
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general route via formamide condensation to prepare atomically dispersed metal–nitrogen–carbon electrocatalysts for energy technologies†
Guoxin
Zhang‡
 ab,
Yin
Jia‡
ac,
Cong
Zhang
a,
Xuya
Xiong
a,
Kai
Sun
ac,
Ruida
Chen
ac,
Wenxing
Chen
d,
Yun
Kuang
ae,
Lirong
Zheng
f,
Haolin
Tang
g,
Wen
Liu
*a,
Junfeng
Liu
*a,
Xiaoming
Sun
*abc,
Wen-Feng
Lin
h and
Hongjie
Dai
e
ab,
Yin
Jia‡
ac,
Cong
Zhang
a,
Xuya
Xiong
a,
Kai
Sun
ac,
Ruida
Chen
ac,
Wenxing
Chen
d,
Yun
Kuang
ae,
Lirong
Zheng
f,
Haolin
Tang
g,
Wen
Liu
*a,
Junfeng
Liu
*a,
Xiaoming
Sun
*abc,
Wen-Feng
Lin
h and
Hongjie
Dai
e
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: wenliu@mail.buct.edu.cn; ljf@mail.buct.edu.cn; sunxm@mail.buct.edu.cn
bShandong University of Science and Technology, Electrical Engineering and Automation, Tsingtao, 266590, China
cCollege of Energy, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, China
dDepartment of Materials, Beijing Institute of Technology, Beijing, 100081, China
eDepartment of Chemistry, Stanford University, Stanford, CA 94305-5080, USA
fBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China
gState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, 430070, China
hDepartment of Chemical Engineering, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
First published on 25th March 2019
Abstract
Single-atom electrocatalysts (SAECs) have gained tremendous attention due to their unique active sites and strong metal–substrate interactions. However, the current synthesis of SAECs mostly relies on costly precursors and rigid synthetic conditions and often results in very low content of single-site metal atoms. Herein, we report an efficient synthesis method to prepare metal–nitrogen–carbon SAECs based on formamide condensation and carbonization, featuring a cost-effective general methodology for the mass production of SAECs with high loading of atomically dispersed metal sites. The products with metal inclusion were termed as formamide-converted metal–nitrogen–carbon (shortened as f-MNC) materials. Seven types of single-metallic f-MNC (Fe, Co, Ni, Mn, Zn, Mo and Ir), two bi-metallic (ZnFe and ZnCo) and one tri-metallic (ZnFeCo) SAECs were synthesized to demonstrate the generality of the methodology developed. Remarkably, these f-MNC SAECs can be coated onto various supports with an ultrathin layer as pyrolysis-free electrocatalysts, among which the carbon nanotube-supported f-FeNC and f-NiNC SAECs showed high performance for the O2 reduction reaction (ORR) and the CO2 reduction reaction (CO2RR), respectively. Furthermore, the pyrolysis products of supported f-MNC can still render isolated metallic sites with excellent activity, as exemplified by the bi-metallic f-FeCoNC SAEC, which exhibited outstanding ORR performance in both alkaline and acid electrolytes by delivering ∼70 and ∼20 mV higher half-wave potentials than that of commercial 20 wt% Pt/C, respectively. This work offers a feasible approach to design and manufacture SAECs with tuneable atomic metal components and high density of single-site metal loading, and thus may accelerate the deployment of SAECs for various energy technology applications.
Broader contextAtomically dispersed metal–nitrogen–carbon (MNC) materials have been regarded as the most promising catalysts for many energy conversion and storage technologies, thanks to their unique properties and exceptionally high catalytic performance. However, their state-of-the-art preparation has heavily relied on costly precursors and rigid conditions, yet still resulting in low loadings of the atomically dispersed metal. We proposed and demonstrated a general, simple yet highly efficient and readily scalable synthesis method based on liquid formamide chemistry for low-cost preparation of atomically dispersed MNC nanomaterials, and employed them as electrocatalysts for two exemplified key reactions in fuel cells and carbon dioxide utilization. The generality of the methodology has been validated by the synthesis of a large family of single-, bi- and tri-metallic atomically dispersed MNC materials. Moreover, these formamide-converted MNC materials allow the easy formation of thin electrocatalytic coatings on conductive materials to produce pyrolysis-free, highly efficient electrocatalysts which exhibited superior catalytic performance compared to commercial Pt/C for the oxygen reduction reaction. The current method can be adapted to prepare vast varieties of atomically dispersed MNC materials, with adjustable metal components and coated on various substrates, for a wide range of energy and environmental electrocatalysis and more general heterogeneous catalysis applications. |
Introduction
Single-atom electrocatalysts (SAECs) have the merits of largely increased surface to bulk ratio, highly unsaturated coordination, and strong catalyst-support interaction enabled by the well defined, homogeneously distributed atomic active sites.1–3 The past several years have witnessed explosive developments of SAECS in the fast-growing fields of renewable energy technologies such as fuel cells, water electrolysis, and metal–air batteries.4–11 Carbon supported SAECs, especially the family of metal-nitrogen complexed carbon (MNC) materials, have high conductivity and unique metal–ligand interaction, which render 100% atom utilization and stimulate strong quantum effects.1–3,12 Therefore, extraordinary catalytic activity and selectivity can be expected from the MNC-based SAECs due to their largely altered and promoted reaction pathway.13–16Despite their extremely promising potential, most of the SAECs are currently synthesized through pyrolysis of metal salts with nitrogen and carbon sources,17–21 including small molecules (such as pyrrole,22 sodium alginate,23 and dopamine9), polymers (such as polyaniline,24 polyphthalocyanine8), carbon materials (such as graphene oxide,17 heteroatom-doped carbon,19 C3N410,25) and MXene.26 However, the weak coordination bonding between the metal cations and N/C sources, and the inevitable high-temperature annealing for acquiring conductivity often result in low-quality SAECs that suffer from metal aggregation.27–29
To facilitate more controllable SAEC synthesis, metal organic frameworks (MOFs) with pre-embedded atomic metal sites in the N-containing organic surroundings are widely employed as a precursor for SAEC production.28,30–32 However, most applicable ligands for MOFs such as 2-methylimidazole33–35 and 2,2-bipyridine36 are costly and toxic, and the latter significantly impedes the scalable production of SAECs and their practical applications. Moreover, the rigid crystal structure of MOFs requires specific metal–ligand interaction, which limits the choice of metal types and surrounding ligands.33,37,38 Therefore, there is an urgent demand to develop a universal and cost-effective method to produce SAECs.
Herein, we proposed and developed a general route for mass production of SAECs using the inexpensive and low-toxicity formamide (FA) as both the carbon and nitrogen sources. Introducing transition metal salt into the condensation/carbonization of FA allows the atomic dispersion of dense metal sites isolated by rich multiple N-dentate ligands. We demonstrated the versatility of this method by synthesizing seven types of single-metallic (Fe, Co, Ni, Mn, Zn, Mo, and Ir), one bi-metallic (Fe/Co) and one tri-metallic (ZnFeCo) SAEC. Moreover, all the SAECs prepared can be uniformly coated onto various supports as ultrathin skin layers. Among them, the pyrolysis-free carbon nanotube-supported f-FeNC and f-NiNC SAECs exhibited excellent performance towards the oxygen reduction reaction (ORR) and the carbon dioxide reduction reaction (CO2RR), respectively. Remarkably, the f-FeCoNC SAEC after annealing treatment, containing isolated Fe–Co dimers as confirmed by the extended X-ray absorption fine structure (EXAFS), showed extraordinary ORR performance in both alkaline and acidic media. The proposed synthesis strategy is evidenced to open up the mass production of SAECs and their skin derivatives for broad implantable electrocatalysis as well as rich heterogeneous catalysis applications.
