
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-anionic and -cationic compounds: new high entropy materials for advanced Li-ion batteries†
Qingsong
Wang
 *a,
Abhishek
Sarkar
ab,
Di
Wang
ac,
Leonardo
Velasco
a,
Raheleh
Azmi
d,
Subramshu S.
Bhattacharya
e,
Thomas
Bergfeldt
c,
Andre
Düvel
f,
Paul
Heitjans
f,
Torsten
Brezesinski
a,
Horst
Hahn
*abg and
Ben
Breitung
*ac
*a,
Abhishek
Sarkar
ab,
Di
Wang
ac,
Leonardo
Velasco
a,
Raheleh
Azmi
d,
Subramshu S.
Bhattacharya
e,
Thomas
Bergfeldt
c,
Andre
Düvel
f,
Paul
Heitjans
f,
Torsten
Brezesinski
a,
Horst
Hahn
*abg and
Ben
Breitung
*ac
aInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: qingsong.wang@kit.edu; horst.hahn@kit.edu; ben.breitung@kit.edu
bJoint Research Laboratory Nanomaterials – Technische Universität Darmstadt and Karlsruhe Institute of Technology (KIT), Otto-Berndt-Str. 3, 64206 Darmstadt, Germany
cKarlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitute for Applied Materials, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
eDepartment of Metallurgical and Materials Engineering, Nano Functional Materials Technology Centre (NFMTC), Indian Institute of Technology Madras, Chennai, 600036, India
fInstitute of Physical Chemistry and Electrochemistry, and ZFM – Center for Solid State Chemistry and New Materials, Leibniz University Hannover, Callinstr. 3a, 30167 Hannover, Germany
gHelmholtz Institute Ulm for Electrochemical Energy Storage, Helmholtzstr. 11, 89081 Ulm, Germany
First published on 2nd May 2019
Abstract
In the present work, a new class of high entropy materials for energy storage applications is introduced. Multi-anionic and -cationic compounds are prepared by facile mechanochemistry using a recently designed multi-cationic transition-metal-based high entropy oxide as the precursor and LiF or NaCl as the reactant, leading to formation of lithiated or sodiated materials. Notably, the Li-containing entropy-stabilized oxyfluoride described herein (Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OFx) exhibits a working potential of 3.4 V vs. Li+/Li, enabling its use as a cathode active material. Unlike conventional (non-entropy-stabilized) oxyfluorides, this new material shows enhanced Li storage properties due to entropy stabilization, which, in general, facilitates tailoring the cycling performance by varying the constituent elements in yet unprecedented ways. In addition, we demonstrate that the concept of entropy stabilization is also applicable to Na-containing oxychlorides with a rock-salt structure, thus paving the way toward development of (next-generation) post-Li battery technologies.
Broader contextThe continuous pursuit of new and advanced materials to enable next-generation applications is a grand challenge facing materials science. In the electrochemical energy storage field, lithium-ion batteries are being considered indispensable for large-scale applications such as in hybrid and full-electric vehicles. Cathode active materials are crucial for further improving the energy density to allow for longer driving ranges of battery-powered vehicles. Herein, a new class of high entropy materials is introduced, with the anion and cation lattices contributing to the configurational entropy. Specifically, Li-containing high entropy oxyfluoride, exhibiting a working potential of 3.4 V vs. Li+/Li, shows enhanced lithium storage properties due to entropy stabilization. Notably, mechanochemical synthesis enabled the incorporation of not only lithium but also sodium, thus paving the way toward rational design of post-lithium battery technologies. Taken together, multi-anionic and multi-cationic high entropy compounds represent a potentially transformative materials class with unprecedented properties. |
Introduction
High entropy materials (HEMs) are gaining significant interest due to their novel and often unexpected and unprecedented properties in many different areas of application.1–4 HEMs are based on the premise of introducing a high configurational entropy (Sconfig) to stabilize a single-phase structure. A prominent and already well-established group of HEMs are the metallic high entropy alloys (HEAs).5–9 Recently, the same concept was applied to ionic compounds where multiple cations occupy the same lattice, thus increasing Sconfig.1,10–12 A large number of such high entropy compounds have been synthesized and reported, including carbides,11,13 diborides,2 nitrides,14,15 chalcogenides12,16 and oxides,1,17–19 with wide-ranging applications as thermoelectrics,12,16 dielectrics19 and for lithium-ion batteries.20,21 The latter HEMs, named high entropy oxides (HEOs), have been reported only since 2015 by the pioneering work of Rost et al.1However, until now, there are no literature reports on HEM compounds with more than one anion. Hence, the stabilizing Sconfig effect only results from the cations present in the crystal structure (eqn (S1), ESI†), since the contribution from the anion site is zero. For this reason, the preparation of a multi-anionic and multi-cationic single-phase structure, showing clear indications of entropy stabilization, is of great interest, especially considering that the configurational entropy gain would be even larger compared to the transition-metal-based HEO systems.
Herein, we report, to the best of our knowledge for the first time, on multi-anionic and multi-cationic high entropy oxyhalides with application in electrochemical energy storage. A multi-cationic transition-metal-based HEO (i.e., only oxygen ions occupy the anion site) was used as the precursor and additional halide (X) and alkali metal ions were introduced to produce a multi-anionic and multi-cationic rock-salt type compound (HEOX). The introduction of monovalent fluorine into the anion lattice of HEO, occupied by divalent oxygen, is charge compensated by incorporation of monovalent lithium (or sodium) into the cation lattice. Since fluorine and oxygen have similar ionic radii (1.33 Å vs. 1.40 Å, respectively),22 such substitution can be achieved without inducing significant strain in the single-phase rock-salt structure. Despite their chemical complexity, the preparation of HEOXs is simple and easily scalable, using a facile mechanochemical (ball-milling) process with HEO and LiF or NaCl as the precursor and reactant, respectively. Specifically, Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OFx and Nay(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OCly, referred to as Li(HEO)F and Na(HEO)Cl in the following, were synthesized and characterized, with special emphasis placed on the Li(HEO)F compound.
Considering the crystal field concept, metal fluorides, in general, possess higher electrochemical potential than oxides due to the strongly ionic character of M–F bonds.23,24 This is also why metal oxides and fluorides have been explored as potential anode and cathode active materials, respectively, in the past.25–29 Here, the incorporation of fluorine increased the working potential to 3.4 V vs. Li+/Li, compared to around 1.0 V for the respective transition-metal-based HEOs,20,21,30,31 thus rendering Li(HEO)F interesting for application as a (next-generation) positive electrode material. The overall redox mechanism appears to be similar to that observed for Li-rich disordered rock-salt oxides, as indicated by X-ray diffraction, transmission electron microscopy and X-ray photoelectron spectroscopy.32
Results and discussion
Materials characterization
The most commonly known HEOs are transition-metal-based rock-salt oxides such as (Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)O.1,20,33,34 Very recently, HEOs were utilized as anode active materials in Li-ion cells, showing high specific capacity while maintaining good capacity retention.20,21 It is assumed that this materials system undergoes a conversion reaction within an entropy-stabilized matrix, facilitating the re-integration of redox-active ions into the lattice during delithiation. Based on the work on HEOs, both Li(HEO)F and Na(HEO)Cl were synthesized by means of high-energy ball-milling. Because none of the late 3d transition metals (Mn, Co, Ni, Cu, Zn) has been reported to form oxyfluorides through soft chemistry or solid-state reaction routes,35,36 ball-milling appears to be the most efficient way to prepare such solid solution phases.37–39
Fig. 1a shows X-ray diffraction (XRD) patterns obtained on the as-prepared Li(HEO)F and a mixture of pristine HEO and LiF. The latter compounds exhibit a rock-salt structure with lattice parameter aHEO = 4.233 Å and aLiF = 4.035 Å (from Rietveld analysis, Fig. S1, ESI†). After 24 h of ball-milling a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of HEO and LiF, single-phase Li(HEO)F was obtained. The diffraction pattern can still be indexed to a cubic rock-salt phase, but with a lattice parameter a = 4.185 Å, clearly lying between that of pristine HEO and LiF. For the Rietveld refinement, the instrumental parameters were taken into account using reference data obtained on LaB6 (NIST 660a). The microstructural parameters (crystallite size and micro-strain) were adequately refined to adjust the peak shapes. Thermal displacement parameters were constrained to be the same for all of the atoms to minimize quantification errors. As shown in the inset of Fig. 1 and in Table S1 (ESI†), the Li and F atoms are randomly distributed on the respective cation and anion sub-lattices of the rock-salt HEO. The substitution of transition-metal cations by Li is evident from the decrease in intensity of the (111) reflection. This is due to the lower atomic number of Li compared to the other transition metals. For O and F, the atomic numbers and ionic radii are similar, and in a rock-salt lattice, there is typically only one site (4b) where the F− ions can reside. Overall, XRD indicates that the LiF is successfully incorporated into the HEO structure.
