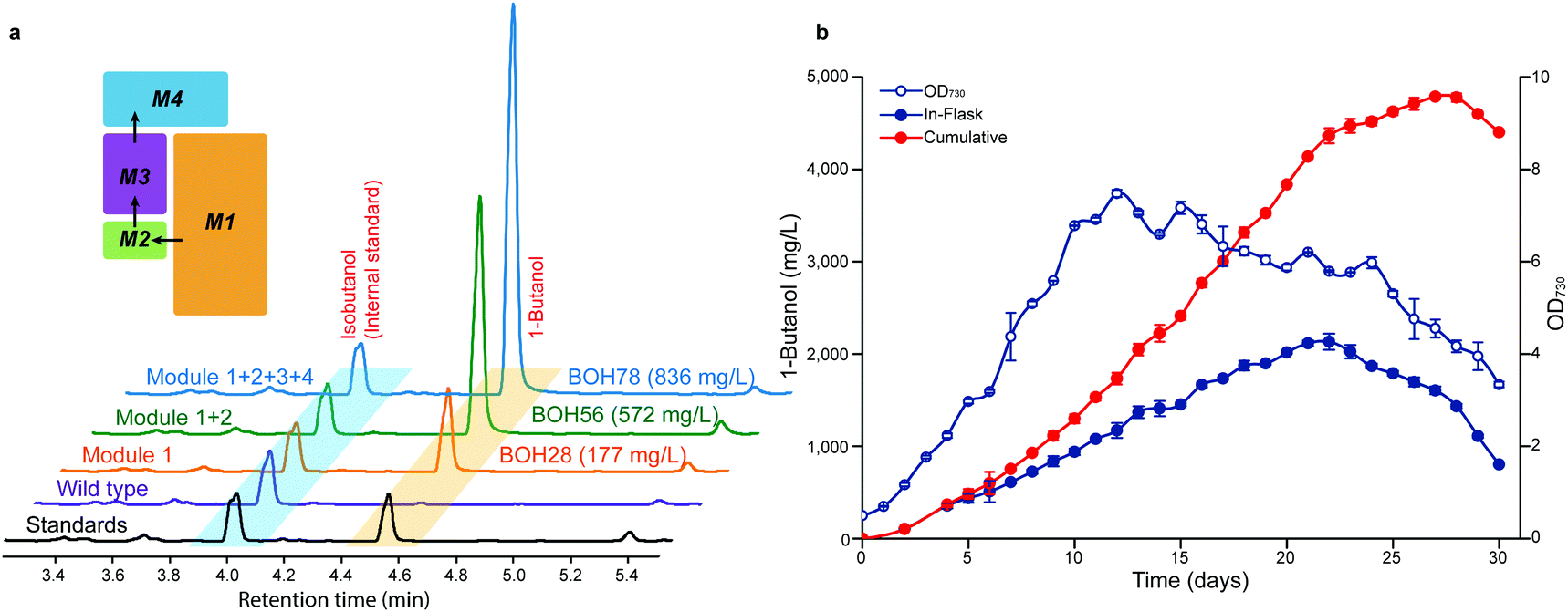 | ||
| Fig. 5 Sequential improvements of 1-butanol production with four consecutive modules assembled and a long-term cultivation experiment for Synechocystis PCC 6803. (a) Representative gas chromatogram (GC) profiles of 1-butanol production from the best engineered strains in different combinatorial modules. The Synechocystis wild type is used as the negative control. The shown chromatogram is one of three similar traces from triplicate cultures. (b) Time course of OD730, in-flask and cumulative 1-butanol production of strain BOH78 cultivated under a light intensity of 50 μmol photons m−2 s−1 with feeding NaHCO3 every day for 30 days. The cumulative titer of 1-butanol takes into account the dilutions made to the cyanobacteria culture broth by feeding NaHCO3. All data are presented as mean ± SD of biological triplicates. | ||
The maximal 1-butanol production of 4.8 g L−1 is 11 times higher than previously reported in cyanobacteria, from engineered cells of Synechococcus elongatus PCC 7942, and even higher than the observed production when using engineered Saccharomyces cerevisiae (ESI,† Table S1). The average 1-butanol productivity during the 28 day cultivation period was 171 mg L−1 day−1, with a maximal rate of 302 mg L−1 day−1 observed between days 15 and 18. The titer and rates of 1-butanol production reported in the present study are comparable with ethanol production (5.5 g L−1, 212 mg L−1 day−1 during 26 days),5 but higher than 2,3-butanediol production (2.38 g L−1, 119 mg L−1 day−1 during 20 days)13 and isoprene production (1.26 g L−1, 60 mg L−1 day−1 during 21 days)8 in cyanobacteria. However, ethanol, 2,3-butanediol and partly isoprene are pyruvate-derived products, whereas a higher alcohol like 1-butanol uses acetyl-CoA as the building block.