Results and discussion
Synthesis of f-MNC SAECs through formamide condensation and carbonization
The synthesis of f-MNC SAECs was realized via the condensation and carbonization of FA upon the existence of metal ions under solvothermal treatment (Fig. S1, ESI†). FA, which contains both a primary amino group and a carbonyl group, can build C/N bonds with each other and thus form 1D molecular chains by the Schiff base reactions39,40 (Fig. S2a, ESI†). More interestingly, the high N-content ligand sites on the chains can be utilized for efficient chelating of metal cations, making it promising for constructing electrochemically active metal-nitrogen moieties.41 With the existence of metal ions, the cross-linking of the as-formed C/N chains was promoted by the chelated metal cations, resulting in atomically dispersed f-MNC, as schematically depicted in Fig. 1a. f-CoNC was first synthesized as a proof-of-concept example by the solvothermal treatment of Co/FA solution at 180 °C for 12 h. The f-NC structure derived from FA without the promotion of metal ions was also prepared for comparison (Fig. S2b, ESI†). f-CoNC exhibits a particulate morphology without any specific orientation (Fig. 1e), which is in agreement with the wide peak in the XRD pattern (Fig. 1b), implying its roughly amorphous nature. In contrast, NC exhibits a sharp peak at 2θ = 27°, corresponding to the typical (002) peak of the carbon nitride phase.42 The peak intensity decreased after introducing Co2+, suggesting that the metal ions decrease the degree of crystallinity of the f-NC materials (Fig. 1b). Raman spectra (Fig. 1c) of f-NC and f-CoNC reveal peaks located at ∼1370 and 1544 cm−1, respectively, ascribed to the characteristic D and G bands of carbonaceous materials derived from the organic precursors.43–45 The main functional groups of C–N (∼1395 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (∼1610 cm−1) in both f-NC and f-CoNC were also verified by the FTIR data (Fig. S2e, ESI†). The XPS C1s spectra of f-NC and f-CoNC further confirmed the considerable presence of NC(–N)–N moieties (Fig. S2d, ESI†). All the N species in f-NC and f-CoNC mainly take pyridinic and pyrrolic forms (Fig. 1d), which favour the coordination with metal cations.42 Elemental mapping images in Fig. 1f reveal the uniform distribution of dense loading of N and Co. The XPS analysis of f-CoNC indicated that ∼1.5 at% Co (equal to ∼6.49 wt%, Table S1, ESI†) had been introduced into the f-CoNC matrix and more pyridinic N (∼74.7% relative to 71.2% in NC, Fig. 1d and Table S1, ESI†) and higher carbon content (∼63.0% relative to 60.6% in f-NC, Table S1, ESI†) were revealed under the Co2+ promotion. Meanwhile, the XPS Co2p spectrum (Fig. 1g) shows that all the Co species are present in their oxidation state (Co2+) other than metallic Co(0), further confirming their isolated coordination configuration.
N (∼1610 cm−1) in both f-NC and f-CoNC were also verified by the FTIR data (Fig. S2e, ESI†). The XPS C1s spectra of f-NC and f-CoNC further confirmed the considerable presence of NC(–N)–N moieties (Fig. S2d, ESI†). All the N species in f-NC and f-CoNC mainly take pyridinic and pyrrolic forms (Fig. 1d), which favour the coordination with metal cations.42 Elemental mapping images in Fig. 1f reveal the uniform distribution of dense loading of N and Co. The XPS analysis of f-CoNC indicated that ∼1.5 at% Co (equal to ∼6.49 wt%, Table S1, ESI†) had been introduced into the f-CoNC matrix and more pyridinic N (∼74.7% relative to 71.2% in NC, Fig. 1d and Table S1, ESI†) and higher carbon content (∼63.0% relative to 60.6% in f-NC, Table S1, ESI†) were revealed under the Co2+ promotion. Meanwhile, the XPS Co2p spectrum (Fig. 1g) shows that all the Co species are present in their oxidation state (Co2+) other than metallic Co(0), further confirming their isolated coordination configuration.