:1 molar mixture of HEO and LiF, single-phase Li(HEO)F was obtained. The diffraction pattern can still be indexed to a cubic rock-salt phase, but with a lattice parameter a = 4.185 Å, clearly lying between that of pristine HEO and LiF. For the Rietveld refinement, the instrumental parameters were taken into account using reference data obtained on LaB6 (NIST 660a). The microstructural parameters (crystallite size and micro-strain) were adequately refined to adjust the peak shapes. Thermal displacement parameters were constrained to be the same for all of the atoms to minimize quantification errors. As shown in the inset of Fig. 1 and in Table S1 (ESI†), the Li and F atoms are randomly distributed on the respective cation and anion sub-lattices of the rock-salt HEO. The substitution of transition-metal cations by Li is evident from the decrease in intensity of the (111) reflection. This is due to the lower atomic number of Li compared to the other transition metals. For O and F, the atomic numbers and ionic radii are similar, and in a rock-salt lattice, there is typically only one site (4b) where the F− ions can reside. Overall, XRD indicates that the LiF is successfully incorporated into the HEO structure.
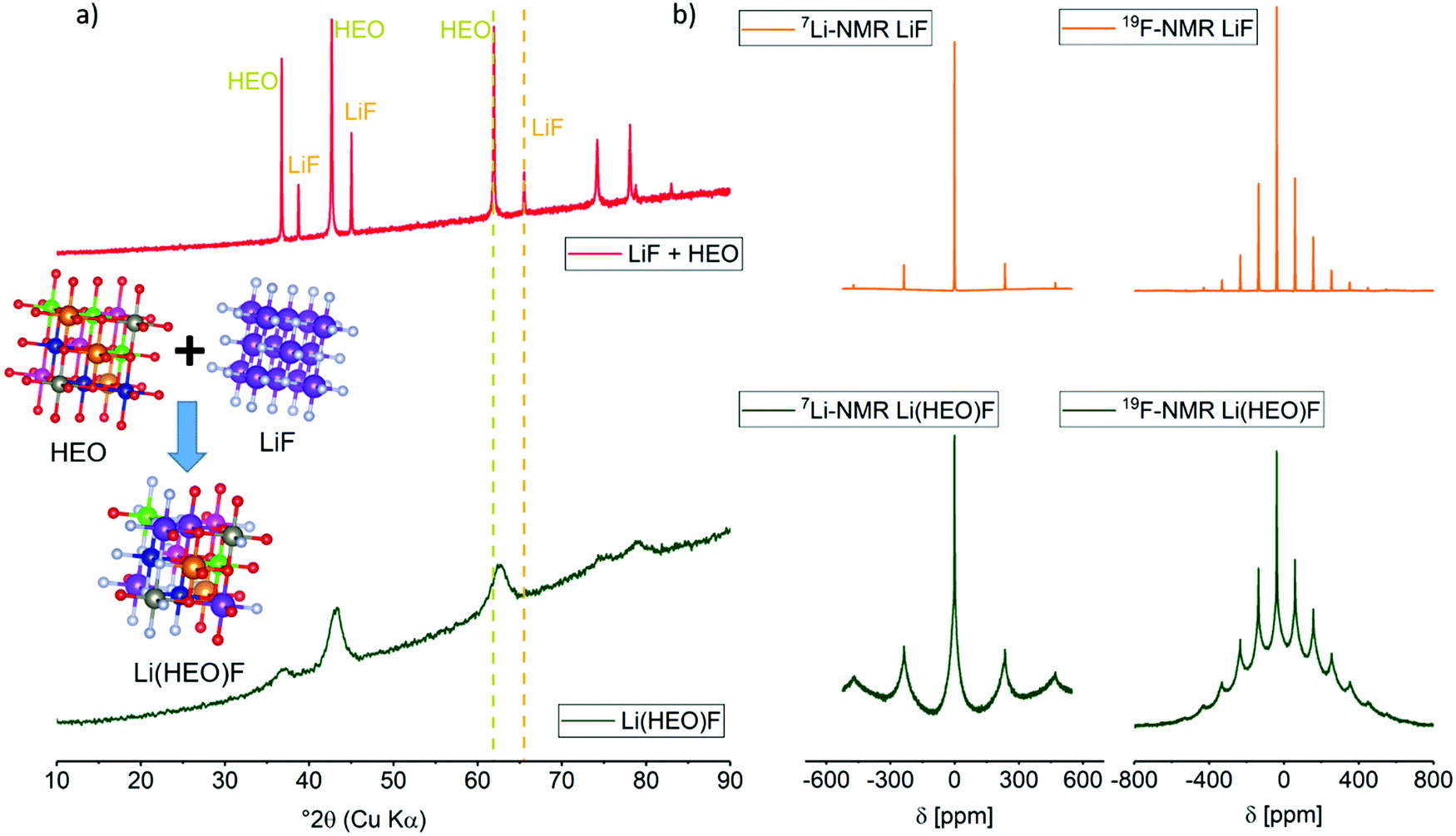 | ||
| Fig. 1 (a) XRD pattern obtained on both a physical mixture of LiF and HEO and the as-prepared Li(HEO)F. Dashed lines denote the position of the (220) reflection of LiF and HEO. For Li(HEO)F, the (220) reflection is located between that of LiF and HEO, indicating successful incorporation of Li+ and F− into the HEO lattice. Oxygen is in red, fluorine in white, nickel in pink, cobalt in blue, magnesium in orange, copper in green, zinc in grey and lithium in violet in the cubic structures shown in the inset. (b) 7Li- and 19F-MAS-NMR of LiF and the as-prepared Li(HEO)F (recorded with νrot = 55 kHz, ν0(7Li) = 233.3 MHz, ν0(19F) = 564.8 MHz). Distinct line broadening is observed in the Li(HEO)F case due to changes in chemical environment. | ||
In order to confirm the presence of lithium and fluorine in the lattice structure, solid-state magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy measurements were conducted on the as-prepared material. Fig. 1b shows the 7Li- and 19F-MAS-NMR spectra of both Li(HEO)F and a physical mixture of HEO and LiF (see ref. 40–42 for reviews of 7Li- and 19F-NMR on energy-related materials). The incorporation of Li+ and F− into the HEO leads to a different chemical environment compared to the LiF reference material. Because the local elemental distribution in Li(HEO)F is random, the number of chemical environments is rather large, therefore, line broadening occurs. In addition, the paramagnetic Co2+ causes a strong line broadening. The narrow lines at −38 ppm and −1.05 ppm in the 19F- and 7Li-MAS-NMR spectrum, respectively, can be assigned to unreacted LiF. Curve fitting revealed the presence of around 8% LiF (Fig. S2, ESI†). Because LiF was neither detected by selected-area electron diffraction (SAED) nor by XRD, it seems reasonable to assume that the ball-milling process left behind some amorphous material, as also reported for Li2VO2F compounds.37 High-resolution X-ray