Conclusions
Throughout this work, our multi-level modular strategy transformed wild type Synechocystis cells into engineered Synechocystis strains with sequential improvements in 1-butanol production reaching 4.8 g L−1 with a maximal rate of 302 mg L−1 day−1 (Fig. 5a and 6). Various genetic modifications and multiple enzyme components, building on previous knowledge, were introduced in each work module and combined synergistically, allowing for fine design of the overall 1-butanol biosynthetic pathway. The designs used here are not limited to enhancing 1-butanol biosynthesis in cyanobacteria but can also be applied in metabolic engineering of other microorganisms, including E. coli and S. cerevisiae, for relevant chemicals production. Since some of the 1-butanol forming enzymes are widely applicable for the biosynthesis of other higher alcohols, acids or alkanes,3,4,16,18,22,28,41 the genes and enzymes explored in our study can also be used in the biosynthesis of these acyl-CoA dependent products. The results from introducing and screening a large number of phosphoketolases (PK) will be useful for all applications requiring acetyl-CoA as a substrate.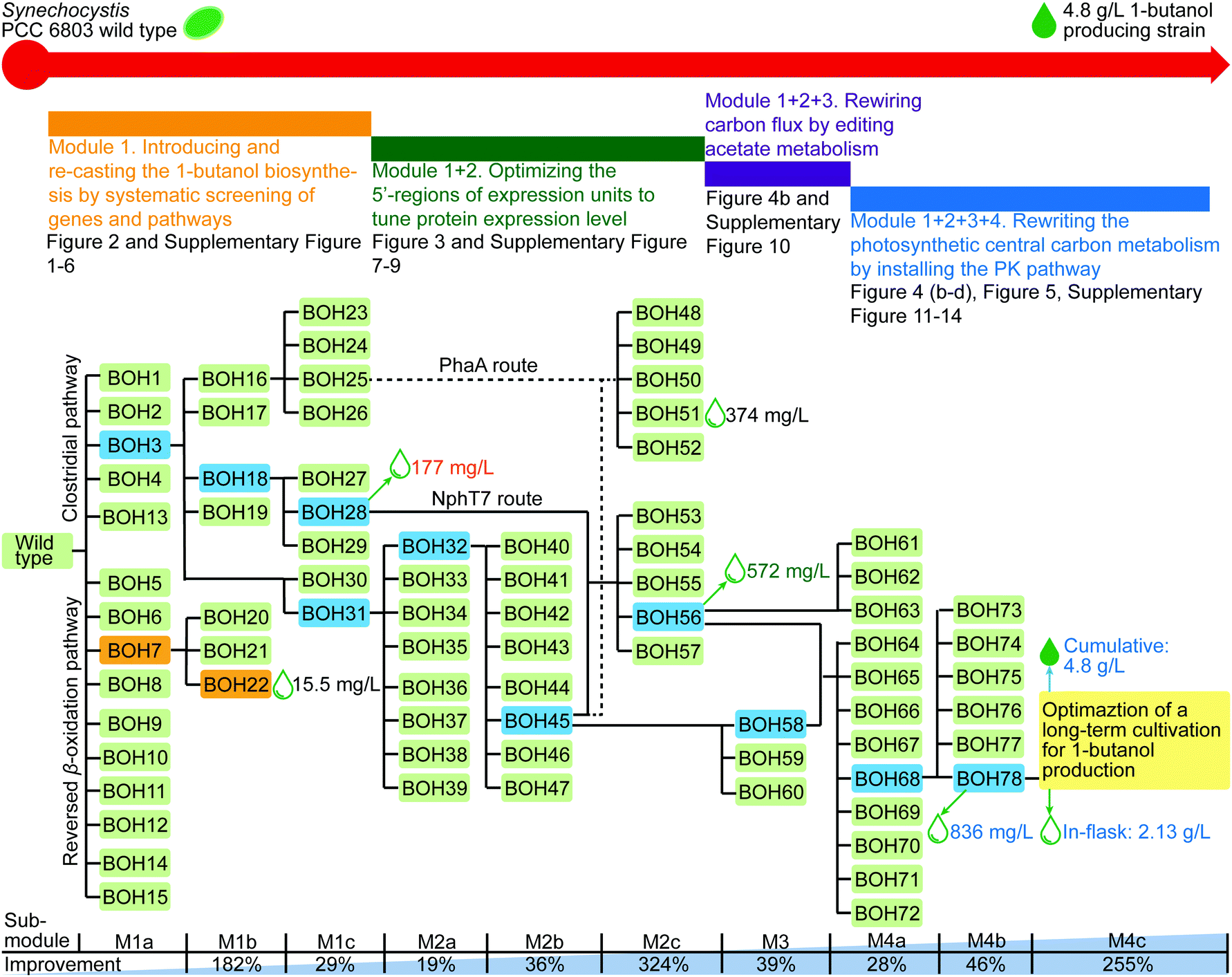 | ||
| Fig. 6 Experimental development of 1-butanol producing Synechocystis strains through our multi-level modular engineering strategy. The blueprint of the process from the Synechocystis wild type to a 4.8 g L−1 1-butanol producing strain is shown at the top, with the composition of combinatorial Modules 1, 2, 3 and 4. The related figures are listed below each Module part. The flowchart of strains with “laboratory evolution” is shown in the middle. This “genealogy map” include all the strains constructed in this study, by dividing them into different pathway and route schemes. The engineered strains connected to the next developed engineering stage or strains with the highest 1-butanol production in each step are highlighted in colors different from green. The target strains in the clostridial pathway and reversed β-oxidation pathway are shown in blue and orange, respectively. The detailed 1-butanol production of the best strains in the different pathways or combinatorial modules is also described beside. Sequential modifications in modules can be divided into different steps, called sub-modules. A diagram summarizing the sub-modules is shown in the lower part, together with a summary of stepwise improvements for in-flask 1-butanol production (in %) in Synechocystis PCC 6803. | ||
Finally, we demonstrated the potential of cyanobacteria to be engineered as microbial cell factories, with a resulting product titer comparable with those achieved in heterotrophs. Our study contributes new insights to the field of metabolic engineering of cyanobacteria. Additional optimizations and use of dedicated automatic and continuous photobioreactor growth systems, operated in e.g. chemostat or turbidostat mode, in combination with product specific harvesting technologies including continuous removal of the products,51 will make industrial biotechnological applications of cyanobacteria for direct production of chemicals and biofuels achievable.