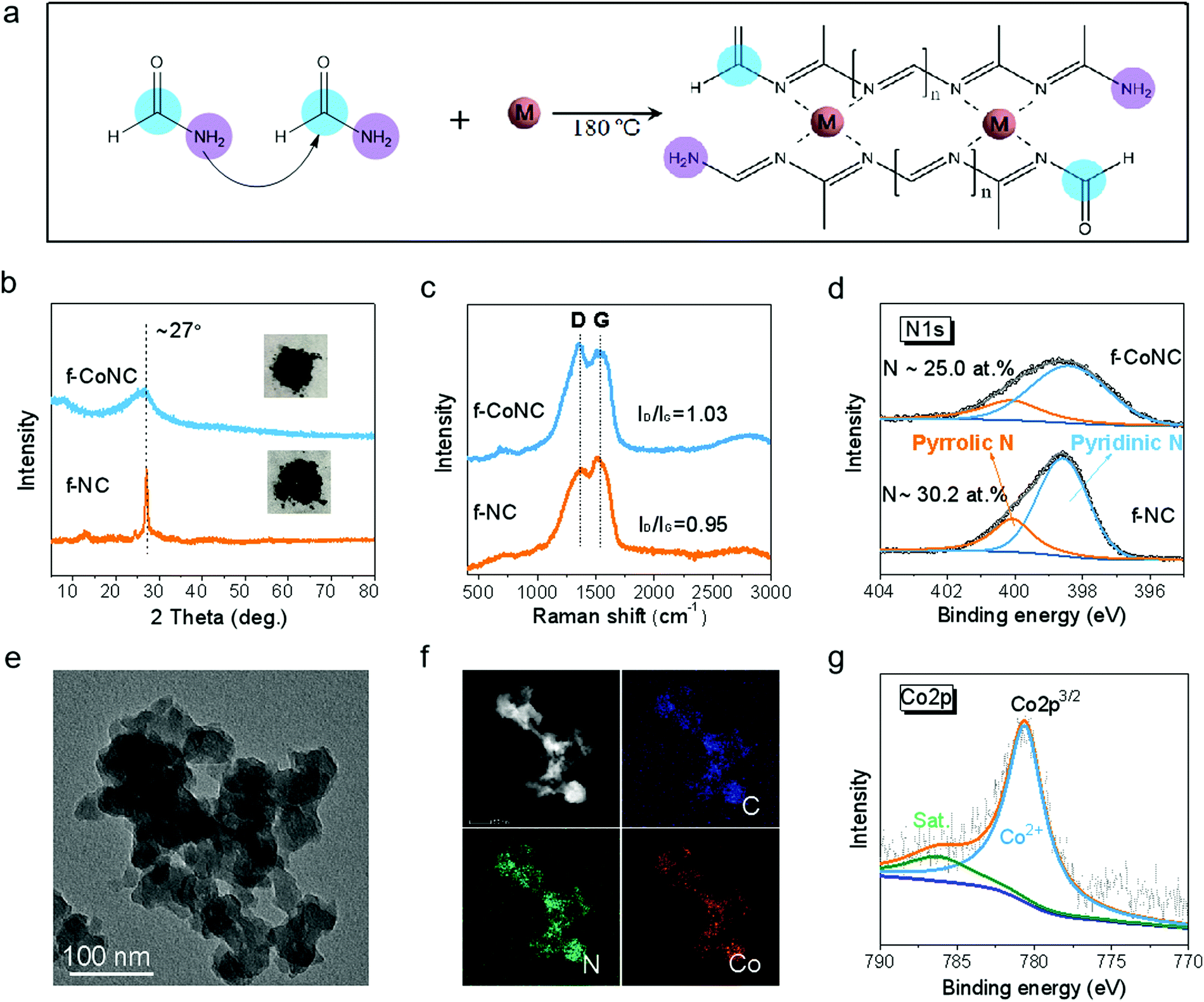 | ||
| Fig. 1 Preparation scheme and characterizations for f-NC and f-CoNC. (a) A schematic illustration of the synthetic process of f-CoNC. (b) The XRD profiles and corresponding digital images of f-NC and f-CoNC. (c) Raman spectra and (d) XPS N1s spectra of f-NC and f-CoNC. (e) A TEM image of f-CoNC synthesized at 180 °C. (f) Element mapping images of f-CoNC, revealing the uniform distributions of C, N and Co. (g) XPS Co2p spectra of f-CoNC. | ||
The atomic-resolution high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray absorption fine structure (XAFS) characterizations were employed to confirm the atomic structure of f-CoNC. Fig. 2a shows the dense distribution of isolated bright dots, which correspond to the atomically dispersed Co atoms. In the Fourier transform (FT) curve (Fig. 2g), a strong peak is located at ca. 1.51 Å, indicating that the sample mainly comprised of Co–N configured coordination. To explore the proposed general preparation method, six other types of atomically dispersed f-MNCs, including f-FeNC, f-MnNC, f-NiNC, f-ZnNC, f-MoNC, and f-IrNC were successfully synthesized. As shown in Fig. 2b–e and Fig. S3 (ESI†), the HAADF-STEM images of f-FeNC, f-MnNC, f-NiNC, f-ZnNC, f-MoNC, and f-IrNC indicated that their metallic components were in an atomic scale high dispersion state. XAFS in Fig. 2g–i and Fig. S4 (ESI†) further confirmed that the Co, Fe and Zn atoms are coordinated with N, by showing the M–N configured coordination. Other essential characterizations including HRTEM, XRD, and XPS for these f-MNCs were also carried out to confirm the isolated state of metal atoms (Fig. S5–S10, ESI†) and the absence of crystalline metal or metal compounds. As in f-CoNC, N species were overwhelmingly presented in the pyridinic N form (72 ± 5%). The XPS spectra also confirm that all the metal species are presented in their ionic forms, and the metal mass loadings of f-CoNC, f-FeNC, f-MnNC, f-ZnNC, f-MoNC, f-NiNC and f-IrNC are 6.49, 4.50, 5.36, 4.40, 6.04, 5.0 and 3.06 wt%, respectively. The abovementioned results strongly suggest that our method is versatile to achieve atomically dispersed f-MNC materials with high metal mass loading.
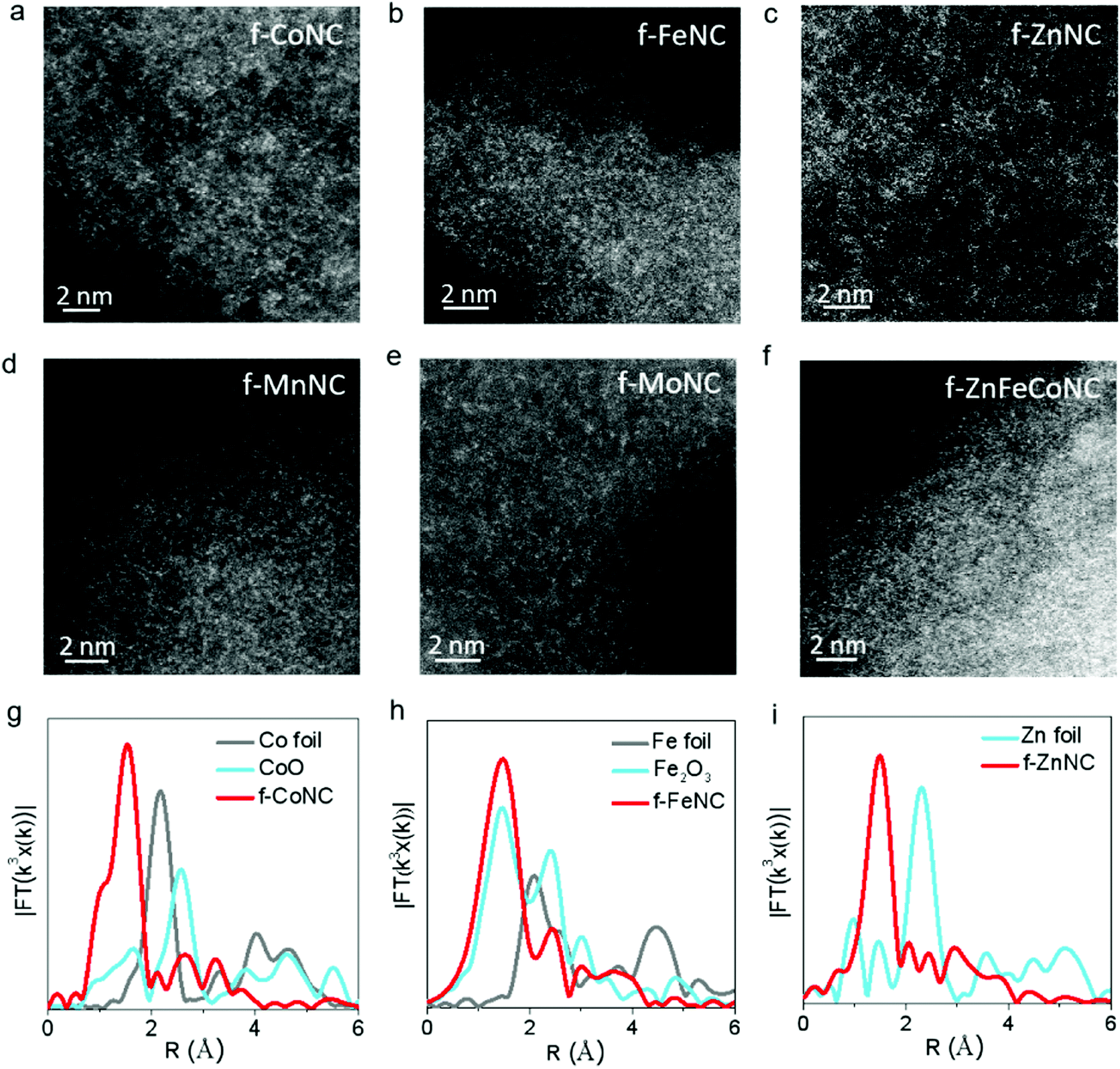 | ||
| Fig. 2 Characterizations of f-MNC. (a–f) HAADF-STEM images of f-CoNC, f-FeNC, f-ZnNC, f-MnNC, f-MoNC and f-ZnFeCoNC. (g–i) A Fourier transformed (FT) k3-weighted χ(k)-function of the EXAFS spectra for the Co K-edge of f-CoNC, Fe K-edge of f-FeNC, Zn K-edge of f-ZnNC and their reference samples. | ||
Atomically dispersed f-MNCs with multiple-metal components were also fabricated using a similar synthetic procedure for a single-metal f-MNC. Bi-metal f-ZnFeNC and f-ZnCoNC, and tri-metal f-ZnFeCoNC, were successfully prepared. Their HRTEM images are shown in Fig. S11a–c (ESI†), which indicate the absence of aggregated metals. Typically, as shown in the elemental mapping images of f-ZnFeCoNC (Fig. S11d–i, ESI†), the uniform distribution of bright dots that represent the highly dispersed metallic components (i.e., Fe, Co, Zn, Fe) can be observed. The isolated states of the Zn, Fe, and Co atoms are also confirmed by the HAADF-STEM image (Fig. 2f).