photoelectron spectroscopy (XPS) measurements were conducted on a powder sample to gain further insights into the oxidation state(s) of the respective elements. The F 1s peaks (Fig. S3, ESI†) at 685.2 eV and 686.5 eV seem to indicate the successful incorporation of fluorine into the HEO structure, since these binding energies can be referred to MOF, MF and MOF2 binding motifs.43,44 The Li 1s spectrum reveals two peaks at binding energies of 55.2 eV and 56.1 eV, indicating two different Li+ chemical environments, namely, oxygen and fluorine. The Co species are identified as being mainly Co2+ (91% Co2+/9% Co3+), while Ni exists as mixture of Ni2+ (37% of total Ni ions) and Ni3+ (63% of total Ni ions).45,46 The Cu 2p spectrum is found to only exhibit a single peak at 933 eV. The lack of a satellite peak hints at either Cu1+ or Cu0. However, a clear distinction using the Cu LMM Auger peak was not possible, since it overlaps with that of the Zn LMM.47 Nevertheless, Cu1+ is assumed to be present in the Li(HEO)F compound. The peak at 1303.8 eV in the Mg 1s spectrum can be attributed to Mg2+,48,49 and the Zn 2p3/2 at 1021.3 eV and Zn LMM at 988.6 eV are indicative of Zn2+.50–52
‘NaCl’ has also been successfully added to the HEO structure through an identical ball-milling approach. Because the ionic radius of Cl− is much larger than that of O2− or F−, and Na+ is larger than Li+ (Cl− = 1.81 Å, O2− = 1.40 Å, F− = 1.33 Å, Na+ = 1.02 Å, Li+ = 0.76 Å),22 it is more difficult to achieve an entropy-stabilized single-phase structure. The incorporation of a reasonable fraction of different size ions (Na+, Cl−) would lead to a major distortion of the lattice, destabilizing the structure. Hence, in the NaCl case, the maximum fraction that can be added to the entropy-stabilized HEO material is expected to be much lower than for LiF. According to XRD (Fig. 2), the Na(HEO)Cl compound also exhibits a single-phase rock-salt structure with the characteristic reflections shifted toward smaller Bragg angles as compared to pristine HEO (12:1 molar ratio of HEO and NaCl, aNa(HEO)Cl = 4.244 Å, aNaCl = 5.642 Å, see Fig. S1, ESI†).
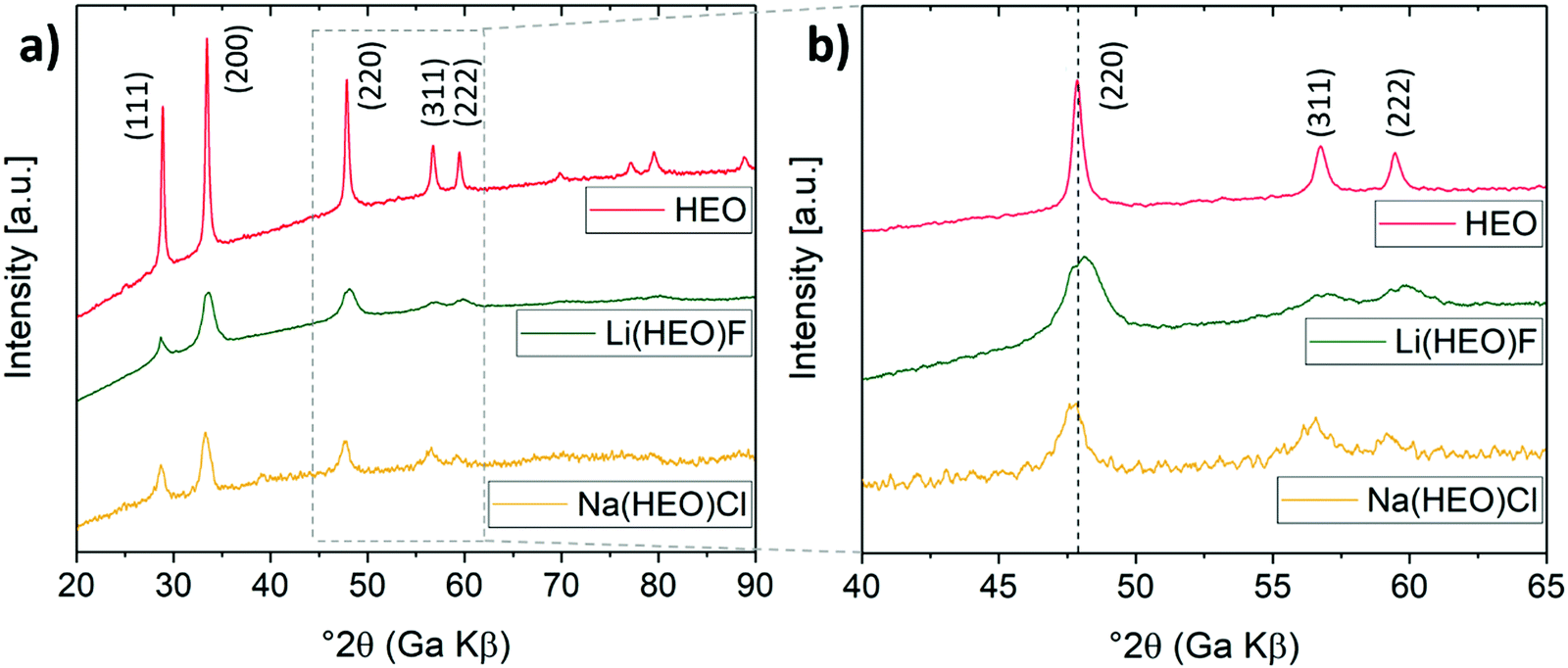 | ||
| Fig. 2 (a) XRD patterns of HEO, Li(HEO)F and Na(HEO)Cl. The shift in reflection position toward smaller and larger Bragg angles for Na(HEO)Cl and Li(HEO)F, respectively, relative to pristine HEO, is denoted by a dashed line in (b). | ||
The concept of entropy stabilization was evaluated for Li(HEO)F in the present work. Because a single-phase entropy-stabilized structure segregates into multiple phases at low temperature (TΔS is small), but will re-form a single phase at high temperature (TΔS is large), as shown previously,1,53 this behavior can be used to corroborate the entropy stabilization effect. To this end, the XRD pattern obtained on single-phase Li(HEO)F heated to 750 °C was analyzed in some more detail (Fig. S1, ESI†). As it is evident from the splitting of reflections (apparently, LiF and different metal oxide phases form), phase segregation indeed occurs. This provides clear evidence that entropy plays a major role in stabilizing the chemically complex system Lix(Co0.2Cu0.2Mg0.2Ni0.2Zn0.2)OxFx.