Materials and methods
Plasmid construction
All the heterogenous genes and some endogenous genes were codon optimized and synthesized by GenScript. Other endogenous genes and DNA fragments (homologous recombination regions and Pcpc560) were PCR amplified from the wild-type Synechocystis PCC 6803 genome using gene specific primers and Phusion Polymerase (Thermo Fisher Scientific). The annotations of all genes, 5′-regions and other genetic elements are listed in the ESI,† Table S2. The sequences of codon optimized synthetic genes are listed in the ESI,† Table S7. PpsbA2, RBS* and Terminator BBa_B0015 fragments were from pEERM1.52 Ptrc2O, PtrccoreBCD and PtrccoreRiboJ promotor fragments were amplified from pEEC series vectors.53 The plasmids constructed in this study are listed in the ESI,† Table S4. The primers used in this study are listed in the ESI,† Table S6.E. coli strain DH5α was used for constructing the plasmids containing PpsbA2 and Pcpc560. E. coli strain DH5αZ1 (Invitrogen) was used for constructing the kanamycin or erythromycin resistance cassette carrying plasmids containing Ptrc2O, PtrccoreBCD or PtrccoreRiboJ, while E. coli strain T7 Express (NEB) was used for constructing the spectinomycin resistance cassette carrying plasmids containing Ptrc2O, PtrccoreBCD or PtrccoreRiboJ. All the strains were grown at 37 °C in LB medium (liquid or agar Petri dish) supplemented with the corresponding antibiotics (Sigma-Aldrich). Kanamycin, spectinomycin and erythromycin were used at a concentration of 50, 50, and 200 μg mL−1 for strains with the corresponding resistance marker, respectively. All the backbone vectors were made based on the recently reported pEERM series of vectors52 by changing the homologous regions, promoters and antibiotic resistance cassettes with restriction enzyme digestion and ligation (ESI,† Table S4). The homologous recombination regions in all plasmids are the around 1000 bp upstream sequence and 1000 bp downstream sequence of the integrated gene locus. Further versions of the integrated plasmids were made based on existing backbone vectors with the BioBrick suffix and prefix for multi-step cloning52 (ESI,† Table S4). Strong ribosome binding site RBS* was added in front of each gene, except the first gene behind the promoter, to allow initiation of translation. Bgl II, EcoR I, Xba I, Spe I, Pst I, BamHI and Sal I were the restriction cloning sites used to construct all the plasmids in this study. All digestion enzymes to cut the restriction cloning sites were fast digestion enzymes from Thermo Fisher Scientific. E. coli colonies with positive plasmids were confirmed with colony PCR using gene specific primers (ESI,† Table S6) and DreamTaq DNA polymerase (Thermo Fisher Scientific). Plasmids purified with miniprep (Thermo Fisher Scientific) were checked by enzyme digestion before Synechocystis transformation.