Ultrathin f-MNC SAEC skin layer on various supports
The as-synthesized f-CoNC showed limited electron conductivity due to its carbon nitride phase, but it can be developed into an electrocatalyst by thinly depositing it on conductive substrates.46–48 Three commonly used substrates were chosen to fabricate the supported f-MNC: graphene oxide (GO), carbon nanotubes (CNTs), and activated carbon (AC). Co-NC was first tentatively coated on GO by simply adding GO in the Co2+/FA solution before the reactions were initiated. Fig. 3a shows the TEM image of GO supported Co-NC (denoted as GO@f-CoNC) with evenly distributed contrast, suggesting uniform deposition of f-CoNC on the GO layer. The AFM image reveals that the thickness of the deposited f-CoNC layer on GO is ∼2 nm, considering both-side deposition of f-CoNC on roughly 1 nm thick GO (Fig. 3b). Such a uniform and thin layer of f-CoNC was mainly ascribed to the intimate affinity between the as-formed f-CoNC and hydrophilic GO.49 In addition, the uniform distribution of C, N, and Co can also be proved using element mapping, as visualized in Fig. S12 (ESI†). The HAADF-STEM image (Fig. 3c) displays the densely and uniformly distributed bright dots on GO, suggesting atomically dispersed metal atoms. | ||
| Fig. 3 Characterizations of supported f-MNC. (a) TEM, (b) AFM, and (c) HAADF-STEM images of GO@f-CoNC (inset in b shows the measured depth variation of the marked line). (d) TEM images of CNT@f-FeNC prepared at 170 °C, 180 °C and 200 °C for 12 h, respectively. (e) Element mapping images of CNT@f-FeNC170 prepared for 12 h. (f) The LSV curves of Pt/C, CNT@f-FeNC170, and a mixture of CNT and f-FeNC catalysts in O2-saturated 0.1 M KOH electrolyte with a potential sweep rate of 5 mV s−1 and electrode rotation rate of 1600 rpm. | ||
Furthermore, other thin f-MNC films on the 2nd nanocarbon substrate chosen, f-FeNC on CNT, were fabricated. Fig. S13a (ESI†) indicates a clear trend of thickness increasing from ∼1 to ∼2 nm then to ∼5 nm with respect to the time duration of 8, 12, and 24 h, respectively. In addition, the reaction temperature also shows a great effect on the formation of the f-FeNC layer on CNT, unlike the sheath growth mode, protuberances begin to emerge and grow perpendicular to the CNT surface at 180 °C, and these protuberances grow sharper and more prominent at 200 °C (Fig. 3d). The element mapping analysis confirms the uniform distribution of C, N, and Fe on CNT (Fig. 3e). Similarly, more thin f-MNC films on various supports were achieved, e.g., ∼10 nm f-MnNC on AC, and ∼5 nm f-ZnNC on SiO2 (Fig. S14, ESI†); which grant us more tools to optimize the properties of f-MNC based SAECs.
Electrocatalytic performance of CNT@f-FeNC and CNT@f-NiNC.
Loading the f-MNC skin films on conductive supports enables them to be used as electrocatalysts for important electrochemical applications.50 As examined, the f-MNC skinned nanocarbon materials are extremely sensitive to catalyst layer thickness. The CNT@f-FeNC170 sample with an ultrathin f-FeNC skin layer shows almost the same ORR performance as Pt/C in 0.1 M KOH by delivering a high onset potential of 0.97 V (cf. Pt/C of ∼1.0 V), a high half-wave potential of 0.84 V (cf. Pt/C of ∼0.84 V), and a remarkable 6.2 mA cm−2 limiting current density (cf. Pt/C of ∼5.5 mA cm−2), as shown in Fig. 3f. This performance is among the best for the non-pyrolysis catalyst system in alkaline media,23,51–54 though the performance in acid media needs further improvement. Meanwhile, the much better performance of CNT@f-FeNC170 than that of the mixture of CNT and f-FeNC also verifies the importance of the intimate affinity between f-FeNC and the support/substrate. In contrast, the ORR performance of CNT@f-FeNC at a higher temperature (CNT@f-FeNC180 and CNT@f-FeNC200) displayed a fast decay in terms of both activity (Fig. S13b, ESI†) and efficiency (Fig. S13c and d, ESI†), emphasizing the effectiveness of the deposition thickness control of the f-MNC skin layer.The excellent ORR performance of CNT@f-FeNC170 enlightens us to explore the potentials of conductive substrate-supported ultrathin f-MNC electrocatalysts in other important electrochemical reactions.55 f-NiNC SAECs were prepared by heat treatment of Ni salt/CNT/FA solution, then tested for the carbon dioxide reduction reaction (CO2RR). The same typical morphology of CNT@f-MNC is shown in Fig. S15a (ESI†). As marked in the magnified view (Fig. S15b, ESI†), the thickness of the deposited f-NiNC layer is ∼1 nm. The element mapping image (Fig. S15c, ESI†) reveals the uniform distribution of C, N, O, and Ni elements. As shown in Fig. S15d (ESI†), CNT@f-NiNC exhibits an onset potential of −0.57 V towards the CO2RR in the CO2-saturated KHCO3 solution, which is obviously higher than those in the N2-saturated solution (dashed curve in Fig. S15d, ESI†). The excellent selectivity and activity of CNT@f-NiNC towards the CO2RR were confirmed by monitoring the gas products formed at different applied potentials using gas chromatography. CO and H2 were confirmed as the dominating products in the potential range from −0.4 to −1.0 V (Fig. S15e, ESI†). Moreover, the CNT@f-NiNC electrocatalyst remained stable at a current density of ∼−4.5 ± 0.5 mA cm−2 and showed a CO-yield faradaic efficiency of ∼80 ± 3% at a potential of −0.6 V for over 10 h (Fig. S15f, ESI†). Table S2 (ESI†) shows the key performance data of the CNT@f-NiNC in comparison to the state-of-the-art non-pyrolyzed nanocarbon-supported f-MNC catalysts. It is to be noted that for the CO2RR the onset potentials of the state-of-the-art non-pyrolyzed nanocarbon-supported MNC catalysts fall in the potential range of −0.6 to −1.2 V.56–64 Clearly, CNT@f-NiNC yielded both a higher onset potential and a higher conversion efficiency for the CO2RR to the CO-product than that of the documented non-pyrolyzed M-NC-based catalysts (Table S2, ESI†).