Fig. 3 shows results from high-resolution transmission electron microscopy (HRTEM) and SAED on the Li(HEO)F. The aggregates have an average size of 100 nm (Fig. 3a), with good crystallinity (Fig. 3b) of the rock-salt structure, while SAED indicates that the as-prepared material is obtained without other (crystalline) secondary phases.
 | ||
| Fig. 3 (a and b) TEM images at different magnification of the as-prepared Li(HEO)F showing crystalline particles around 100 nm in size. The inset in (b) is an SAED pattern obtained on the same material. The (200) and (220) reflections are denoted by red and blue circles, respectively. (c) EFTEM mapping results revealing uniform element distribution. | ||
The lattice parameter of the rock-salt structure was calculated from both the SAED and HRTEM data and agrees well with the Rietveld refinement result (Fig. S1, ESI†). The homogeneous distribution of elements, as mentioned above, was confirmed by energy-filtered TEM (EFTEM) mapping (Fig. 3c). Of note, both Mg and Zn are not depicted because of the lack of sufficient signal intensity for the corresponding core-loss edges. Nevertheless, the uniform distribution of each single element in the Li(HEO)F was also corroborated by spectrum imaging using energy dispersive X-ray (EDX) spectroscopy in scanning TEM (STEM) mode (Fig. S4, ESI†). Within the resolution of the two methods (both approaching the sub-nanometer level), there is no notable elemental segregation or clustering, both of which would lead to a decrease in the number of possible microstates, and therefore, lower the configurational entropy. Finally, inductively-coupled plasma optical emission spectroscopy (ICP-OES) measurements were conducted on the Li(HEO)F to examine the chemical composition. The combined results from ICP-OES, NMR and EDX reveal an overall composition of Li0.94(Co0.21Ni0.21Zn0.2Mg0.2Cu0.18)O1F0.87, as shown and discussed in Fig. S5 (ESI†).
Electrochemical characterization
The introduction of the multi-anionic concept enabled the use of the high entropy oxyfluoride as a new cathode active material for rechargeable Li-ion cells. Notably, the incorporation of fluorine into the structure increased the working potential significantly. In the present work, we show, to the best of our knowledge for the first time, the utilization of an entropy-stabilized compound as a cathode active material for reversible energy storage. The benefits arising from the high entropy crystal structure with respect to the electrochemical performance comprise the capacity retention and Li-extraction ability of the compound. In general, oxyfluorides represent an important class of electrode materials, owing to their ability to suppress oxygen loss during cycling operation. The latter is often observed for state-of-the-art cathode materials and can lead to capacity degradation.32,54–57 The incorporated fluorine renders the material resistant against HF etching. Note that HF is usually formed in the electrolyte due to reaction of trace water (or protic species) with LiPF6/PF5.58 Therefore, oxyfluoride-based electrode materials have been the subject of intense investigations in recent years.59–61 With our approach, we aim at combining the advantages of oxyfluorides with the benefits arising from the entropy stabilization. However, the objective of this study was not to optimize the cycling performance, but rather to demonstrate the applicability of high entropy cathode active materials in the forefront field of electrochemical energy storage. Optimization regarding choice of binder, electrolyte, material preparation, additives etc. needs to be done in the next step. Here, the electrochemical properties of Li(HEO)F are compared with rock-salt type LiNiOF, which was prepared in the same way. Because LiNiOF shows a much lower Sconfig than Li(HEO)F, but also contains Ni as the redox active cation, it is an ideal reference compound to evaluate the entropy stabilization concept for Li-insertion materials.Electrochemical testing of the as-prepared Li(HEO)F and pure HEO was performed on half-cells with a Li metal counter electrode. For HEO, cyclic voltammetry (CV) shows the already reported cathodic peak around 0.5 V in the initial cycle and around 0.8 V in the subsequent cycles, characteristic of the anode conversion reaction upon lithiation (Fig. 4a).20 In contrast, for Li(HEO)F, a cathodic peak around 3.4 V is observed from the CV curves (Fig. 4b), which is in agreement with the galvanostatic discharge profiles shown in Fig. 4c. Regarding energy density, a middle discharge potential of 3.4 V is superior to most disordered rock-salt oxyfluoride materials reported in the literature, showing potentials closer to 2.5 V.36,37,62,63 The higher working potential compared to pure HEO is due in part to the fact that the overall (de)lithiation mechanism changed from a conversion to an insertion type reaction. More importantly, the incorporation of fluorine leads to an increase in the Gibbs free energy of the system, and therefore, to a higher theoretical potential of the redox reaction (see discussion in the ESI†).64 Note that the application of Li(HEO)F in rechargeable Li-ion batteries is also fostered by its relatively high lithium content. Overall, this entropy-stabilized oxyfluoride compound fulfills one of the most important requirements of a cathode active material for graphite-based cells—it is capable of serving as the lithium source.
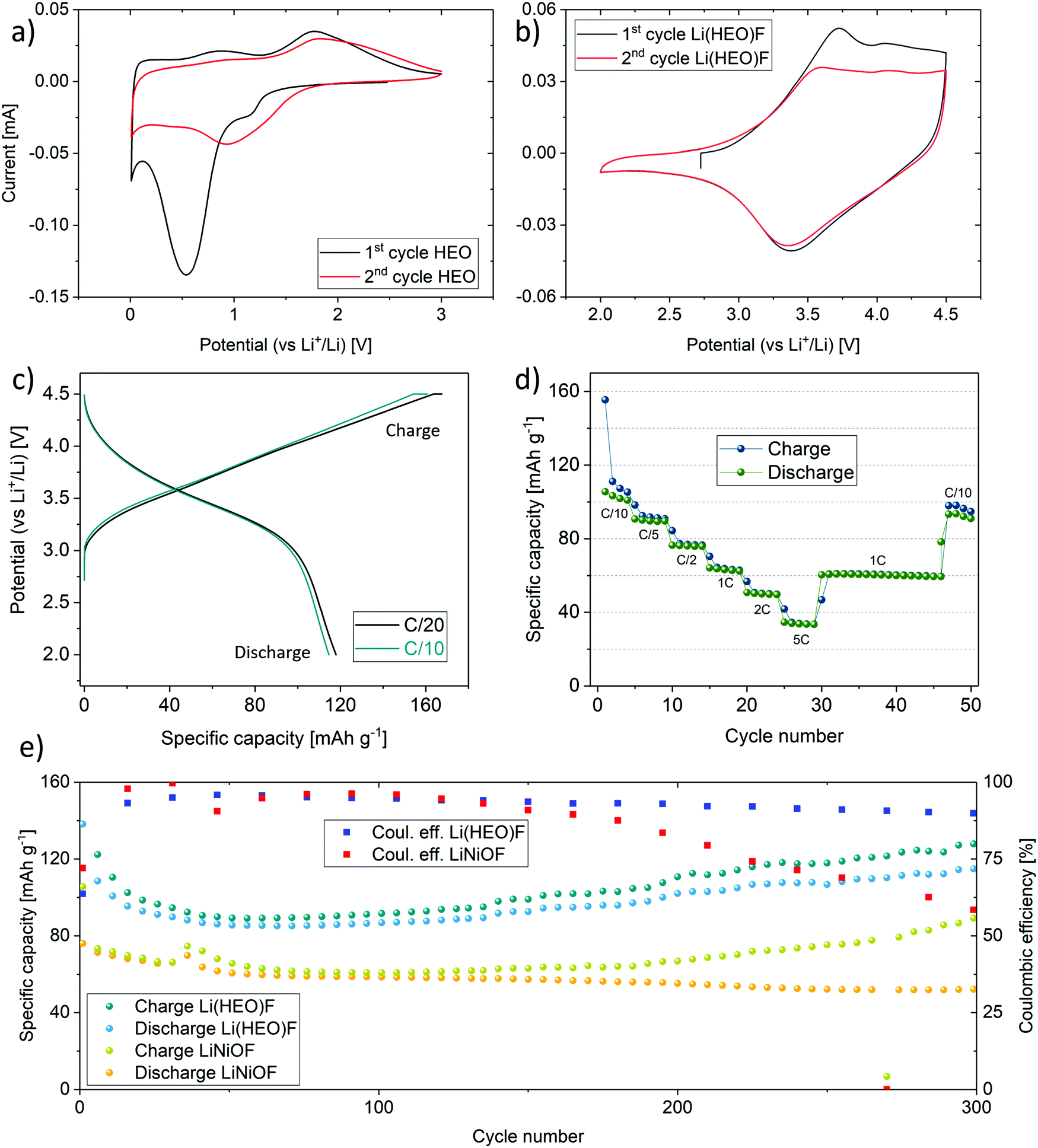 | ||
| Fig. 4 Cyclic voltammetric curves at a sweep rate of 0.1 mV s−1 for (a) pure HEO cycled in the voltage range between 0.01 and 3.0 V vs. Li+/Li and (b) Li(HEO)F between 2.5 and 4.5 V vs. Li+/Li. (c) Charge/discharge curves for Li(HEO)F-based half-cells at rates of C/20 (10 mA g−1) and C/10 (20 mA g−1). The cut-off potentials were set at 2.0 and 4.5 V vs. Li+/Li. (d) Specific capacities as a function of cycle number for rates ranging from C/10 to 5C. (e) Comparison of the capacity retention at C/10 of Li(HEO)F and LiNiOF. | ||
As shown in Fig. 4c, specific capacities of 168 and 161 mA h g−1 were achieved in the initial charge cycle (with a constant voltage step at 4.5 V) at C/20 and C/10, respectively. The specific discharge capacity was around 120 mA h g−1 at both rates. The initial capacity loss is due in part to electrolyte degradation and formation of a solid electrolyte interphase (SEI) layer.36 During the course of cycling, the specific capacity, especially at low C-rates, decreases slightly, as can be seen from the rate performance data in Fig. 4d. However, other side reactions associated with the cathode active material cannot be ruled out, accounting for the difference in the charge and discharge capacities at low C-rates. It should be noted that the cell chemistry has not been optimized yet. Nevertheless, the specific capacity at C/10 can be recovered virtually completely after applying high rates of up to 5C.
A direct comparison of the cyclability at C/10 of Li(HEO)F to LiNiOF is presented in Fig. 4e. From the data shown, it is evident that the Li(HEO)F delivers higher specific capacities than LiNiOF. This is noteworthy because the redox-active nickel has been replaced by other elements; both cobalt and nickel are likely the only species to be oxidized during delithiation. The molar fraction of Co and Ni together amounts to only 0.4 instead of 1 for LiNiOF. This means that despite the decrease in redox-active ion fraction, the specific capacity is increased.
After around 150 cycles, the Coulombic efficiency clearly shows differences between the entropy-stabilized Li(HEO)F and the non-entropy stabilized LiNiOF. While the Coulombic efficiency tends to decrease rapidly for the LiNiOF system, in the Li(HEO)F, it remains fairly stable. The entropy stabilization seems to play a key role in maintaining high reversible capacities and stabilizing the Coulombic efficiency. In general, oxide and/or oxyfluoride cathode active materials show some oxygen (anion) redox activity at high potential, leading to oxygen evolution at the particle/electrolyte interface, and therefore, formation of an oxygen deficient material.32,56,59 Note that oxygen release adversely affects the cycling stability of any electrode material. In a nutshell, the entropy stabilization of the Li(HEO)F compound is capable of somewhat counteracting this kind of degradation. Nevertheless, in this regard, it is important to note that the Coulombic efficiencies for both materials are lower compared to state-of-the-art cathode active materials.
A higher reversible capacity directly relates to the capability of the material in providing access to mobile Li in the structure. Most commonly, not all of the Li in the lattice is accessible, since it is contributing to some degree to the structural stability. Using the entropy stabilization approach, it seems that more Li can be extracted, thus leading to the observed increase in specific capacity compared to LiNiOF. The theoretical specific capacities of Li(HEO)F and LiNiOF are similar with 278 and 266 mA h g−1, respectively, when assuming a one-electron redox process per formula unit. However, because only 0.8 Li+ can presumably be extracted from the Li(HEO)F, the theoretical specific capacity drops to 222 mA h g−1. This can be explained as follows: Cu2+ (after the initial cycle; see section on redox mechanism below), Mg2+ and Zn2+ in the Li(HEO)F structure cannot be oxidized, and therefore, Ni2+ and Co2+ are the only remaining redox-reactive cations in the potential range applied (based on the assumption that the divalent state of the cations is prevailing). Finally, we note that improvements in cycling performance, especially the specific capacity, can be expected when even more lithium is incorporated into the rock-salt lattice, as shown recently for Li-rich disordered rock-salt materials.65,66
Reaction mechanism
XRD and TEM experiments were conducted on the Li(HEO)F at different states of charge (SOC) during cycling to gain more insight into the reaction mechanism (Fig. 5a). The parent rock-salt structure was preserved throughout cycling and no secondary (crystalline) phases were detected. As can be seen in Fig. 5a, the reflections in the ex situ XRD pattern clearly shift toward larger Bragg angles (smaller lattice parameter) in the charge cycle, thus hinting at an insertion mechanism.32 Li extraction from the Li(HEO)F is accompanied by transition-metal oxidation (presumably Ni2+ and/or Co2+), leading to a decrease in ionic radius, and therefore, to a smaller unit cell. Yet, the reflections shift back to their original position upon subsequent lithiation, emphasizing the reversibility of the Li insertion/extraction process. These findings are also supported by SAED. The patterns shown in Fig. 5a confirm the preservation of the rock-salt lattice upon cycling.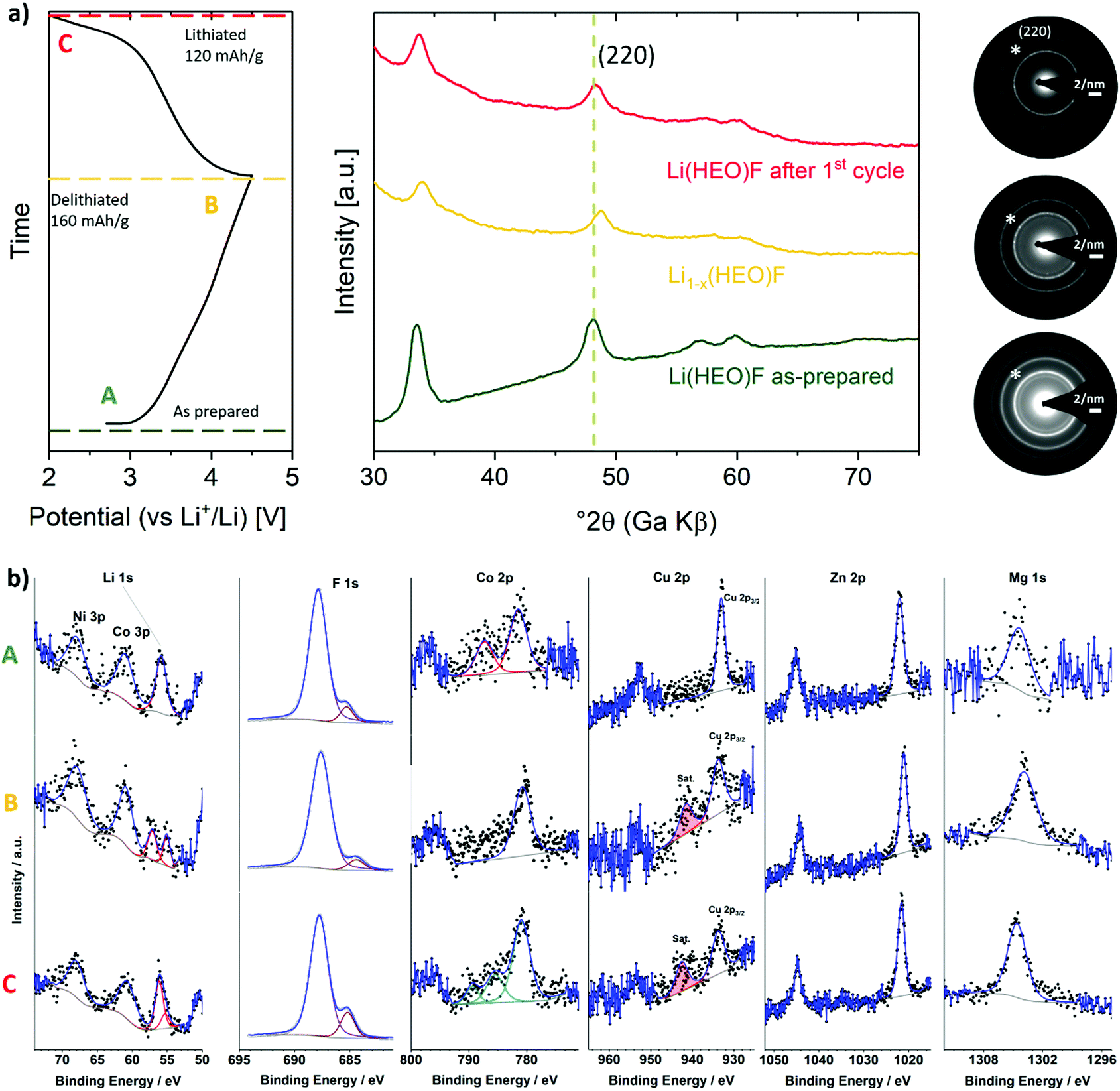 | ||
| Fig. 5 (a) Initial charge/discharge curve at C/10 (20 mA g−1) in the voltage range between 2.0 and 4.5 V (left) and the corresponding ex situ XRD patterns collected under an Ar atmosphere using a protective Kapton film (middle) as well as SAED data (right). The (220) reflection is denoted by an asterisk in the SAED patterns. (b) XPS spectra of the Li 1s, F 1s, Co 2p, Cu 2p, Zn 2p and Mg 1s core levels – before and after cycling (A: pristine state, B: after delithiation and C: after re-lithiation). | ||
The changes in oxidation state of the elements upon cycling were examined by ex situ XPS. The corresponding measurements were performed on electrodes with different states of charge/discharge (equivalent to ex situ XRD in Fig. 5a). The Li 1s, F 1s and Co 2p spectra in Fig. 5b emphasize the reversibility of the Li extraction/insertion during charge/discharge. Note that utilizing a multiplet splitting approach for quantified oxidation state analysis of Co ions was hampered by the low concentration of active material at the top surface of the electrode, leading to the presence of very low intensity peaks. Qualitative analysis reveals that in the pristine electrode, the Co ions are predominantly present as Co2+. This is due to the existence of a strong satellite peak about 6 eV higher in binding energy than the main peak, which can be considered characteristic of Co2+.46 This satellite peak disappears after delithiation, thus indicating oxidation of Co2+ to Co3+. After lithiation, the re-appearance of a less intense satellite peak suggests co-existence of Co3+ and Co2+ ions.46,47,51,52 In the F 1s spectra, the peak at 685.2 eV can be assigned to the fluorine ions in the Li(HEO)F rock-salt structure, as discussed above (Fig. S3, ESI†). This peak slightly shifts to lower binding energy (684.4 eV) after delithiation, probably because of a lower electron density around the fluorine atoms after Li extraction. As expected, it shifts back to 685.2 eV with re-lithiation. The major peak located at around 687.9 eV in the F 1s spectra is due to the polyvinylidene difluoride (PVdF) binder used, which remains unaltered during cycling.67 In the Cu 2p spectra, the appearance of a satellite peak (∼942 eV) after delithiation and subsequently re-lithiation is indicative of the oxidation of Cu1+ to Cu2+.47 Such (surface) oxidation, which apparently is not reversible, seems to contribute to some extent to the irreversible capacity loss in the initial cycle (Fig. 4c). As expected, both Mg and Zn remain in divalent state during cycling, thus providing evidence that they are indeed redox-inactive. Unfortunately, the analysis of Ni oxidation state was hampered by superposition of the Ni 2p and F KLL peaks (Fig. S6, ESI†). The XPS results were also confirmed by electron energy loss spectroscopy (EELS, data not shown). In addition, EELS indicated that the bulk Cu in the as-prepared material has a mixed oxidation state of Cu1+/Cu2+.
Conclusions
With the incorporation of multiple anions into an entropy-stabilized multi-cationic compound, we were able to show, for the first time, that not only the cations but also the anions can be varied while preserving a single-phase rock-salt structure. These compounds constitute a new class of entropy-stabilized materials, with the anion lattice also contributing to the configurational entropy, resulting in an additional structural stabilization gain. Using this approach, we successfully synthesized an oxyfluoride cathode active material with a rock-salt structure for next-generation Li-ion battery applications. Notably, the entropy stabilization improved the cycling performance considerably. Additionally, this approach enables the reduction of toxic and costly elements in battery cathodes, without significantly affecting the energy density. Taken together, the concept of multi-anionic and multi-cationic high entropy compounds introduces a new class of energy storage materials with unprecedented properties.Methods
Synthesis
HEO was prepared by a reverse co-precipitation process, followed by high-temperature calcination, the details of which can be found elsewhere.53 For the synthesis of Li(HEO)F, a 1:1 molar ratio of LiF (Alfa Aesar, 99.99%) and HEO was ball-milled using a high-energy planetary ball-mill (Retsch PM 100, Retsch GmbH). Na(HEO)Cl was prepared in the same way but with a 12:1 molar ratio of HEO and NaCl (Alfa Aesar, >99%). Specifically, WC vials (50 ml in volume) and WC balls (4 mm in diameter) were used, with a 40:1 ball-to-powder weight ratio. The milling process was carried out at 500 rpm for 24 h in an Ar atmosphere. The as-prepared powder was collected and stored inside an Ar-filled glovebox.
Electrochemistry
For the cathode preparation, active material (100 mg), Super C65 carbon black (28.6 mg, Timcal) and polyvinylidene difluoride (14.3 mg, Solef 5130) were uniformly dispersed into N-methyl-2-pyrrolidone. The resulting slurry was doctor-bladed onto an Al current collector and dried in a vacuum oven at 80 °C for 12 h. Circular electrodes (13 mm in diameter) were cut from the cathode sheet, assembled in an Ar-filled glovebox and tested in CR2032 coin cells. 1 M LiPF6 in a 3:7 weight mixture of ethylene carbonate/ethyl methyl carbonate (Selectilyte LP57, BASF SE), glass microfiber filter paper (GF/C, Whatman™) and Li metal foil (Gelon LIB Co., Ltd) were used as the electrolyte, separator and counter electrode, respectively. Galvanostatic cycling measurements were performed on an Arbin battery test system (BT-2000) at room temperature. Cyclic voltammetry was performed on a Bio-logic potentiostat (VMP3). The areal cathode active material loading was about 1 mg cm−2. The specific capacity was calculated based on the total weight of cathode active material in the electrode.
Characterization
XRD patterns for the as-prepared compounds were recorded using a STOE Stadi P diffractometer with Cu-Kα radiation. The powder was filled into borosilicate glass capillaries (Hilgenberg) with inner diameter of 0.48 mm and wall thickness of 0.01 mm. For the cycled samples, the powder was collected from the electrode inside an Ar-filled glovebox and measured under inert conditions using a STOE Stadi P diffractometer, equipped with a Ga-jet X-ray source (Ga-Kβ radiation).ICP-OES spectroscopy (iCAP 7600 ICP-OES DUO, Thermo Fisher Scientific) was conducted on 5 to 6 mg samples (±0.05 mg accuracy) dissolved into hydrochloric and nitric acid. More details can be found in the ESI.†
TEM measurements were conducted on powder samples dispersed on a holey carbon-coated gold grid. The samples were loaded onto a Gatan vacuum-transfer holder inside an Ar-filled glovebox and transferred to the TEM without exposure to air. TEM, SAED, EFTEM and STEM-EDX/EELS data were collected using a FEI Titan 80-300 microscope, equipped with a CEOS image spherical aberration corrector, a HAADF STEM detector (Fischione model 3000), EDAX SUTW EDX detector and a Tridiem Gatan image filter. The microscope was operated at an accelerating voltage of 300 kV.
7Li- and 19F-NMR data were acquired at room temperature on a Bruker Avance III 600 spectrometer (14 T) at Larmor frequencies of 233.3 and 564.8 MHz, respectively. A 1.3 mm high-speed probe was used for magic-angle spinning experiments. The samples were spun at 55 kHz. For the 19F-MAS-NMR measurements, a Hahn-Echo experiment was performed (pi/2 pulse: 1 μs), while the 7Li-MAS-NMR spectra were recorded with a single-pulse experiment (pi/2 pulse: 1.1 μs). As reference substances for the chemical shifts, both a dilute solution of LiCl and liquid C6F6 were used.
XPS measurements on the as-prepared powder and cycled samples at different states of charge were performed directly on the powder and recovered electrode tape using a K-Alpha+ instrument (Thermo Fisher Scientific) with a monochromatic Al-Kα X-ray source (1486.6 eV) with 400 μm spot size. The K-Alpha+ charge compensation system was applied to prevent localized charge buildup during analysis, using 8 eV electrons and low-energy Ar ions. Data acquisition and processing were carried out using the software Thermo Avantage.68 The spectra were fitted with one or more Voigt profiles. The binding energies are reported with respect to the binding energy of C 1s for hydrocarbon at 285.0 eV. The analyzer transmission function, Scofield sensitivity factors69 and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism.70
Data availability
The data used for this study are available from the corresponding authors upon request.Author contributions
Q. W. prepared the materials and electrodes and performed the electrochemical experiments and analysis. A. S. prepared the materials and performed Rietveld refinement of the XRD data. D. W. performed the TEM measurements and analyzed the data. L. V. helped in the preparation of the materials. R. A. collected the XPS spectra and analyzed the data. S. S. B. developed the HEO synthesis and supported the synthesis efforts. Th. B. conducted the ICP measurements. A. D. and P. H. performed the NMR measurements and analyzed the data. T. B. co-wrote the manuscript and helped with the electrochemical characterization. Q. W., H. H. and B. B. supervised the synthesis and experiments, directed the project and co-wrote the manuscript. All the authors contributed to the data analysis, discussion and manuscript preparation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Q. W. acknowledges financial support by EnABLES. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 730957. H. H. and A. S. acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, under contract HA 1344/43-1). P. H. is grateful to Niedersächsisches Ministerium für Wissenschaft und Kultur (MWK) for a Niedersachsen Professorship (VWZN3095) and A. D. acknowledges financial support by DFG (DU 1668/3-1). The K-Alpha+ was financially supported by the Federal Ministry of Economics and Energy (BMWi). Dr Julia Maibach (from IAM-ESS, KIT) is acknowledged for access to XPS laboratory.References
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- J. Gild, Y. Zhang, T. Harrington, S. Jiang, T. Hu, M. C. Quinn, W. M. Mellor, N. Zhou, K. Vecchio and J. Luo, Sci. Rep., 2016, 6, 37946 CrossRef CAS PubMed.
- D. B. Miracle and O. N. Senkov, Acta Mater., 2017, 122, 448–511 CrossRef CAS.
- J. W. Yeh and S. J. Lin, J. Mater. Res., 2018, 33, 3129–3137 CrossRef CAS.
- J.-W. Yeh, S.-K. Chen, S.-J. Lin, J.-Y. Gan, T.-S. Chin, T.-T. Shun, C.-H. Tsau and S.-Y. Chang, Adv. Eng. Mater., 2004, 6, 299–303 CrossRef CAS.
- Y. J. Zhou, Y. Zhang, Y. L. Wang and G. L. Chen, Appl. Phys. Lett., 2007, 90, 181904 CrossRef.
- M.-H. Tsai, C.-W. Wang, C.-W. Tsai, W.-J. Shen, J.-W. Yeh, J.-Y. Gan and W.-W. Wu, J. Electrochem. Soc., 2011, 158, H1161 CrossRef CAS.
- W.-Y. Tang, M.-H. Chuang, H.-Y. Chen and J.-W. Yeh, Surf. Coat. Technol., 2010, 204, 3118–3124 CrossRef CAS.
- F. Tian, L. K. Varga, N. Chen, J. Shen and L. Vitos, Intermetallics, 2015, 58, 1–6 CrossRef CAS.
- W. J. Shen, M. H. Tsai, K. Y. Tsai, C. C. Juan, C. W. Tsai, J. W. Yeh and Y. S. Chang, J. Electrochem. Soc., 2013, 160, C531–C535 CrossRef CAS.
- E. Castle, T. Csanádi, S. Grasso, J. Dusza and M. Reece, Sci. Rep., 2018, 8, 8609 CrossRef PubMed.
- R. Z. Zhang, F. Gucci, H. Zhu, K. Chen and M. J. Reece, Inorg. Chem., 2018, 57, 13027–13033 CrossRef CAS PubMed.
- J. Zhou, J. Zhang, F. Zhang, B. Niu, L. Lei and W. Wang, Ceram. Int., 2018, 44, 22014–22018 CrossRef CAS.
- C.-W. Tsai, S.-W. Lai, K.-H. Cheng, M.-H. Tsai, A. Davison, C.-H. Tsau and J.-W. Yeh, Thin Solid Films, 2012, 520, 2613–2618 CrossRef CAS.
- T. Jin, X. Sang, R. R. Unocic, R. T. Kinch, X. Liu, J. Hu, H. Liu and S. Dai, Adv. Mater., 2018, 30, 1707512 CrossRef PubMed.
- R. Liu, H. Chen, K. Zhao, Y. Qin, B. Jiang, T. Zhang, G. Sha, X. Shi, C. Uher, W. Zhang and L. Chen, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- A. Sarkar, R. Djenadic, D. Wang, C. Hein, R. Kautenburger, O. Clemens and H. Hahn, J. Eur. Ceram. Soc., 2018, 38, 2318–2327 CrossRef CAS.
- J. Dąbrowa, M. Stygar, A. Mikuła, A. Knapik, K. Mroczka, W. Tejchman, M. Danielewski and M. Martin, Mater. Lett., 2018, 216, 32–36 CrossRef.
- D. Bérardan, S. Franger, D. Dragoe, A. K. Meena and N. Dragoe, Phys. Status Solidi RRL, 2016, 10, 328–333 CrossRef.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya, H. Hahn and B. Breitung, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- H. Chen, N. Qiu, S. Sun, Y. Wang, Y. Cui and Z. Yang, J. Alloys Compd., 2018, 777, 767–774 Search PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- J. B. Goodenough, Advances in Lithium-Ion Batteries, Springer US, Boston, MA, 2002, pp. 135–154 Search PubMed.
- G. G. Amatucci and N. Pereira, J. Fluorine Chem., 2007, 128, 243–262 CrossRef CAS.
- X. L. Huang, X. Zhao, Z. L. Wang, L. M. Wang and X. B. Zhang, J. Mater. Chem., 2012, 22, 3764–3769 RSC.
- Y. Huang, X. L. Huang, J. S. Lian, D. Xu, L. M. Wang and X. B. Zhang, J. Mater. Chem., 2012, 22, 2844–2847 RSC.
- X. L. Huang, R. Z. Wang, D. Xu, Z. L. Wang, H. G. Wang, J. J. Xu, Z. Wu, Q. C. Liu, Y. Zhang and X. B. Zhang, Adv. Funct. Mater., 2013, 23, 4345–4353 CrossRef CAS.
- D. L. Ma, Z. Y. Cao, H. G. Wang, X. L. Huang, L. M. Wang and X. B. Zhang, Energy Environ. Sci., 2012, 5, 8538–8542 RSC.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2018, 57, 102–120 CrossRef CAS PubMed.
- Q. Wang, A. Sarkar, Z. Li, Y. Lu, L. Velasco, S. S. Bhattacharya, T. Brezesinski, H. Hahn and B. Breitung, Electrochem. Commun., 2019, 100, 121–125 CrossRef CAS.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 1806236 CrossRef PubMed.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Förstermann, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Energy Environ. Sci., 2018, 11, 926–932 RSC.
- D. Bérardan, S. Franger, A. K. Meena and N. Dragoe, J. Mater. Chem. A, 2016, 4, 9536–9541 RSC.
- H. Chen, J. Fu, P. Zhang, H. Peng, C. W. Abney, K. Jie, X. Liu, M. Chi and S. Dai, J. Mater. Chem. A, 2018, 6, 11129–11133 RSC.
- B. L. Chamberland, A. W. Sleight and W. H. Cloud, J. Solid State Chem., 1970, 2, 49–54 CrossRef CAS.
- L. Zhang, D. Dambournet, A. Iadecola, D. Batuk, O. J. Borkiewicz, K. M. Wiaderek, E. Salager, M. Shao, G. Chen and J. M. Tarascon, Chem. Mater., 2018, 30, 5362–5372 CrossRef CAS.
- R. Chen, S. Ren, M. Knapp, D. Wang, R. Witter, M. Fichtner and H. Hahn, Adv. Energy Mater., 2015, 5, 1401814 CrossRef.
- Y. Tomita, H. Nasu, Y. Izumi, J. Arai, S. Otsuka, Y. Yamane, K. Yamada, Y. Kohno and K. Kobayashi, J. Power Sources, 2016, 329, 406–411 CrossRef CAS.
- Y. Tomita, N. Kimura, H. Nasu, Y. Izumi, J. Arai, Y. Yamane, K. Yamada, Y. Kohno and K. Kobayashi, J. Appl. Electrochem., 2017, 47, 1057–1063 CrossRef CAS.
- C. P. Grey and N. Dupré, Chem. Rev., 2004, 104, 4493–4512 CrossRef CAS.
- C. V. Chandran and P. Heitjans, Annu. Rep. NMR Spectrosc., 2016, 89, 1–102 CrossRef CAS.
- M. Wilkening, A. Düvel, F. Preishuber-Pflügl, K. Da Silva, S. Breuer, V. Šepelák and P. Heitjans, Z. Kristallogr. – Cryst. Mater., 2017, 232, 107–127 CAS.
- Y. T. Sul, Int. J. Nanomed., 2010, 5, 87–100 CrossRef CAS.
- K. I. Murai, Y. Suzuki, T. Moriga and A. Yoshiasa, AIP Conf. Proc., 2007, 882, 463–465 CrossRef CAS.
- A. W. Moses, H. G. G. Flores, J. G. Kim and M. A. Langell, Appl. Surf. Sci., 2007, 253, 4782–4791 CrossRef CAS.
- R. Azmi, V. Trouillet, M. Strafela, S. Ulrich, H. Ehrenberg and M. Bruns, Surf. Interface Anal., 2018, 50, 43–51 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- J. Y. Zhang, G. Yang, S. G. Wang, Y. W. Liu, Z. D. Zhao, Z. L. Wu, S. L. Zhang, X. Chen, C. Feng and G. H. Yu, J. Appl. Phys., 2014, 116, 1–6 Search PubMed.
- M. Morales, Int. J. Green Technol., 2018, 3, 51–62 CrossRef.
- Z. Zhao, G. Tian, A. Sarapulova, V. Trouillet, Q. Fu, U. Geckle, H. Ehrenberg and S. Dsoke, J. Mater. Chem. A, 2018, 6, 19381–19392 RSC.
- V. Kumar, C. R. Mariappan, R. Azmi, D. Moock, S. Indris, M. Bruns, H. Ehrenberg and G. Vijaya Prakash, ACS Omega, 2017, 2, 6003–6013 CrossRef CAS.
- C. R. Mariappan, V. Kumar, R. Azmi, L. Esmezjan, S. Indris, M. Bruns and H. Ehrenberg, CrystEngComm, 2018, 20, 2159–2168 RSC.
- A. Sarkar, R. Djenadic, N. J. Usharani, K. P. Sanghvi, V. S. K. Chakravadhanula, A. S. Gandhi, H. Hahn and S. S. Bhattacharya, J. Eur. Ceram. Soc., 2017, 37, 747–754 CrossRef CAS.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4, 2223–2233 RSC.
- T. Bartsch, F. Strauss, T. Hatsukade, A. Schiele, A.-Y. Kim, P. Hartmann, J. Janek and T. Brezesinski, ACS Energy Lett., 2018, 3, 2539–2543 CrossRef CAS.
- T. Hatsukade, A. Schiele, P. Hartmann, T. Brezesinski and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 38892–38899 CrossRef CAS PubMed.
- L. de Biasi, A. Schiele, M. Roca-Ayats, G. Garcia, T. Brezesinski, P. Hartmann and J. Janek, ChemSusChem, 2019 DOI:10.1002/cssc.201900032.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327–A2334 CrossRef CAS.
- S. K. Jung, H. Gwon, J. Hong, K. Y. Park, D. H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1–7 Search PubMed.
- L. Li, B. H. Song, Y. L. Chang, H. Xia, J. R. Yang, K. S. Lee and L. Lu, J. Power Sources, 2015, 283, 162–170 CrossRef CAS.
- Z. Lun, B. Ouyang, D. A. Kitchaev, R. J. Clément, J. K. Papp, M. Balasubramanian, Y. Tian, T. Lei, T. Shi, B. D. McCloskey, J. Lee and G. Ceder, Adv. Energy Mater., 2019, 9, 1802959 CrossRef.
- S. W. Kim, N. Pereira, N. A. Chernova, F. Omenya, P. Gao, M. S. Whittingham, G. G. Amatucci, D. Su and F. Wang, ACS Nano, 2015, 9, 10076–10084 CrossRef CAS PubMed.
- R. Chen, E. Maawad, M. Knapp, S. Ren, P. Beran, R. Witter and R. Hempelmann, RSC Adv., 2016, 6, 65112–65118 RSC.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 CrossRef.
- J. Lee, D. A. Kitchaev, D. H. Kwon, C. W. Lee, J. K. Papp, Y. S. Liu, Z. Lun, R. J. Clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS PubMed.
- T. Zhang, Y. He, F. Wang, H. Li, C. Duan and C. Wu, Sep. Purif. Technol., 2014, 138, 21–27 CrossRef CAS.
- K. L. Parry, A. G. Shard, R. D. Short, R. G. White, J. D. Whittle and A. Wright, Surf. Interface Anal., 2006, 38, 1497–1504 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2011, 43, 689–713 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee00368a |
| This journal is © The Royal Society of Chemistry 2019 |