Transformation of Synechocystis
The glucose-tolerant Synechocystis PCC 6803 (Synechocystis) strain was used in this study27 (ESI,† Table S5). Transformation was performed by using a double homologous-recombination system, and the genes were integrated into the target site of the Synechocystis genomic DNA. The Synechocystis strains constructed in this study are listed in the ESI,† Table S5. Background strains (Synechocystis strains transformed with the corresponding backbone vectors) were used as a control for the engineered strains with genetic constructs on the chromosome. Kanamycin was used at a concentration of 50 μg mL−1 for strains with only the kanamycin resistance marker. For strains with multiple resistance markers, kanamycin, spectinomycin and erythromycin were used at 25 μg mL−1.Natural transformation with plasmids was performed via an optimized protocol based on previous reports.54–60Synechocystis cells were grown to the mid-log phase with OD730 = 0.5–1.2 in liquid BG11 medium. Then, the cell culture was collected by centrifugation at 5000 × g for 5 min and washed twice with fresh liquid BG11 medium without antibiotics, and finally resuspended into fresh liquid BG11 medium at a density of 1 × 109 cells·mL−1 (OD730 = 2.5). A total of 400 μL concentrated Synechocystis cells were mixed with 4 μg target plasmid DNA. After incubating under constant 50 μmol photons m−2 s−1 illumination at 30 °C for 4–5 hours, the cells were spread on membrane filters on BG11 agar plates without antibiotics for another 18–24 hours incubation under constant 50 μmol photons m−2 s−1 illumination at 30 °C. For colony selection and maintenance, the filters were changed onto new BG11 agar plates with the corresponding antibiotics and incubated under the same conditions. Isolated single colonies (transformants) were streaked on a new BG11 agar plate with the corresponding antibiotics, and were analyzed by PCR using gene specific primers (ESI,† Table S6) and DreamTaq DNA polymerase (Thermo Fisher Scientific) to verify successful integration of the expression cassette into the genomic DNA. Positive homologous recombinants were inoculated into 6-well plates and further cultivated in liquid BG11 medium with the corresponding antibiotics and propagated continuously until fully segregated. The segregation of each strain was examined by PCR using gene specific primers (ESI,† Table S6) and DreamTaq DNA polymerase (Thermo Fisher Scientific) on the extracted genome DNA prepared via an established protocol.61
Cultivation conditions for 1-butanol production
All experimental cultures were grown in BioLite 25 cm2 plug-sealed tissue culture flasks (Thermo Fisher Scientific) to avoid 1-butanol evaporation during cultivation.27 The NaHCO3 supplemented was the inorganic carbon source for photosynthesis in Synechocystis. All experimental cultures were prepared in triplicate. Light was measured using an LI-190SB Quantum sensor.Seed cultures of strains were grown under constant 50 μmol photons m−2 s−1 illumination at 30 °C in liquid BG11 medium with the corresponding antibiotics in 100 mL Erlenmeyer flasks (VWR) until the mid-log phase. For the HEPES-buffered cultures, each seed culture was then inoculated to OD730 = 0.1 with a total volume of 25 mL liquid medium in plug-sealed tissue culture flasks. The medium used was BG11 medium with 50 mM NaHCO3 (Sigma-Aldrich), 25 mM HEPES (pH 7.5, Sigma-Aldrich) and the corresponding antibiotics. The initial OD730 was changed to 0.5 and the NaHCO3 concentration was reduced to 20 mM for the cultivation of strains BOH48–57, BOH61–63 and BOH73–78 due to the specific phenotype of these strains. The flasks were shaken at 120 rpm, under constant 50 μmol photons m−2 s−1 illumination at 30 °C. 100 μL culture was sampled from each flask daily for OD730 measurements. An additional 2.3 mL culture was sampled from each flask every second day for 1-butanol extraction while 2.5 mL fresh BG11 medium with 500 mM NaHCO3, 25 mM HEPES (pH 7.5) and the corresponding antibiotics was added back for filling up to the original volume. This procedure ensures a sustained carbon supply for Synechocystis. The cultivation was terminated when the 1-butanol titer in the culture began to decrease, and then the maximal in-flask 1-butanol titer was obtained.