Pyrolyzed atomically dispersed f-MNC towards the ORR
The as-prepared f-MNC materials were pyrolyzed to further crosslink or solidify the structure, and to increase the conductivity at the same time. The assistance of Zn was found to have a significant role for single-site or dual-site f-MNC electrocatalysts to further enhance the performance of f-MNC beyond Pt/C.5,9,65 In order to clarify the important role of Zn, f-FeCoNC900 was also prepared without Zn by adding different concentrations of the Fe/Co salts (C[Fe2+] or C[Co2+] = 0.001, 0.003, and 0.005 M) with Fe/Co mole ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The TEM images of f-FeCoNC900 obtained using different Fe/Co concentrations are shown in Fig. S16a–c (ESI†). Minor metal aggregation can be observed in the TEM image of 0.001 M f-FeCoNC900, when the Fe/Co concentration increases to 0.003 M and above, obvious metal particles start to form. The severe metal aggregation in the 0.005 M case cannot be eliminated after being leached in 1.0 M H2SO4 at 80 °C for 12 h (Fig. S16d, ESI†). The XRD profiles in Fig. S16e (ESI†) confirm the easy formation of CoFe alloys when the Zn components were absent. Due to the “fence effect”, carbon materials decorated with isolated f-Fe/CoNC moieties were steadily achieved after annealing at 900 °C (named f-FeCoNC900), when f-ZnFeCoNC can be readily transformed with Zn components mostly vaporized. Similarly, atomically dispersed f-FeNC and f-CoNC were also obtained following the same route as for f-FeCoNC900, which were denoted as f-FeNC900 and f-CoNC900, respectively. Fig. S17a (ESI†) shows the typical TEM image of f-FeCoNC900 with irregularly shaped morphology. There is no obvious aggregation for f-FeCoNC900 and the control samples of f-FeNC900 and f-CoNC900 (Fig. S18a and d, ESI†). The elemental mapping in Fig. S17b (ESI†) reveals the uniform distribution of N, Fe, and Co elements after the high-temperature pyrolysis. Fig. 4a exhibits the HAADF-STEM image of f-FeCoNC900, confirming the rich content of atomically dispersed Fe/Co species. XPS (Fig. S17c, ESI†) confirms mainly five elements of C, O, N, Fe, and Co in f-FeCoNC900, which take overwhelming contents, reaching ∼86.2, ∼7.7, ∼5.0, ∼0.53, and ∼0.54 at%, respectively. As shown in Fig. S17d (ESI†), about 27.6% of N species were pyrrolic N with N-metal configuration. The XPS spectra on Fe2p and Co2p confirm that all the Fe/Co components were present in their oxidation states, and no signal of zero-valence metal is deconvoluted, as shown in Fig. S17e and f (ESI†).
:1. The TEM images of f-FeCoNC900 obtained using different Fe/Co concentrations are shown in Fig. S16a–c (ESI†). Minor metal aggregation can be observed in the TEM image of 0.001 M f-FeCoNC900, when the Fe/Co concentration increases to 0.003 M and above, obvious metal particles start to form. The severe metal aggregation in the 0.005 M case cannot be eliminated after being leached in 1.0 M H2SO4 at 80 °C for 12 h (Fig. S16d, ESI†). The XRD profiles in Fig. S16e (ESI†) confirm the easy formation of CoFe alloys when the Zn components were absent. Due to the “fence effect”, carbon materials decorated with isolated f-Fe/CoNC moieties were steadily achieved after annealing at 900 °C (named f-FeCoNC900), when f-ZnFeCoNC can be readily transformed with Zn components mostly vaporized. Similarly, atomically dispersed f-FeNC and f-CoNC were also obtained following the same route as for f-FeCoNC900, which were denoted as f-FeNC900 and f-CoNC900, respectively. Fig. S17a (ESI†) shows the typical TEM image of f-FeCoNC900 with irregularly shaped morphology. There is no obvious aggregation for f-FeCoNC900 and the control samples of f-FeNC900 and f-CoNC900 (Fig. S18a and d, ESI†). The elemental mapping in Fig. S17b (ESI†) reveals the uniform distribution of N, Fe, and Co elements after the high-temperature