In the long-term cultivation experiment, each seed culture was then inoculated to OD730 = 0.5 with a total volume of 25 mL liquid modified BG11 medium with 20 mM NaHCO3 and the corresponding antibiotics. For examining different light intensities, the flasks were shaken at 120 rpm, at 30 °C, under constant 20, 50, and 100 μmol photons m−2 s−1, respectively. 2.5 mL culture was sampled from each flask every day or every second day for OD730 measurements and 1-butanol extraction while 2.5 mL fresh BG11 medium with 500 mM NaHCO3 and the corresponding antibiotics was added back for filling up to the original volume. For the HCl titrated cultures, a certain amount of 37% HCl (Sigma-Aldrich) was added every day to adjust the pH of the culture to around 7.5, which is close to the theoretical optimal pH for Synechocystis.62 The pH was measured using MColorpHast™ pH-indicator strips (pH 6.5–10) (Merck). The detailed experimental procedure is shown in the ESI,† Fig. S13.
Optical density measurements and 1-butanol extraction
The cell growth was monitored by measuring the optical density at 730 nm (OD730) of each culture. The absorbance at 730 nm was measured for 200 μL diluted culture every day in 96-well plates using a micro-plate reader (HIDEX, Plate Chameleon).For 1-butanol extraction, 2.3 mL culture was centrifuged at 5000 rpm, for 10 min. Then, 2.07 mL supernatant was transferred into a 15 mL screw cap tube, and mixed with 30 μL 7000 mg L−1 internal standard isobutanol (Sigma-Aldrich) and 700 μL (1/3 volume) extraction solvent DCM (dichloromethane, Sigma-Aldrich). The mixture was shaken on a Multi-Tube Vortexer VX-2500 (VWR) at maximum speed for 5 min and then centrifuged at 5000 rpm, 4 °C, for 10 min, to get a faster and clearer separation of the two phases. The DCM phase (bottom phase) was transferred into 1.5 mL clear glass gas chromatography (GC) vials (VWR).
1-Butanol quantification by gas chromatography
The detection method was described in a previous study.27 In short, the extracted samples were analyzed on a PerkinElmer GC 580 system equipped with a flame ionization detector (FID) and an Elite-WAX polyethylene glycol series capillary column, 30 m × 0.25 mm × 0.25 μm (PerkinElmer). Nitrogen was the carrier gas, with a 10 mL min−1 flow rate. The temperatures of the injector and detector were 220 °C and 240 °C, respectively. The initial oven temperature was 50 °C and then it was raised to 100 °C with a rate of 10 °C min−1 followed by a rise to 180 °C with a rate of 20 °C min−1. The GC results were analyzed using TotalChrom Navigator version 6.3.2.The retention time was determined using 100 mg L−1 isobutanol (Sigma-Aldrich) and 1-butanol standards (VWR). The chromatographic peaks of 1-butanol and isobutanol were indicated at a retention time of 4.55 min and 4 min, respectively, as shown in Fig. 5a. The presence of 1-butanol was identified by the retention time and comparison with the 1-butanol standard. All samples were spiked with the same amounts of isobutanol (100 mg L−1), which was used as the internal standard for quantitative determination. Thus, a standard curve was made by measuring extracts (extracted in the same way as our samples) from liquid BG11 medium with nine different concentrations of the 1-butanol standard (10 mg L−1, 25 mg L−1, 50 mg L−1, 75 mg L−1, 100 mg L−1, 250 mg L−1, 500 mg L−1, 750 mg L−1 and 1000 mg L−1) and 100 mg L−1 of isobutanol as an internal standard. The amount of 1-butanol in the culture (in-flask titer of 1-butanol) was calculated based on the ratio of its signal peak area and that of the internal standard. The cumulative titer of 1-butanol takes into account the dilutions made to the cyanobacteria culture broth by feeding.
Crude protein extraction and Western-immunoblotting
The analyzed protein samples were the soluble fractions in the crude extract from the cell lysate of the engineered Synechocystis strains. Proteins extraction was done via a modified protocol based on a previous study.63 5 mL of culture was harvested from an OD750 = 2 culture by centrifugation at 4500 rpm, 4 °C, for 5 min. The pellet was washed in 2 mL PBS buffer and collected again by centrifugation at 4500 rpm, 4 °C, for 5 min. Then, 200 μL PBS buffer supplemented with 4 μL of 50× Protease Arrest (GBioscience) was used to resuspend the pellet and this mixture was frozen at −80 °C for 4 min followed by heating at 37 °C for 4 min. For the remainder of the procedure, the samples were kept on ice. Acid-washed glass beads (425–600 μm diameter, Sigma-Aldrich) were mixed with the thawed cells. The cells were disrupted using a Precellys-24 Beadbeater (Bertin Instruments); a 5000 rpm program for 4 × 30 s with 2 minutes of rest on ice between the runs is sufficient. 100 μL PBS buffer was added and the total lysates were briefly centrifuged twice at 1000 × g, 4 °C, 30 s, to get a supernatant containing soluble proteins.The protein concentration was determined by the DC protein assay (Bio-Rad). 20 μg total soluble proteins from each strain were loaded in each lane and separated by running SDS-PAGE, using Mini-PROTEAN TGX™ gels (Bio-Rad), and transferred to a “Trans-Blot Turbo Transfer Pack” membrane (Bio-Rad) by a membrane transfer machine (Bio-Rad). Commercially available anti-Strep-tag antibody (abcam), anti-Flag-tag antibody (Sigma) and anti-His-tag antibody (Genscript) were used to detect the corresponding tagged protein through the standard techniques of immunoblotting. The annotations including the N-terminal tag of all enzymes are in the ESI,† Table S2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Xuefeng Lv (Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences) for kindly providing the spectinomycin resistance cassette. This work was supported by the European Union Horizon 2020 Framework Programme under the grant agreement number 640720 (Photofuel), the Swedish Energy Agency (CyanoFuels, project number P46607-1), and the NordForsk NCoE program “NordAqua” (project number 82845).References
- A. Deneyer, E. Peeters, T. Renders, S. Van den Bosch, N. Van Oeckel, T. Ennaert, T. Szarvas, T. I. Korányi, M. Dusselier and B. F. Sels, Nat. Energy, 2018, 3, 969–977 CrossRef CAS.
- Y. J. Zhou, E. J. Kerkhoven and J. Nielsen, Nat. Energy, 2018, 3, 925–935 CrossRef CAS.
- S. Atsumi, W. Higashide and J. C. Liao, Nat. Biotechnol., 2009, 27, 1177–1180 CrossRef CAS PubMed.
- C. R. Shen and J. C. Liao, Energy Environ. Sci., 2012, 5, 9574–9583 RSC.
- Z. Gao, H. Zhao, Z. Li, X. Tan and X. Lu, Energy Environ. Sci., 2012, 5, 9857–9865 RSC.
- E. I. Lan, S. Y. Ro and J. C. Liao, Energy Environ. Sci., 2013, 6, 2672–2681 RSC.
- W. Xiong, J. A. Morgan, J. Ungerer, B. Wang, P.-C. Maness and J. Yu, Nat. Plants, 2015, 1, 15053 CrossRef CAS.
- X. Gao, F. Gao, D. Liu, H. Zhang, X. Nie and C. Yang, Energy Environ. Sci., 2016, 9, 1400–1411 RSC.
- M. Kanno, A. L. Carroll and S. Atsumi, Nat. Commun., 2017, 8, 14724 CrossRef CAS PubMed.
- A. Wegelius, N. Khanna, C. Esmieu, G. D. Barone, F. Pinto, P. Tamagnini, G. Berggren and P. Lindblad, Energy Environ. Sci., 2018, 11, 3163–3167 RSC.
- D. Lips, J. M. Schuurmans, F. Branco dos Santos and K. J. Hellingwerf, Energy Environ. Sci., 2018, 11, 10–22 RSC.
- J. W. Lee, D. Na, J. M. Park, J. Lee, S. Choi and S. Y. Lee, Nat. Chem. Biol., 2012, 8, 536–546 CrossRef CAS PubMed.
- J. W. Oliver, I. M. Machado, H. Yoneda and S. Atsumi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1249–1254 CrossRef CAS PubMed.
- P. K. Ajikumar, W. H. Xiao, K. E. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS PubMed.
- F. Zhang, J. M. Carothers and J. D. Keasling, Nat. Biotechnol., 2012, 30, 354–359 CrossRef CAS PubMed.
- H. C. Tseng and K. L. Prather, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17925–17930 CrossRef CAS PubMed.