pyrolysis. Fig. 4a exhibits the HAADF-STEM image of f-FeCoNC900, confirming the rich content of atomically dispersed Fe/Co species. XPS (Fig. S17c, ESI†) confirms mainly five elements of C, O, N, Fe, and Co in f-FeCoNC900, which take overwhelming contents, reaching ∼86.2, ∼7.7, ∼5.0, ∼0.53, and ∼0.54 at%, respectively. As shown in Fig. S17d (ESI†), about 27.6% of N species were pyrrolic N with N-metal configuration. The XPS spectra on Fe2p and Co2p confirm that all the Fe/Co components were present in their oxidation states, and no signal of zero-valence metal is deconvoluted, as shown in Fig. S17e and f (ESI†).
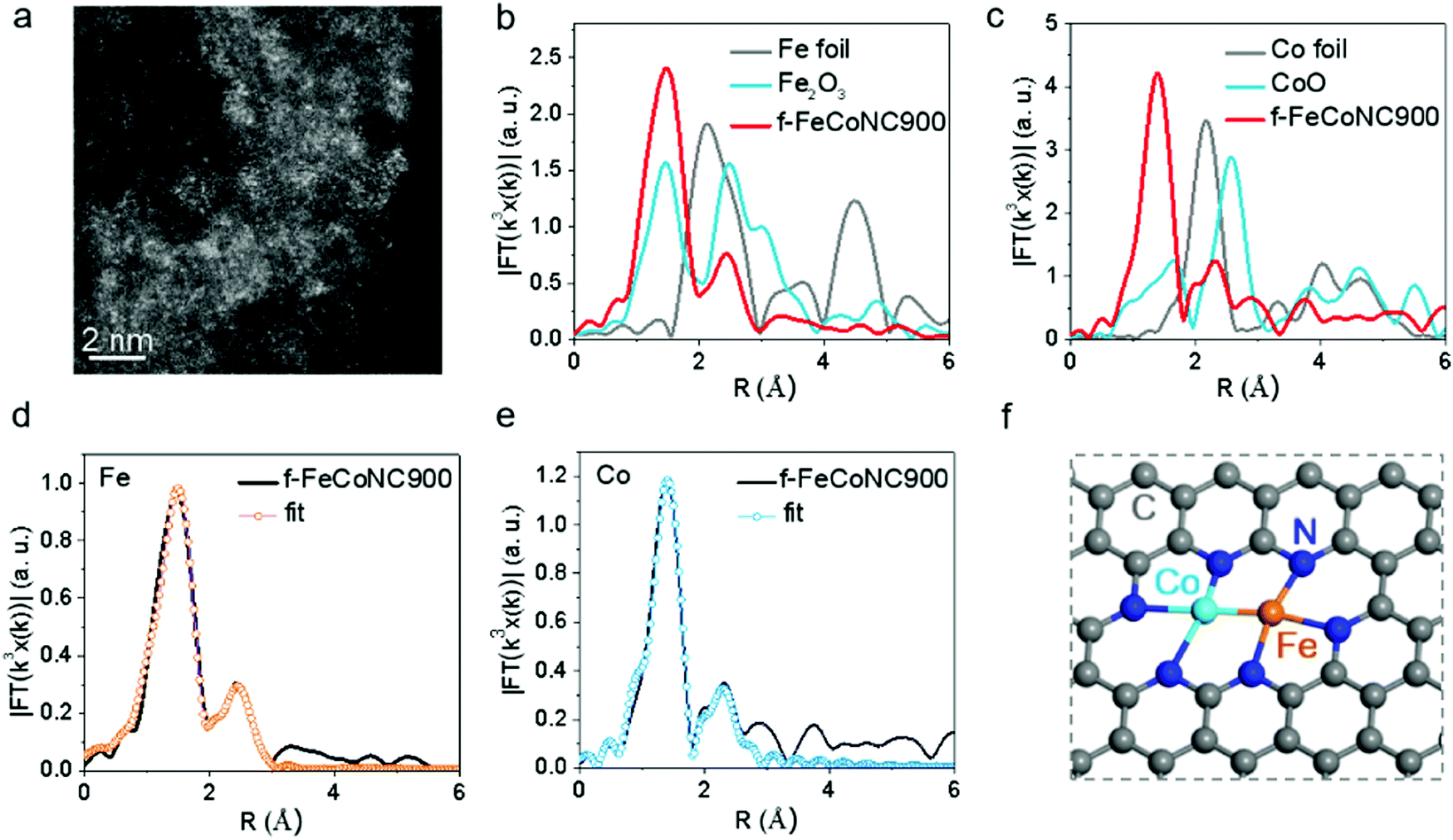 | ||
| Fig. 4 Chemical state and coordination information of atomically dispersed f-FeCoNC900. (a) The HAADF-STEM image of f-FeCoNC900. (b) The Fourier transformed (FT) k3-weighted χ(k)-function of the EXAFS spectra for the Fe K-edge of f-FeCoNC900 and reference samples. (c) The Fourier transformed (FT) k3-weighted χ(k)-function of the EXAFS spectra for Co K-edge of f-FeCoNC900 and reference samples. (d) The corresponding Fe K-edge EXAFS fitting curves of f-FeCoNC900. (e) The corresponding Co K-edge EXAFS fitting curves of f-FeCoNC900. (f) The proposed structure of the Fe–Co dual sites. | ||
The X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were employed to investigate the local coordination environment of the Fe and Co atoms. Fig. S19a (ESI†) shows the Fe k-edge XANES spectra of the f-FeCoNC900 sample, which falls in between that of Fe foil and Fe2O3, suggesting that the Fe atoms carry positive charges. The FT k3-weighted EXAFS spectrum in R space of Fe is shown in Fig. 4b, in which a strong peak at ca. 1.56 Å indicates that the main coordination structure of f-FeCoNC900 is in the Fe–N path. Meanwhile, the secondary peak at ca. 2.30 Å indicates the presence of the M–M bond. Similarly, the Co atoms also carry positive charges, as shown in Fig. S19b (ESI†). The strong FT curve of the Co K-edge at ca. 1.40 Å contributes to the main Co–N path of f-FeCoNC900. A small peak at ca. 2.30 Å contributing to the M–M bond is also observed in Fig. 4c. Based on the fitting curve in Fig. 4d and e and Fig. S19c and d (ESI†), the coordination numbers of Co–N and Co–Fe are 2.8 and 0.7, as summarized in Table S3 (ESI†). The coordination numbers of Fe–N and Fe–Co are 3.2 and 1.1, respectively. Thus, the possible structure of f-FeCoNC900 can be deduced as Fe(N3)–Co(N3), as displayed in Fig. 4f. The FT curves of f-CoNC900 and f-FeNC900 are shown in Fig. S18 (ESI†), which indicate that the samples are mainly composed of Co–N and Fe–N coordination paths, respectively. Such coordination nature is very similar to those of M-NC SAECs (M includes mainly Fe and Co) with either single or dual active sites obtained by pyrolyzing the MOF,28,38,66 but herein the starting materials are cheaper and more metals are feasible.
The scalable production of the abovementioned f-FeCoNC900 was realized through pre-deposition of the precursor f-ZnFeCoNC on the widely available inexpensive activated carbon (AC), followed by annealing at 900 °C.50 Fig. S20a (ESI†) displays the typical TEM image of the AC-supported f-FeCoNC900 (denoted as AC@f-FeCoNC900), revealing the similar irregular shape of the AC support. The HAADF-STEM image indicates the intimate contact between the AC and f-FeCoNC900 skin layer, as demonstrated in Fig. 5a. The atomically dispersed Fe/Co sites were well-kept after being loaded onto AC (Fig. 5a and Fig. S20b, ESI†). More importantly, AC@f-FeCoNC900 can be easily scaled up. For instance, by using a 300 mL autoclave, we obtained 17.0 g AC@f-FeCoNC900 in a single batch (inset of Fig. 5a). The EXAFS and FT k3-weighted χ(k)-function of AC@f-FeCoNC900 (Fig. S20c–f, ESI†) identified the same intrinsic structure: Co–N and Fe–N coordination along with bits of Fe–Co bonds.