- P. Xu, Q. Gu, W. Wang, L. Wong, A. G. Bower, C. H. Collins and M. A. Koffas, Nat. Commun., 2013, 4, 1409 CrossRef PubMed.
- M. J. Sheppard, A. M. Kunjapur, S. J. Wenck and K. L. Prather, Nat. Commun., 2014, 5, 5031 CrossRef CAS PubMed.
- J. Qin, Y. J. Zhou, A. Krivoruchko, M. Huang, L. Liu, S. Khoomrung, V. Siewers, B. Jiang and J. Nielsen, Nat. Commun., 2015, 6, 8224 CrossRef PubMed.
- C. R. Shen, E. I. Lan, Y. Dekishima, A. Baez, K. M. Cho and J. C. Liao, Appl. Environ. Microbiol., 2011, 77, 2905–2915 CrossRef CAS PubMed.
- E. I. Lan and J. C. Liao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6018–6023 CrossRef CAS PubMed.
- C. Dellomonaco, J. M. Clomburg, E. N. Miller and R. Gonzalez, Nature, 2011, 476, 355–359 CrossRef CAS PubMed.
- G. Taroncher-Oldenburg, K. Nishina and G. Stephanopoulos, Appl. Environ. Microbiol., 2000, 66, 4440–4448 CrossRef CAS PubMed.
- C. Fan, S. Cheng, Y. Liu, C. M. Escobar, C. S. Crowley, R. E. Jefferson, T. O. Yeates and T. A. Bobik, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7509–7514 CrossRef CAS PubMed.
- C. Fan, S. Cheng, S. Sinha and T. A. Bobik, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14995–15000 CrossRef CAS PubMed.
- J. W. Cooley and W. F. Vermaas, J. Bacteriol., 2001, 183, 4251–4258 CrossRef CAS PubMed.
- R. Miao, X. Liu, E. Englund, P. Lindberg and P. Lindblad, Metab. Eng. Commun., 2017, 5, 45–53 CrossRef PubMed.
- Y. X. Huo, K. M. Cho, J. G. Rivera, E. Monte, C. R. Shen, Y. Yan and J. C. Liao, Nat. Biotechnol., 2011, 29, 346–351 CrossRef CAS PubMed.
- H. Liu, K. K. Wallace and K. A. Reynolds, J. Am. Chem. Soc., 1997, 119, 2973–2979 CrossRef CAS.
- S. Slater, T. A. Mitsky, K. L. Houmiel, M. Hao, S. E. Reiser, N. B. Taylor, M. Tran, H. E. Valentin, D. J. Rodriguez, D. A. Stone, S. R. Padgette, G. Kishore and K. J. Gruys, Nat. Biotechnol., 1999, 17, 1011–1016 CrossRef CAS PubMed.
- S. J. Sim, K. D. Snell, S. A. Hogan, J. Stubbe, C. Rha and A. J. Sinskey, Nat. Biotechnol., 1997, 15, 63–67 CrossRef CAS PubMed.
- A. Pohlmann, W. F. Fricke, F. Reinecke, B. Kusian, H. Liesegang, R. Cramm, T. Eitinger, C. Ewering, M. Potter, E. Schwartz, A. Strittmatter, I. Voss, G. Gottschalk, A. Steinbuchel, B. Friedrich and B. Bowien, Nat. Biotechnol., 2006, 24, 1257–1262 CrossRef PubMed.
- E. Okamura, T. Tomita, R. Sawa, M. Nishiyama and T. Kuzuyama, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11265–11270 CrossRef CAS PubMed.
- K. Matsumoto, Y. Tanaka, T. Watanabe, R. Motohashi, K. Ikeda, K. Tobitani, M. Yao, I. Tanaka and S. Taguchi, Appl. Environ. Microbiol., 2013, 79, 6134–6139 CrossRef CAS PubMed.
- E. J. Kim and K. J. Kim, Biochem. Biophys. Res. Commun., 2014, 452, 124–129 CrossRef CAS PubMed.
- P. Lindberg, S. Park and A. Melis, Metab. Eng., 2010, 12, 70–79 CrossRef CAS PubMed.
- H. H. Huang, D. Camsund, P. Lindblad and T. Heidorn, Nucleic Acids Res., 2010, 38, 2577–2593 CrossRef CAS PubMed.
- J. Zhou, H. Zhang, H. Meng, Y. Zhu, G. Bao, Y. Zhang, Y. Li and Y. Ma, Sci. Rep., 2014, 4, 4500 CrossRef PubMed.
- V. K. Mutalik, J. C. Guimaraes, G. Cambray, C. Lam, M. J. Christoffersen, Q. A. Mai, A. B. Tran, M. Paull, J. D. Keasling, A. P. Arkin and D. Endy, Nat. Methods, 2013, 10, 354–360 CrossRef CAS PubMed.
- C. Lou, B. Stanton, Y. J. Chen, B. Munsky and C. A. Voigt, Nat. Biotechnol., 2012, 30, 1137–1142 CrossRef CAS PubMed.
- Y. Dekishima, E. I. Lan, C. R. Shen, K. M. Cho and J. C. Liao, J. Am. Chem. Soc., 2011, 133, 11399–11401 CrossRef CAS PubMed.
- R. Carpine, W. Du, G. Olivieri, A. Pollio, K. J. Hellingwerf, A. Marzocchella and F. B. Santos, Algal Res., 2017, 25, 117–127 CrossRef.
- A. Kunert, M. Hagemann and N. Erdmann, J. Microbiol. Methods, 2000, 41, 185–194 CrossRef CAS PubMed.
- M. Azuma, T. Osanai, M. Y. Hirai and K. Tanaka, Plant Cell Physiol., 2011, 52, 404–412 CrossRef CAS PubMed.
- I. W. Bogorad, T. S. Lin and J. C. Liao, Nature, 2013, 502, 693–697 CrossRef CAS PubMed.
- W. Xiong, T. C. Lee, S. Rommelfanger, E. Gjersing, M. Cano, P. C. Maness, M. Ghirardi and J. Yu, Nat. Plants, 2015, 2, 15187 CrossRef PubMed.
- J. Anfelt, D. Kaczmarzyk, K. Shabestary, B. Renberg, J. Rockberg, J. Nielsen, M. Uhlen and E. P. Hudson, Microb. Cell Fact., 2015, 14, 167 CrossRef PubMed.
- E. Presecan-Siedel, A. Galinier, R. Longin, J. Deutscher, A. Danchin, P. Glaser and I. Martin-Verstraete, J. Bacteriol., 1999, 181, 6889–6897 CAS.
- R. Miao, H. Xie and P. Lindblad, Biotechnol. Biofuels, 2018, 11, 267 CrossRef CAS PubMed.
- J. Anfelt, B. Hallstrom, J. Nielsen, M. Uhlen and E. P. Hudson, Appl. Environ. Microbiol., 2013, 79, 7419–7427 CrossRef CAS PubMed.
- J. Wagner, D. Lee-Lane, M. Monaghan, M. Sharifzadeh and K. Hellgardt, Algal Research, 2018, 37, 92–102 CrossRef.
- E. Englund, J. Andersen-Ranberg, R. Miao, B. Hamberger and P. Lindberg, ACS Synth. Biol., 2015, 4, 1270–1278 CrossRef CAS PubMed.
- E. Englund, K. Shabestary, E. P. Hudson and P. Lindberg, Metab. Eng., 2018, 49, 164–177 CrossRef CAS PubMed.
- S. S. Golden, J. Brusslan and R. Haselkorn, Methods Enzymol., 1987, 153, 215–231 CAS.
- R. D. Porter, Methods Enzymol., 1988, 167, 703–712 CAS.
- J. G. K. Williams, Methods Enzymol., 1988, 167, 766–778 CAS.
- W. Vermaas, J. Appl. Phycol., 1996, 8, 263–273 CrossRef CAS.
- C. Jansson, G. Salih, J. Eriksson, R. Wiklund and H. Ghebramedhin, Methods Enzymol., 1998, 297, 166–182 CAS.
- X. Zang, B. Liu, S. Liu, K. K. Arunakumara and X. Zhang, J. Microbiol., 2007, 45, 241–245 CAS.
- B. Wang, J. Yu, W. Zhang and D. R. Meldrum, Appl. Environ. Microbiol., 2015, 81, 8500–8506 CrossRef CAS PubMed.
- P. Tamagnini, O. Troshina, F. Oxelfelt, R. Salema and P. Lindblad, Appl. Environ. Microbiol., 1997, 63, 1801–1807 CAS.
- J. J. Huang, N. H. Kolodny, J. T. Redfearn and M. M. Allen, Arch. Microbiol., 2002, 177, 486–493 CrossRef CAS PubMed.
- N. B. Ivleva and S. S. Golden, Methods Mol. Biol., 2007, 362, 365–373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee01214a |
| This journal is © The Royal Society of Chemistry 2019 |