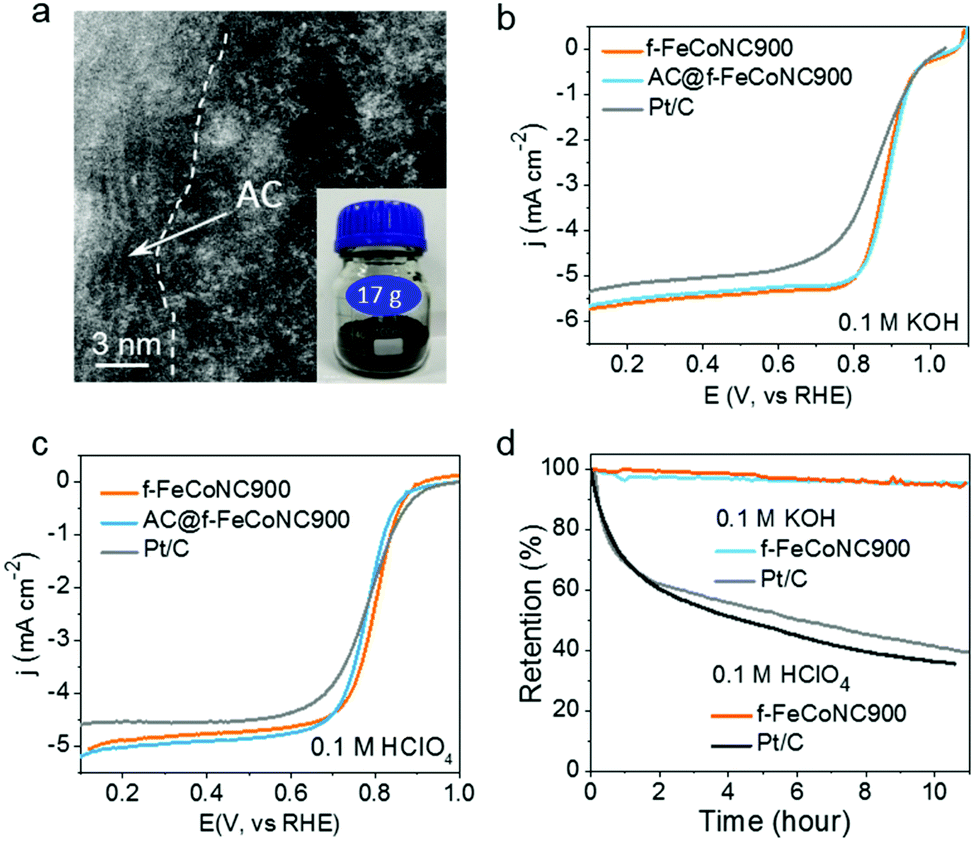 | ||
| Fig. 5 Electrochemical measurements of atomically dispersed f-FeCoNC w/o AC support. (a) The HAADF-STEM image of AC@f-FeCoNC900 with the inset photo showing the production of AC@f-FeCoNC900 in the gram scale. (b and c) The LSV curves of Pt/C, f-FeCoNC900 and AC@f-FeCoNC900 in O2-saturated 0.1 M KOH (b) and 0.1 M HClO4 (c) with a sweep rate of 5 mV s−1 and electrode rotation rate of 1600 rpm. (d) Current stability of f-FeCoNC900 at 0.8 V vs. RHE in O2-saturated 0.1 M KOH and 0.1 M HClO4, respectively. | ||
Excellent ORR performance of f-FeCoNC900
Due to the same intrinsic nature of rich atomically dispersed Fe–Co dimers, the AC@f-FeCoNC900 produced on a large scale has nearly identical high ORR performance to f-FeCoNC900, as submitted to the ORR measurement in 0.1 M KOH (Fig. 5b). It outperformed the Pt/C catalyst in terms of onset potential (1.05 V cf. Pt/C of 1.00 V), half-wave potential (0.89 V cf. Pt/C of 0.84 V) and limiting current density (5.75 mA cm−2 at 1600 rpm cf. Pt/C of 5.5 mA cm−2), indicating its extraordinary catalytic activity. Meanwhile, the significantly enhanced ORR performance of f-FeCoNC900 compared to the single-metal f-FeNC900 and f-CoNC900 confirms the beneficial synergistic effect in between the isolated Fe(N3)–Co(N3) sites (Fig. S20a, ESI†), which matches well with those reported on the atomically dispersed dual Fe and Co sites.9,65,67,68 The LSV curves of f-FeCoNC900 prepared with or without Zn were also collected and compared in Fig. S21 (ESI†). f-FeCoNC900 prepared without Zn showed much worse ORR performance in both alkaline and acid medium relative to the ones with Zn addition and Pt/C as well, which once more confirmed the importance of Zn for effective spatial confinement of the Fe and Co atoms.In the potential range of 0.3 to 0.8 V, f-FeCoNC900 exhibited nearly the 4-electron reaction pathway (Fig. S22b, ESI†). Besides the outstanding catalytic activity, f-FeCoNC900 also exhibited the capability of tolerance to methanol (Fig. S22c, ESI†) and long-term current stability under a practical operating potential of 0.8 V (< 5% activity loss in 11 h. cf. over 60% activity loss for Pt/C, Fig. 5d). Fig. 5c shows the typical polarization curve of f-FeCoNC900, AC@f-FeCoNC900 and a commercial Pt/C catalyst in 0.1 M HClO4. Although the onset potential (0.87 V) of f-FeCoNC is 30 mV lower than that of Pt/C (0.90 V), it shows an impressive half-wave potential of 0.81 V, which is 20 mV higher than that of Pt/C. Meanwhile, a higher limiting current density of ∼5.0 mA cm−2 was measured for f-FeCoNC900 at 0.6 V, compared to 4.5 mA cm−2 of Pt/C. f-FeCoNC900 exhibited a higher ORR activity in acidic medium than both f-FeNC and f-CoNC (Fig. S23a, ESI†). For the ORR, f-FeCoNC900 possesses close-to-4 electron charge transfer number in the wide potential range (0.1 to 0.6 V), which is even higher than Pt/C (Fig. S23b, ESI†). Additionally, f-FeCoNC900 displayed very high stability in an acidic electrolyte, e.g., the activity loss was less than 5% after 11 h measurement while Pt/C showed nearly 65% activity loss (Fig. 5d). Such a high performance could be ascribed to the much-stabilized metal–nitrogen sites as well as the significantly enhanced graphitization degree and conductivity, being similar to that observed on the MOF-pyrolysis derivatives.9,33,34 Combining the merits of benign synthetic procedures, low fabrication cost and high ORR catalytic performance, it is safe to envision that AC@f-FeCoNC900 can serve as a promising alternative to the expensive Pt/C for the ORR cathode catalyst in practical PEMFC applications.
Conclusions
In summary, a scalable and general synthetic method for the fabrication of atomically dispersed formamide-converted transition metal–nitrogen–carbon materials (f-MNCs) has been developed through metal-mediated formamide condensation and carbonization. Seven types of single-metallic f-MNCs including Fe, Co, Ni, Mn, Zn, Mo, and Ir, two types of bi-metallic f-MNCs (ZnFe and ZnCo), and one tri-metallic f-MNC (ZnFeCo) were synthesized to demonstrate the generality of the developed method. Moreover, all the f-MNC single-atom electrocatalysts (SAECs) prepared can be thinly coated onto various carbon and oxide supports to increase the production scale. For instance, simply depositing a thin layer of f-FeNC and f-NiNC on commercially available unmodified CNTs delivered an excellent catalytic performance towards the oxygen reduction reaction (ORR) and the CO2 reduction reaction (CO2RR), respectively. After high temperature pyrolysis, the treated f-FeCoNC sample yielded dense atomically dispersed isolated Fe(N3)–Co(N3) sites, the latter exhibited outstanding ORR activity and stability which were higher than the commercial Pt/C in both alkaline and acid media under the same catalyst loading. More importantly, the extraordinary ORR performance was well retained after the f-FeCoNC was deposited on an inexpensive activated carbon (AC). Considering the capability of accommodating various metallic components, the developed methodology provides numerous new possibilities for the large-scale synthesis of SAECs with isolated single-, dual-, or even triple-metallic sites for relevant electrocatalysis applications in energy technologies such as fuel cells.Author contributions
G. Z. and Y. J. contributed equally to this work, G. Z., and X. S. conceived the idea. G. Z. and Y. J. carried out the sample synthesis, characterization, performance measurement and manuscript writing. C. Z., X. X., and K. S. helped with the sample synthesis. R. C. helped with the CO2RR measurement. W. C. and L. Z. helped with the XAFS measurements and discussion. H. T. helped with the catalyst performance measurement. Y. K., W. L., J. L., X. S., W.-F. L. and H. D. edited the manuscript, W.-F. L. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC, 21520102002, 91622116, 21471014, and 21701101), the National Key Research and Development Project (2016YFF0204402 and 2017YFA0206500), the Fundamental Research Funds for the Central Universities, the Long-Term Subsidy Mechanism from the Ministry of Finance and the Ministry of Education of China, the Shandong Scientific Research Awards Foundation for Outstanding Young Scientists (grant number ZR2018JL010), the Shandong Joint Fund of Outstanding Young Talents (grant number ZR2017BB018) and Loughborough University (H10841). We are also grateful to Prof. Shanfu Lu from BeiHang University for the help with the catalyst performance test.Notes and references
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- B. Bayatsarmadi, Y. Zheng, A. Vasileff and S. Z. Qiao, Small, 2017, 13, 1700191 CrossRef PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2017, 1701343 Search PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D. J. Liu and L. Zhuang, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- Q. Liu, X. Liu, L. Zheng and J. Shui, Angew. Chem., Int. Ed., 2017, 57, 1204–1208 CrossRef PubMed.
- Y. Pan, S. Liu, K. Sun, X. Chen, B. Wang, K. Wu, X. Cao, W. C. Cheong, R. Shen and A. Han, Angew. Chem., Int. Ed., 2018, 130, 8750–8754 CrossRef.
- S. Li, C. Chen, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2017, 57, 1856–1862 CrossRef PubMed.
- L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- G. Zhao, H. Liu and J. Ye, Nano Today, 2018, 19, 108–125 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu and P. Du, Sci. Adv., 2015, 1, e1500462 CrossRef PubMed.
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef PubMed.
- D. Yan, L. Guo, C. Xie, Y. Wang, Y. Li, H. Li and S. Wang, Sci. China Mater., 2018, 61, 679–685 CrossRef CAS.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef.
- K. Jiang and H. Wang, Chem, 2018, 4, 194–195 CAS.
- X. Wang, H. Xiao, A. Li, Z. Li, S. Liu, Q. Zhang, Y. Gong, L. Zheng, Y. Zhu and C. Chen, J. Am. Chem. Soc., 2018, 140, 15336–15341 CrossRef CAS PubMed.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W. C. Cheong and R. Shen, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- Z. Miao, X. Wang, M. C. Tsai, Q. Jin, J. Liang, F. Ma, T. Wang, S. Zheng, B. J. Hwang and Y. Huang, Adv. Energy Mater., 2018, 8, 1801226 CrossRef.
- H. W. Liang, W. Wei, Z. S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 10987 CrossRef.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, M. Shu, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- D. Liu, C. Wu, S. Chen, S. Ding, Y. Xie, C. Wang, T. Wang, Y. A. Haleem, Z. U. Rehman and Y. Sang, Nano Res., 2017, 11, 2217–2228 CrossRef.
- X. Song, S. Chen, L. Guo, Y. Sun, X. Li, X. Cao, Z. Wang, J. Sun, C. Lin and Y. Wang, Adv. Energy Mater., 2018, 8, 1801101 CrossRef.
- Z.-Y. Wu, S.-L. Xu, Q.-Q. Yan, Z.-Q. Chen, Y.-W. Ding, C. Li, H.-W. Liang and S.-H. Yu, Sci. Adv., 2018, 4, eaat0788 CrossRef PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- Z. Liang, C. Qu, D. Xia, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 57, 9604–9633 CrossRef CAS PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu and C. Wu, Angew. Chem., Int. Ed., 2016, 129, 625–629 CrossRef.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng and W. Yan, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- Q.-L. Zhu, W. Xia, L.-R. Zheng, R. Zou, Z. Liu and Q. Xu, ACS Energy Lett., 2017, 2, 504–511 CrossRef CAS.
- J. L. D. L. Fuente, M. Ruiz-Bermejo, C. Menor-Salván and S. Osuna-Esteban, Polym. Degrad. Stab., 2011, 96, 943–948 CrossRef.
- Z. Gong, B. Yang, H. Lin, Y. Tang, Z. Tang, J. Zhang, H. Zhang, Y. Li, Y. Xie and Q. Li, ACS Nano, 2016, 10, 4228–4235 CrossRef CAS PubMed.
- V. S. Nguyen, H. L. Abbott, M. M. Dawley, T. M. Orlando, J. Leszczynski and M. T. Nguyen, J. Phys. Chem. A, 2011, 115, 841–851 CrossRef CAS PubMed.
- Z. Chen, S. Mitchell, E. Vorobyeva, R. K. Leary, R. Hauert, T. Furnival, Q. M. Ramasse, J. M. Thomas, P. A. Midgley, D. Dontsova, M. Antonietti, S. Pogodin, N. López and J. Pérez-Ramírez, Adv. Funct. Mater., 2017, 27, 1605785 CrossRef.
- A. Zitolo, N. Ranjbarsahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil and S. Mukerjee, Nat. Commun., 2017, 8, 957 CrossRef PubMed.
- S. Barman and M. Sadhukhan, J. Mater. Chem., 2012, 22, 21832–21837 RSC.
- J. Xu, J. Zhu, X. Yang, S. Cao, J. Yu, M. Shalom and M. Antonietti, Adv. Mater., 2016, 28, 6727–6733 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham and L. M. Liu, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao and Y. Li, Chem, 2017, 3, 652–664 CAS.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- Y. Wang, Z. Wei, Y. Nie and Y. Zhang, J. Mater. Chem. A, 2016, 5, 1442–1445 RSC.
- N. Bing, O. Chen, X. Xu, Z. Jing and W. Xun, Adv. Mater., 2017, 29, 1701354 CrossRef PubMed.
- J. Masa, K. Elumeeva, M. Kazakova, D. M. Morales, D. Medina, A. Selyutin, G. Golubtsov, Y. Ivanov, V. Kuznetzov and A. Chuvilin, ChemSusChem, 2018, 11, 1204–1214 CrossRef PubMed.
- C. Wang, H. Zhang, J. Wang, Z. Zhao, J. Wang, Y. Zhang, M. Cheng, H. Zhao and J. Wang, Chem. Mater., 2017, 29, 9915–9922 CrossRef CAS.
- P. Song, M. Luo, X. Liu, W. Xing, W. Xu, Z. Jiang and L. Gu, Adv. Funct. Mater., 2017, 27, 1700802 CrossRef.
- X. Chen, L. Yu, S. Wang, D. Deng and X. Bao, Nano Energy, 2017, 32, 353–358 CrossRef CAS.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. J. Dynes, M. Gangishetty and D. Su, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- A. Maurin and M. Robert, Chem. Commun., 2016, 52, 12084–12087 RSC.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- T. Moeller, W. Ju, A. Bagger, X. Wang, F. Luo, T. N. Thanh, A. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- A. Tatin, C. Comminges, B. Kokoh, C. Costentin, M. Robert and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5526–5529 CrossRef CAS PubMed.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2017, 129, 6568–6572 CrossRef.
- X. Zhang, Z. Wu, X. Zhang, L. Li, Y. Li, H. Xu, X. Li, X. Yu, Z. Zhang and Y. Liang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- Z. Zhang, J. Xiao, X. J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 16339–16342 CrossRef CAS PubMed.
- J. E. Pander III, A. Fogg and A. B. Bocarsly, ChemCatChem, 2016, 8, 3536–3545 CrossRef.
- D. Zhang, W. Chen, Z. Li, Y. Chen, L. Zheng, Y. Gong, Q. Li, R. Shen, Y. Han and W. C. Cheong, Chem. Commun., 2018, 54, 4274–4277 RSC.
- R. Zhao, Z. Liang, S. Gao, C. Yang, B. Zhu, J. Zhao, C. Qu, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2018, 131, 1997–2001 CrossRef.
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu and W. He, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang and C. Zhao, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ee00162j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |