
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and evaluation of conjugated polymers with polar side chains as electrode materials for electrochemical energy storage in aqueous electrolytes†
Davide
Moia‡
 *a,
Alexander
Giovannitti‡
*ab,
Anna A.
Szumska
a,
Iuliana P.
Maria
b,
Elham
Rezasoltani
a,
Michael
Sachs
b,
Martin
Schnurr
b,
Piers R. F.
Barnes
a,
Iain
McCulloch
bc and
Jenny
Nelson
*a
*a,
Alexander
Giovannitti‡
*ab,
Anna A.
Szumska
a,
Iuliana P.
Maria
b,
Elham
Rezasoltani
a,
Michael
Sachs
b,
Martin
Schnurr
b,
Piers R. F.
Barnes
a,
Iain
McCulloch
bc and
Jenny
Nelson
*a
aDepartment of Physics, Imperial College London, SW7 2AZ London, UK. E-mail: d.moia@fkf.mpg.de; ag19@stanford.edu; jenny.nelson@imperial.ac.uk
bDepartment of Chemistry, Imperial College London, SW7 2AZ London, UK
cPhysical Sciences and Engineering Division, KAUST Solar Center (KSC), King Abdullah University of Science and Technology (KAUST), KSC Thuwal 23955-6900, Saudi Arabia
First published on 15th March 2019
Abstract
We report the development of redox-active conjugated polymers that have potential applications in electrochemical energy storage. Side chain engineering enables processing of the polymer electrodes from solution, stability in aqueous electrolytes and efficient transport of ionic and electronic charge carriers. We synthesized a 3,3′-dialkoxybithiophene homo-polymer (p-type polymer) with glycol side chains and prepared naphthalene-1,4,5,8-tetracarboxylic-diimide-dialkoxybithiophene (NDI-gT2) copolymers (n-type polymer) with either a glycol or zwitterionic side chain on the NDI unit. For the latter, we developed a post-functionalization synthesis to attach the polar zwitterion side chains to the polymer backbone to avoid challenges of purifying polar intermediates. We demonstrate fast and reversible charging of solution processed electrodes for both the p- and n-type polymers in aqueous electrolytes, without using additives or porous scaffolds and for films up to micrometers thick. We apply spectroelectrochemistry as an in operando technique to probe the state of charge of the electrodes. This reveals that thin films of the p-type polymer and zwitterion n-type polymer can be charged reversibly with up to two electronic charges per repeat unit (bipolaron formation). We combine thin films of these polymers in a two-electrode cell and demonstrate output voltages of up to 1.4 V with high redox-stability. Our findings demonstrate the potential of functionalizing conjugated polymers with appropriate polar side chains to improve the accessible capacity, and to improve reversibility and rate capabilities of polymer electrodes in aqueous electrolytes.
Broader contextThe ideal electrode material for electrochemical energy storage should be able to transport both electrons and ions efficiently and store a large density of these charges at accessible potentials. In high-performance battery electrodes, the requirements of ionic and electronic conductivity and charge storage are commonly fulfilled by combining different materials to implement these functions separately. In this work we show that conjugated polymers can be designed to combine favourable ionic and electronic transport properties in a single-phase material through appropriate design of the polymer backbone and side chains. We find that whilst choice of polymer backbone controls stability, redox-potentials and electronic transport, design of the polar side chain influences the ionic mobility, charging rate capability and capacity. Using solution-processed p- and n-type polymers we demonstrate fast charging (on the second timescale) and reversible behaviour for microns-thick electrodes in aqueous electrolytes, and present a proof-of-concept energy storage device that functions in a salt-water electrolyte. In comparison with conventional lithium-ion electrodes, the new materials show fast charging rates without the need for additives or porous scaffolds, although with lower specific capacity. The new materials thus open up a new approach to the design of solution processable, non-toxic electrodes compatible with aqueous electrolytes. |
Introduction
Polar side chains attached to conjugated polymer backbones have been investigated as a strategy to facilitate ionic transport in the bulk of mixed electronic-ionic conducting polymers.1–4 In particular, ethylene glycol based side chains have been used to facilitate ion transport in conjugated polymer films leading to the successful demonstration of organic electrochemical transistors (OECTs),3–5 electrochromic6,7 and biomedical devices8 operating in aqueous electrolytes. Conjugated polymers with polar side chains are therefore a promising class of materials for the development of electrodes for safe and sustainable electrochemical energy storage devices using water based electrolytes.Prior attempts to develop polymer based electrodes with high energy and power densities have typically targeted electrochemically active polymers9 with a high density of redox sites in combination with conductive additives or scaffolds and inert polymer binders.10–14 Through such approaches, examples of capacities exceeding 150 mA h g−1, efficient charging on the 10–100 second timescale and electrode stability over more than 1000 cycles were reported. Most studies report operation of these electrodes in organic electrolytes that show a wide electrochemical stability window but are usually hazardous and/or flammable. Several examples of conjugated p-type polymers have been previously presented, showing in some cases competitive specific capacities. Significantly fewer reports of n-type conjugated polymer electrodes have been made and, to our knowledge, no previous study successfully demonstrated this class of materials as electrodes in water-based applications. Liang et al. have shown that naphthalene-1,4,5,8-tetracarboxylic-diimide-bithiophene (NDI-T2) copolymers with alkyl side chains can be a promising electrode material (capacity of ∼50 mA h g−1 and charging times in the order of ∼10 seconds) when the copolymer is mixed with conductive carbon and measured in an organic electrolyte.15 Functionalizing conjugated polymer backbones with alkyl side chains increases their solubility in common organic solvents and facilitates processing of electrodes using printing or coating techniques. However, alkyl chains are an unsuitable transport medium for polar hydrated ions3 as shown by previous studies on poly(3-hexylthiophene-2,5-diyl) (P3HT)16 and naphthalene1,4,5,8-tetracarboxylic-diimide-bithiophene copolymers (NDI-T2).4
To achieve the levels of ionic conductivity, specific capacity and stability that are needed for the application of conjugated polymers as electrodes, the polymer backbone and side chains need to be optimized independently. Redox-stability in aqueous electrolytes requires that the oxidation and reduction potentials of the electrode materials lie at voltages within the electrochemical stability window of water.17 Electron-rich polythiophene (p-type), and electron-deficient donor–acceptor and acceptor-type polymer backbones (n-type) show, respectively, low oxidation potentials and low reduction potentials in aqueous electrolytes which makes them suitable structures for electrochemical charging in water. Efficient ionic exchange with aqueous electrolytes has been achieved for p-type polymers based on polythiophenes by using glycol side chains.3 On the other hand, the use of glycol chains in n-type polymers based on NDI-T2 results in low electron mobility, as shown by measurements on OECT devices,4 and is also expected to inhibit cation transport.18 Alternative polar side chains, such as zwitterionic and polyelectrolyte, have been attached to NDI-T2 copolymers to improve interfacial properties in organic electronic devices but have not so far not been tested in electrochemical energy storage devices.19,20 Polyelectrolytes with a mobile cation have been explored as side groups of non-conjugated radical polymers, where the cation is exchanged with the electrolyte to form a zwitterion structure when the polymer is oxidized. This strategy improved the reversibility as well as the capacity of the polymer in water electrolytes, suggesting that the interaction between the polymer redox sites and compensating ionic charge plays an important role on the electrochemical response of the material.21 Appropriate choice of the side chain is expected to also influence the reversibility and specific capacity of n-type polymer structures, but design rules that address this question are currently missing. In addition, while polar side chains are known to facilitate ion transport in polymer films, it is not clear to what extent this strategy can improve the rate capability of compact, solution processed polymer electrodes of micrometer scale thickness.
Here, we report the development of solution processable redox-active polymers with the goal of enabling electrochemical energy storage in aqueous electrolytes. We choose polymer backbones which show high stability during electrochemical redox reactions and engineer the side chains to enable reversible charging in water based electrolytes. We synthesize a p-type polymer based on a homo-3,3′-dialkoxybithiophene polymer (p(gT2), Fig. 1a) with methyl end-capped triethylene glycol side chains (TEG). For the n-type polymer, we prepare donor–acceptor type copolymers with naphthalene-1,4,5,8-tetracarboxylic diimide (NDI) and 3,3′-dialkoxybithiophene22–24 where we either attach zwitterionic side chains (p(ZI-NDI-gT2), Fig. 1b) or linear glycol side chains4 (p(g7NDI-gT2)) to the NDI unit. For the zwitterionic side chain, we place the cation (ammonium ion) in close proximity (C2-spacer) to the NDI unit. This is done with the aim of enabling the electronic charge to interact with the ammonium ion during charging of the polymer rather than requiring charge compensating cations to approach the polymer backbone (Fig. 1c). For all n-type copolymers reported here, TEG side chains are attached on the donor comonomer to ensure high solubility of the polymers in common organic solvents, which allows solution processing of the materials. We characterize the polymer films in sodium chloride aqueous solution using electrochemistry and spectroelectrochemistry, revealing their ability to host bipolarons and to charge/discharge on the second timescale for samples over 1 micron thick. We finally explore their application in a two-electrode electrochemical energy storage device, which can be charged up to 1.4 V, and discuss some of the limitations involved when considering salt water as the electrolyte of the cell.
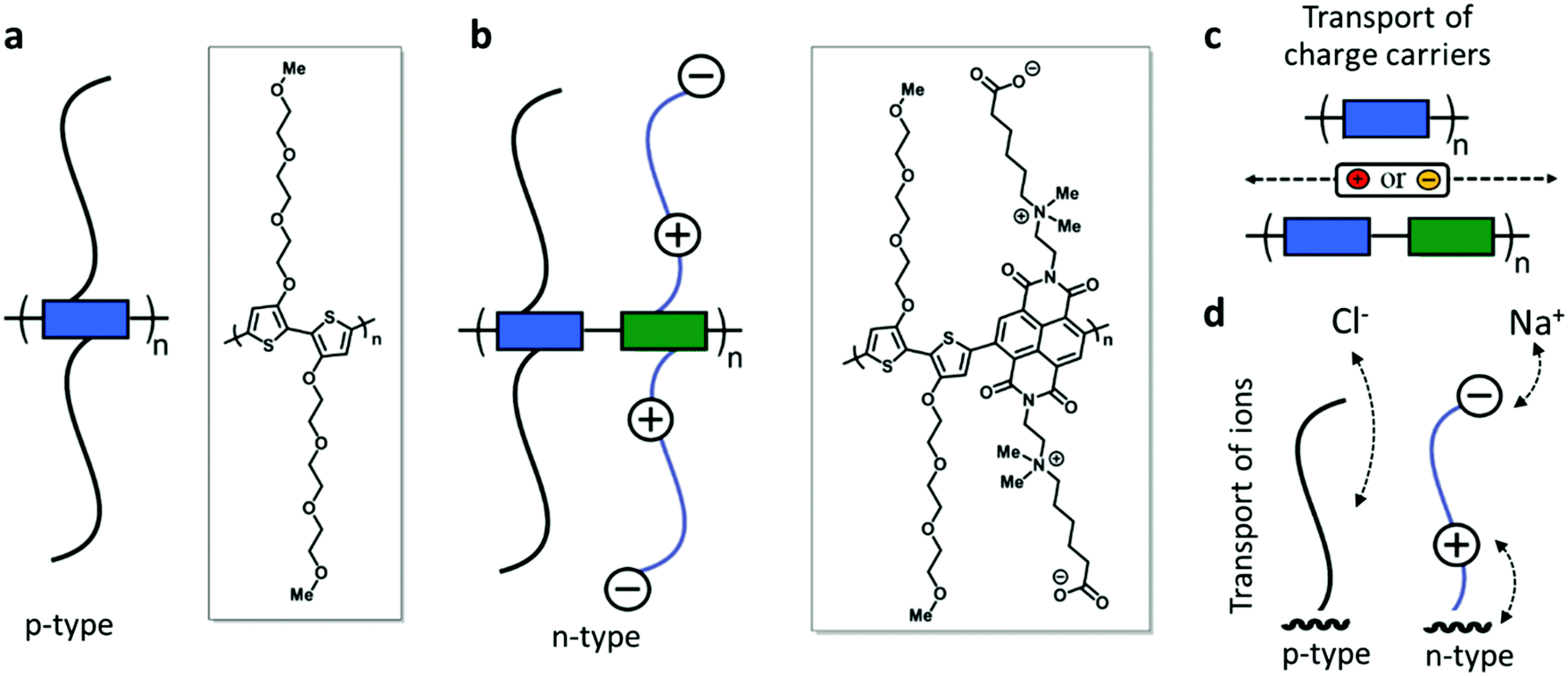 | ||
| Fig. 1 (a) p-type homo-polymer with TEG side chains p(gT2); (b) n-type donor–acceptor copolymer with zwitterion side chains on the acceptor units (marked as green blocks in the schematic) and TEG side chains on the donor units (blue blocks) p(ZI-NDI-gT2). Schematic of the transport of (c) the electronic charges along the backbone of the polymers and (d) ions with the aid of polar side chains. | ||
Results and discussion
The properties of the p- and n-type polymers are summarized in Table 1. Details of synthesis and characterization of monomers and polymers are reported in Sections S1–S7 of the ESI.† Polymer p(gT2) was synthesized by Stille polymerization as previously reported for other copolymers containing glycol side chains.25 The polymer thin film absorption spectrum is presented in Fig. 2a and shows an absorption onset of 755 nm with two vibronic transitions (0–0 and 0–1). The ionization potential (IP) of p(gT2) was measured by photoelectron spectroscopy in air (PESA, 4.5 eV) and cyclic voltammetry (CV, 4.5 eV) in organic electrolytes where the IP of the polymer was calculated as reported in the literature22,26 (Fig. 2b). The CV in acetonitrile electrolyte shows broad peaks and high redox reversibility (proximity for the peak position and approximately equal integrated charge for the charging and discharging curves) for the oxidation of the p(gT2) polymer film. The molecular weight distribution of the polymer was measured by gel permeation chromatography (GPC) in DMF (Mn = 36 kDa and Mw = 60 kDa). To avoid overestimation of the molecular weight distribution mainly due to aggregation of the conjugated polymer with glycol side chains,4 the high molecular weight fraction of the GPC trace was neglected and only the lower molecular weight fraction was considered for molecular weight analysis (Fig. S10 (ESI†), bimoduale eluation). Additionally, mass spectrometry measurements by matrix-assisted laser deposition/ionization time of flight (MALDI-ToF) were carried out and polymer chains length with >35 repeat units were detected (Fig. S11 (ESI†), (corresponding to >18 kDa)). In comparison to other reported 3,3′-dialkoxybithiophene copolymers with glycol side chains on only every other repeat unit,3,25 p(gT2) has a higher solubility in organic solvents such as N,N-dimethylformamide (DMF) or chloroform (CHCl3). Good solubility of the polymer is essential for fabrication of electrodes from solution.| Polymer | IPa [eV] | IPb [eV] | Absorption onset [nm] | Optical band gap [eV] | M n [kDa] | M w [kDa] |
|---|---|---|---|---|---|---|
| a Photoelectron spectroscopy in air (PESA). b CV measurements were carried out in acetonitrile (0.1 M NBu4PF6, 100 mV s−1). c GPC measurements were carried out in DMF with 5 mM NH4BF4. d Bimoduale eluation; high molecular weight fraction (aggregates) were not considered in the molecular weight analysis. | ||||||
| p(gT2) | 4.48 | 4.50 | 755 | 1.64 | 36d | 60d |
| p(ZI-NDI-gT2) | 5.15 | 5.25 | 1049 | 1.18 | 24 | 53 |
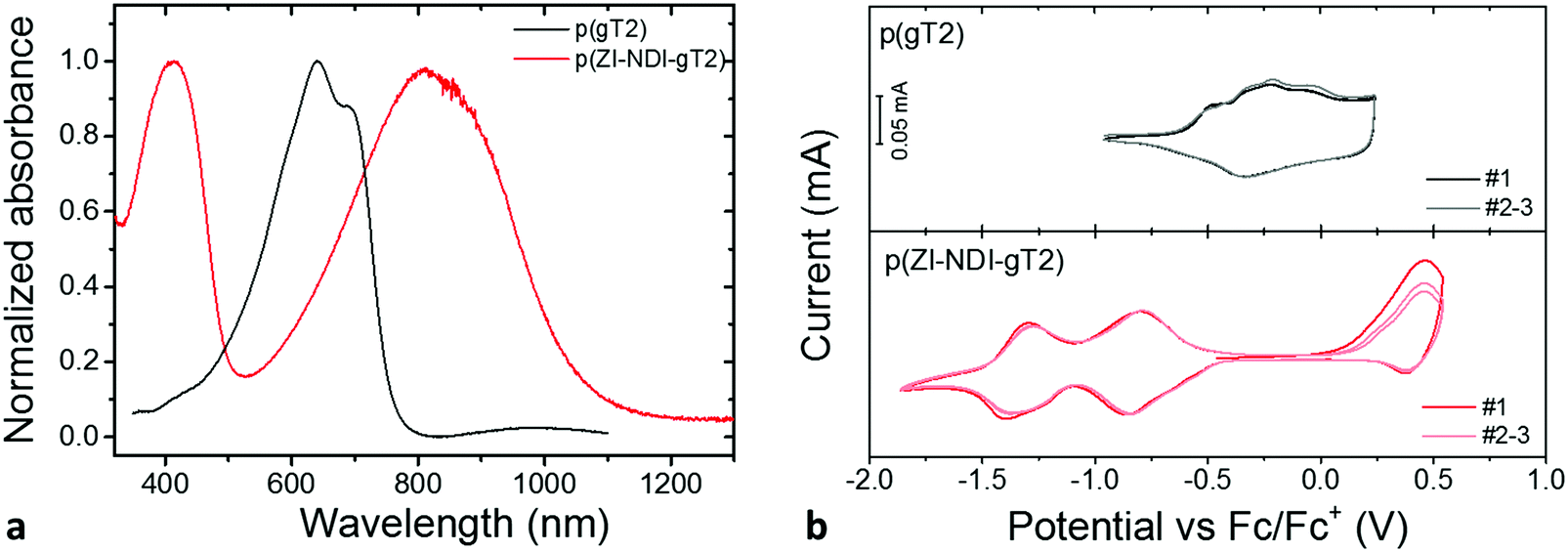 | ||
| Fig. 2 (a) Thin film UV-Vis spectra of a p(gT2) film on fluorine doped tin oxide (FTO) glass and of a p(ZI-NDI-gT2) film on a glass substrate. A potential of −0.3 V vs. Ag/AgCl was applied to the p-type polymer prior to recording of the UV-Vis spectrum (three-electrode setup) (p(gT2) can become oxidized in ambient conditions). (b) Thin film CV measurements of p(gT2) and p(ZI-NDI-gT2) on FTO substrates were recorded by using a degassed 0.1 M NBu4PF6 acetonitrile solution as supporting electrolyte (100 mV s−1, three cycles (#1–3) are presented where the first cycle (#1) is highlighted). | ||
The n-type polymer p(ZI-NDI-gT2) was synthesized following a post-functionalization route as shown in Fig. 3a where the precursor copolymer p((DMA)-NDI-gT2) with dimethylamino groups was prepared by Stille polymerization. The dimethylamino groups of p((DMA)-NDI-gT2) were first converted into ammonium bromides featuring an ester group at the end of the side chain. 1H nuclear magnetic resonance (NMR) spectroscopy and 2D correlation spectroscopy (COSY) were carried out to monitor the formation of the ammonium bromide (Fig. S16, ESI†). In the next step, the ester was cleaved to form the ammonium-carboxylate zwitterionic copolymer p(ZI NDI-gT2). Finally, the polymer was purified by dialysis in deionized water to remove water soluble side products. 1H NMR spectra (Fig. 3b) were recorded to monitor the ester cleavage, showing the disappearance of the signals corresponding to the proton of the ethyl group of the ester (4.1 ppm and 1.2 ppm) in accordance with reactions reported in the literature.28 This shows that the here presented post-functionalization approach can be a viable route to attach functional groups at NDI copolymers. The UV-Vis spectrum of p(ZI-NDI-gT2) is shown in Fig. 2a where the copolymer has two absorption peaks and an absorption onset of 1049 nm. The IP of p(ZI-NDI-gT2) was measured to be 5.15 eV (5.20 eV by CV, Fig. 2b). The CV measurement in acetonitrile based electrolyte shows redox features with high reversibility for the reduction of the p(ZI-NDI-gT2) polymer film, while the oxidation peak shows lower reversibility and a change in magnitude upon cycling, which might be due to structural changes in the film. For the remainder of this work we focus on the properties of the n-type polymer at negative potentials. The molecular weight distribution of p(ZI-NDI-gT2) was measured by GPC in DMF (Mn = 24 kDa and Mw = 53 kDa), which most likely overestimates the molecular weight distribution of the polymer since only oligomers up to 6 repeat units could be detected for the precursor copolymer p((DMA)-NDI-gT2) (see Fig. S13, ESI†). Unfortunately, MALDI-ToF measurements of p(ZI-NDI-gT2) were inconclusive. Polymer p(ZI-NDI-gT2) is soluble in polar organic solvents such as dimethyl sulfoxide (DMSO) and methanol (MeOH). Note that the substitution of the anion from carboxylate to sulfonate resulted in the formation of a water soluble polymer which could therefore not be tested as an electrode material in aqueous electrolytes. The n-type polymer bearing heptakis-ethylene glycol side chains on the NDI unit (p(g7-NDI-gT2)) was synthesized by Stille coupling and the properties of the polymers are reported in Section S5 of the ESI.†
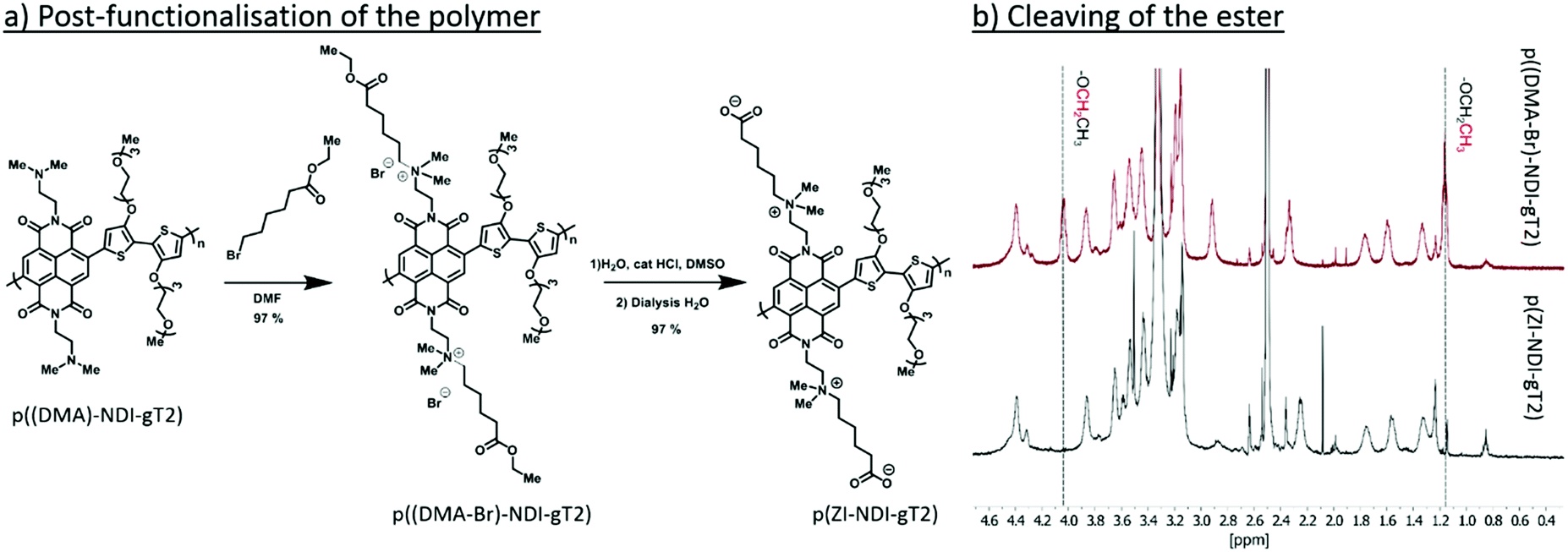 | ||
| Fig. 3 (a) Post-functionalization of p((DMA)-NDI-gT2) to form p(ZI-NDI-gT2) and (b) 1H NMR spectra of p((DMA-Br)-NDI-gT2) and p(ZI-NDI-gT2), highlighting (vertical lines at 4.04 ppm and 1.16 ppm) the protons corresponding to the ester of p((DMA-Br)-NDI-gT2) and their disappearance after cleavage. | ||
We now consider the electrochemical and spectroelectrochemical characterization of thin films of the polymers described above in aqueous electrolytes. Fig. 4a and b show cyclic voltammetry measurements of thin films of both p-type (p(gT2)) and n-type (p(ZI-NDI-gT2)) polymers performed in 0.1 M NaCl aqueous solution in a three electrode cell (more details on the experimental conditions are described in Section S8 of the ESI†). Similarly to the charging in organic electrolytes, both polymers show highly reversible redox features with stable peak positions after the first cycle. The polymer p(gT2) shows two reversible oxidation peaks at relatively low potentials, consistent with previous reports on electrochemistry of polythiophene based polymer films.27 Remarkably, the reduction of p(ZI-NDI-gT2) in the aqueous electrolyte also shows two distinct reduction peaks. This suggests that the NDI unit can be reversibly charged with two electrons in water, in agreement with findings in the literature.15,29 Continuous cycling of polymer films at 25 mV s−1 showed that for p(gT2) and p(ZI-NDI-gT2), 98% and 63% of the initial (2nd scan) capacity is retained after 500 cycles in 0.1 M NaCl aqueous solution (Fig. S37, ESI†).
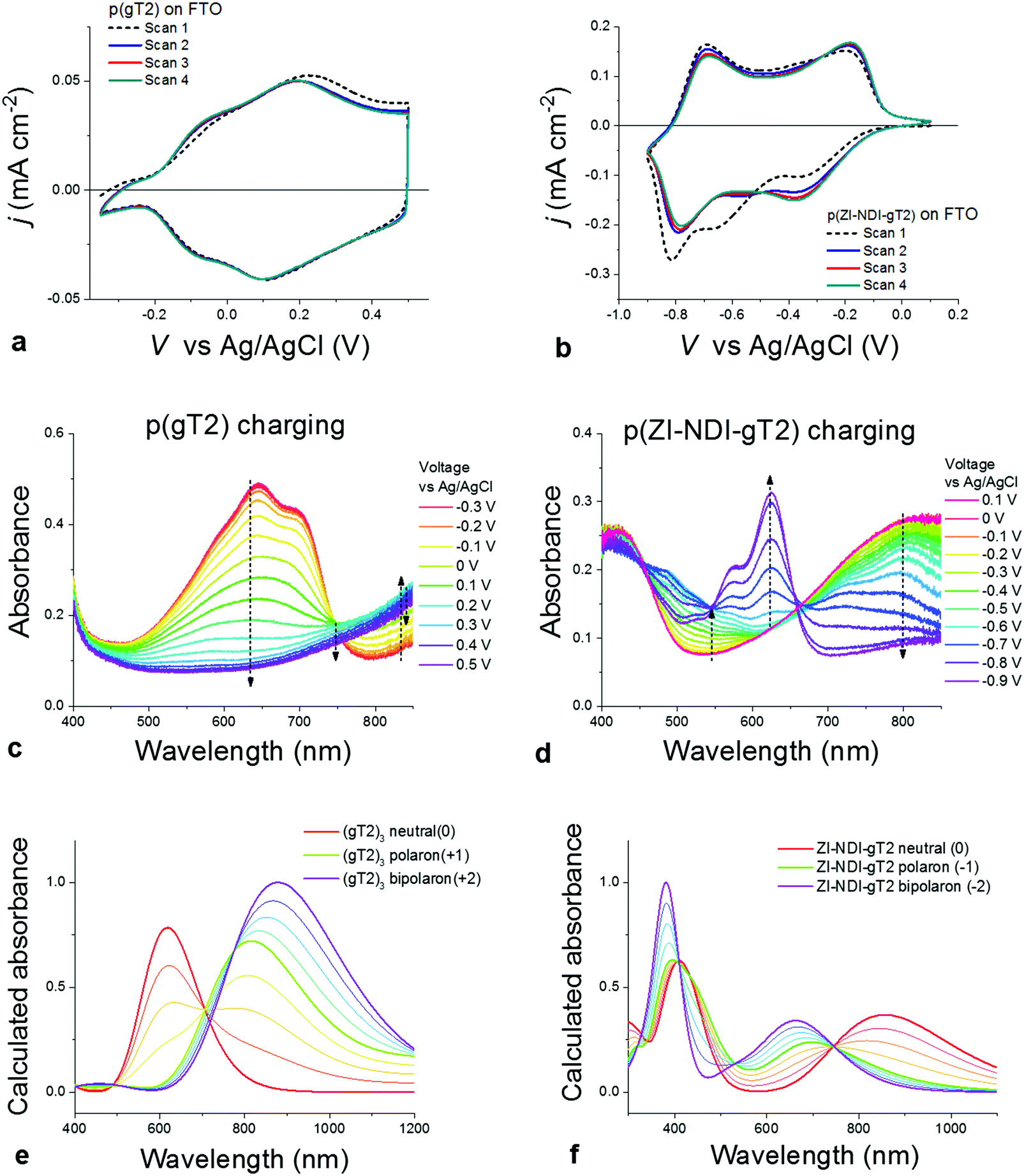 | ||
| Fig. 4 Characterization of p(gT2) (left) and p(ZI-NDI-gT2) (right). CV measurements of (a) a 43 nm p(gT2) film and (b) a 70 nm p(ZI-NDI-gT2) film using scan rate of 50 mV s−1 (cycle #1–4). To avoid side reactions with oxygen, the electrolyte for the n-type polymer was degassed with argon for 15 min prior to performing the measurement. UV vis absorbance spectra of (c) p(gT2) and (d) p(ZI-NDI-gT2) for the charging of the polymer films during the first CV scan shown in (a) and (b). Normalised absorbance spectra, calculated with TD-DFT for (e) the trimer (gT2)3 and (f) the monomer ZI-NDI-gT2 in the neutral and charged (polaron and bipolaron) states (bold lines) in water. Linear combinations of these three spectra were used to plot spectra of intermediate states (light lines). These correspond to a weighted sum of neutral and polaron states’ absorbance, and of polaron and bipolaron states’ absorbance. The weighting factors are x and (1 − x) (x = 0.25, 0.5, 0.75). | ||
Since conjugated polymers show a distinct color change between their neutral and charged state, we used spectroelectrochemical measurements to monitor the optical absorbance of the films during the CV measurements shown in Fig. 4a and b. Fig. 4c shows that the main absorption feature of p(gT2) in the visible region (peak at 645 nm) is completely quenched upon oxidation of the polymer by applying a potential of up to 0.2 V vs. Ag/AgCl and that a new absorption band appears in the NIR. In agreement with reported results for other conjugated polymers,30 we attribute the change of the polymer neutral state absorption to the formation of positive polarons on the polymer backbone. Further oxidation (V > 0.2 V vs. Ag/AgCl) reduces the NIR absorption suggesting that further conversion occurs, which we ascribe to bipolaron formation.27 Calculated spectra of the trimer (gT2)3 in its neutral, singly and doubly charged state in water using time-dependent density functional theory (TD-DFT) shown in Fig. 4e (further details are presented in Section S9 of the ESI†) support our assignment of the spectral changes to polaron and bipolaron formation, as do prior reports on polythiophene spectroelectrochemistry.23,27,31 Importantly, the spectral evolution suggests complete conversion of neutral polymers into the charged state, indicating that the full volume of the electrode undergoes charging. Scanning to more positive potentials shows that higher levels of charging can be achieved, although this compromises the coulombic efficiency and stability of the electrode (see Section S10 of the ESI†). The results for the n-type polymer p(ZI-NDI-gT2) are presented in Fig. 4d and indicate reversible formation of an electron polaron at a potential of −0.4 V and bipolaron between −0.4 V and −0.75 V vs. Ag/AgCl. Our assignment of the spectral features to polaron and bipolaron formation are again supported by calculated absorbance spectra of neutral and charged monomers in water (Fig. 4f). From the comparison of Fig. 4c and d, and the respective film thicknesses, it is clear that the p(gT2) polymer has a higher peak absorption coefficient than the p(ZI-NDI-gT2) polymer. This is consistent with the charge transfer character of the low energy transition in the donor–acceptor n-type polymer as shown by our calculations (see Section S9 of the ESI†). The capacity of the redox-active polymers in aqueous electrolytes was measured to be 25 mA h cm−3 and 37 mA h cm−3 for p(gT2) and p(ZI-NDI-gT2), respectively (see Sections S10 and S11 of the ESI†). Measurements of gravimetric capacities are also shown in Fig. S38 (ESI†). The values reported for the p- and n-type polymers are respectively 21.3 mA h g−1 and 25.9 mA h g−1, which suggest that the density of these two polymers is in the order of 1.2–1.4 g cm−3 (accuracy of these values is discussed in Section S10.4 of the ESI†). We note that for these estimates we are using the volume (mass) of the dry films and neglect volume expansions (mass increase) through swelling in the presence of the aqueous electrolyte.
The rate capabilities of the electrode materials were studied in NaCl aqueous electrolytes by performing galvanostatic charge/discharge measurements of films of between 400 nm and several microns thickness. The results are summarized in Fig. 5. For these measurements, we used gold coated glass substrates and a 5 M NaCl aqueous solution to minimize the series resistance of the cell. The galvanostatic charge–discharge profiles during charging of 400 nm thick p(gT2) and p(ZI-NDI-gT2) films are shown in Fig. 5a. Polymer p(gT2) can be charged reversibly to 0.5 V vs. Ag/AgCl and its specific capacity drops by less than 15% when the current density is increased from 3.6 A cm−3 to 240 A cm−3. For p(ZI-NDI-gT2), the specific capacity drops by 37% when the current density is increased from 2.1 A cm−3 to 139 A cm−1, suggesting lower rate capabilities than the p-type polymer considered here. The p(ZI-NDI-gT2) polymer does not show further charging at more negative voltages, after the formation of the bipolaron at −0.85 V vs. Ag/AgCl, which is in agreement with previous reports on NDI-T2 copolymers.15
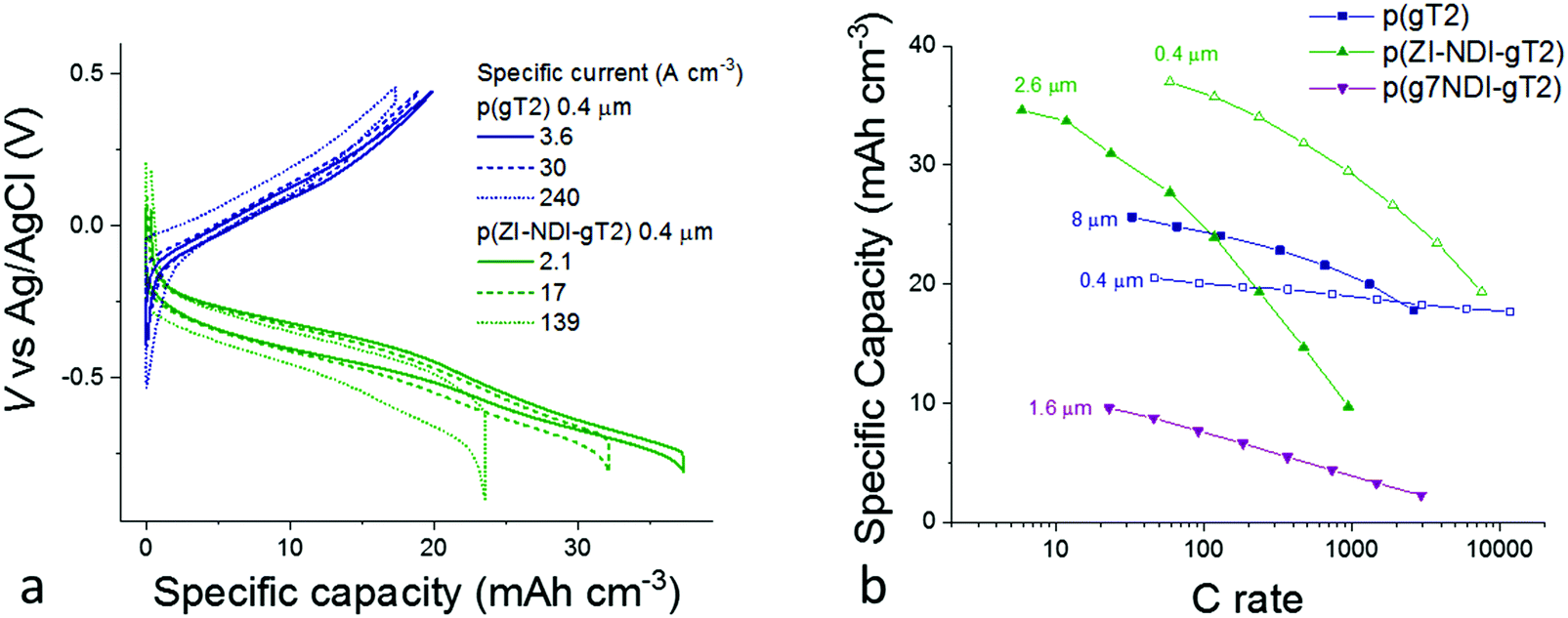 | ||
| Fig. 5 (a) Galvanostatic charge–discharge profiles of p(gT2) (blue curves) and p(ZI-NDI-gT2) (green curves) at different specific currents and (b) specific capacity as a function of C rate of the p-type and n-type polymers on gold coated glass substrates with a degassed 5 M NaCl aqueous solution as the supporting electrolyte. For both graphs, the datasets correspond to the second measurement performed at each specific current (C-rate) condition. The data in (b) display the charge obtained during the discharging of the electrode. Additional data including measurements on the films at lower electrolyte concentration are reported in Fig. S40–S44 (ESI†). | ||
Fig. 5b presents the rate capabilities of thick electrode films of p(gT2) and p(ZI-NDI-gT2) as well as showing the influence on rate capability of replacing the zwitterionic side chains on p(ZI-NDI-gT2) with glycol chains (p(g7NDI-gT2)) polymer. For p(gT2), we observe that the specific capacity for a 8 μm thick film drops by less than 20% when increasing the C rate from 30 to 1000 C (specific capacity per unit area of 0.02 mA h cm−2). This remarkable finding shows that, for p(gT2), ionic transport occurs on the second timescale even for thicknesses on the order of 10 μm. Similarly fast kinetics for solution processed polymer films in aqueous electrolytes were previously reported only for thin films (∼100 nm)32,33 or microns thick p-type polymer films processed through additional acid/base treatments.34,35
Fast charging of p(gT2) can be related to efficient hole and ionic transport properties in the film. Values of hole mobility up to 0.95 cm2 V−1 s−1 were observed in OECT structures for copolymers based on dialkoxybithiophenes such as p(g2T-TT) and suggest that ion transport is the rate limiting factor for the charging/discharging of these electrodes.3,36 It has been shown that ion transport in conjugated polymers depends on the size and type of the anion, where the hydration sphere of the ion is an important factor as reported for P3HT16 and p(g2T-TT).37 Additionally, the concentration of the salt dissolved in the electrolyte plays an important role in the swelling behavior of the polymer film.38 The n-type polymer p(ZI-NDI-gT2) shows higher specific capacity than p(gT2) at low charging rates, however it also shows lower rate capabilities as shown in Fig. 5a. We note that spectroelectrochemistry performed in transmission mode, as shown in Fig. 4, could be used to monitor the drop in capacity shown at fast rates in Fig. 5b. However the technique cannot be applied when considering several hundreds of nanometers thick films, due to the large optical densities of such samples. Interestingly, the n-type polymer with zwitterionic side chains on the NDI repeat unit (p(ZI-NDI-gT2)) shows a specific capacity more than three times higher than that of the same n-type backbone with linear glycol side chains (p(g7NDI-gT2)). For polymer p(g7NDI-gT2), we observe an upper limit to reversible charging when scanning to potentials beyond the first reduction peak (Fig. S36 and S37, ESI†). We hypothesize that this observation is due to stronger interaction of the glycol side chains on the NDI unit with alkali-metal ions18 and water molecules4 compared to the ZI side chain. Such interactions might also induce a more pronounced uptake of water in the film and induce the observed faster capacity fading of thin films of this polymer during continuous cycling. In particular, during charging of the polymer, additional water molecules migrate into the structure as part of the alkali metal ions’ hydration shell. Water molecules can then also interact with the glycol side chains via hydrogen bonding. As a result, more water molecules are likely to remain in the polymer structure after discharging of the polymer, and could underlie the observed change in electrochemical response of the film. This interpretation emphasizes the importance of ensuring structural stability of the polymer electrode upon cycling. This can be achieved by designing chemical structures that allow sufficient swelling when exposed to water electrolytes while preserving the electrode's mechanical integrity.
The specific capacities measured for the three materials at slow C rate shown in Fig. 5b are consistent with the values measured for thin films (see Fig. S39, ESI†), suggesting that, even for >1 μm thick films, complete charging of the bulk occurs. We note that the specific capacity per unit area of the thickest film reported here (0.02 mA h cm−2) is low compared to other reported electrochemical energy storage electrodes using non-conjugated, side-chain-free organic materials processed with conductive additives.39 This is a direct consequence of using redox-inactive polar side chains to enable fast charging of the electrodes in neutral aqueous electrolytes. However, the observation that side chain engineering of the n-type polymer can be used to improve the electrode specific capacity suggests that side chain optimization could allow the theoretical specific capacity of these materials to be reached. Importantly, this is shown here for non-porous single-phase films. Practically relevant electrodes may require thicker (on the order of 100 μm) polymer films, also depending on the energy and power specifications of the application. Based on our results, 1–10 μm thick films of these polymers can already provide a competitive combination of capacity/rate performance.
Based on the promising results obtained from the galvanostatic measurements for individual electrodes, thin films of p(ZI-NDI-gT2) and p(gT2) with similar capacity were prepared and tested in a two-electrode electrochemical cell with a 0.1 M NaCl aqueous solution as the supporting electrolyte. The charging process of the two-electrode cell is illustrated in Fig. 6a. When a positive potential Vcell > 0 V is applied to the cathode with respect to the anode, Na+ ions (Cl-ions) from the electrolyte migrate into the bulk of the polymer to compensate for the negative (positive) electronic charge on the p-type (n-type) polymer backbone. The redox reactions of both polymers with corresponding potentials vs. Ag/AgCl are expressed in Fig. 6b. In the same figure we also illustrate the electrochemical window of neutral water in ambient conditions including electrochemical redox-reactions involving oxygen. Electrochemical side reactions, including reduction of oxygen and water splitting, can occur at relatively low potentials in aqueous electrolytes and it is therefore important to design the electrode materials accordingly to avoid them.
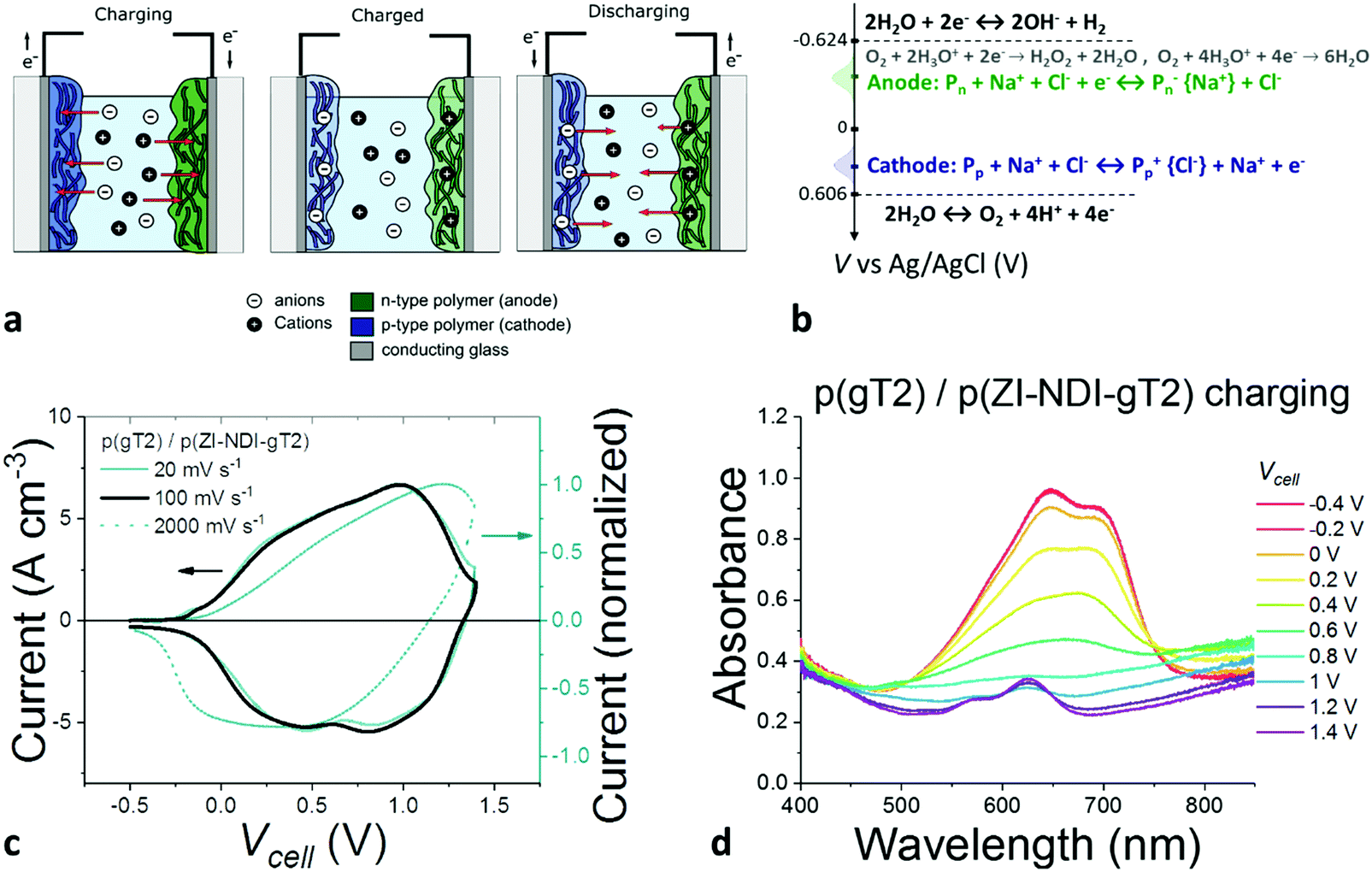 | ||
| Fig. 6 (a) Schematics of structure and charge distribution at different charge states of the electrochemical energy storage cell using films of the p- and the n-type polymers as the two electrodes, separated by an aqueous electrolyte. (b) Reactions at the cathode and at the anode and electrochemical window of water at neutral pH. (c and d) Characterization of a two electrode cell with structure FTO/p(gT2) (87 nm)/0.1 M NaCl:DIW/p(ZI-NDI-gT2) (70 nm)/FTO. The voltage (Vcell) is applied/measured at the cathode (p(gT2) electrode) with respect to the anode (p(ZI-NDI-gT2) electrode). (c) Cyclic voltammetry measurements performed at different scan rates. The left y-axis shows the current density normalized by the sum of the dry volumes of both p- and n-type films. (d) Evolution of the optical absorbance for the two-film assembly when charging the cell to 1.4 V at 100 mV s−1. | ||
The CV measurements of the cell are presented in Fig. 6c. The device is charged up to 1.4 V, which corresponds to the potential range used to characterize the p-type (0.5 V vs. Ag/AgCl) and n-type (−0.9 V vs. Ag/AgCl) polymers (see also Fig. S45, ESI†). The spectroelectrochemical response resembles the contribution of the individual electrode spectra and shows that electrochemical charging of the cell produces the bipolaron in both films as shown separately for the two films (Fig. 4c and d). Further characterization of the cell is shown in Section S13 of the ESI.†
We carried out charge-retention experiments in degassed 0.1 M NaCl aqueous solutions to investigate electrochemical side-reactions of the charged electrodes and the aqueous electrolyte. During this experiment, we observed a charge retention of 25% after 5000 seconds (see Fig. S47 and S48, ESI†), and discovered that oxygen in the aqueous electrolyte plays an important role, influencing the retention of electronic charges in the electrodes. When oxygen is present in the electrolyte, a larger charge is measured during charging than during discharging of the cell. This results in a lower coulombic efficiency and can compromise the stability of the polymer films. Electron transfer from either the neutral p-type p(gT2) or the reduced n-type polymer p(ZI-NDI-gT2) to oxygen is an expected side reaction.17 For this case, oxygen can either be converted into hydrogen peroxide (two electron process) or water (four electron process).17,40,41 Based on recent findings of efficient electrocatalytic production of hydrogen peroxide of electrodes made of organic polymers,40 we hypothesized that one pathway for the loss of the charges is an electron transfer from the reduced n-type polymers to oxygen dissolved in the electrolyte. To test this hypothesis, we charged the electrochemical cell in ambient conditions and used the horseradish peroxidase/3,3′,5,5′-tetramethylbenzidine (dye/enzyme) system42 to detect potential generated hydrogen peroxide. We observed hydrogen peroxide formation after charging the electrode in the presence of oxygen and the results are presented in Fig. S50 (ESI†). Additionally, we monitored hydrogen and oxygen evolution in degassed aqueous electrolyte to verify whether other faradaic electrochemical side reactions such as water splitting occur in our system. We observed no changes of oxygen or hydrogen concentrations during the charging of the cell (Fig. S49, ESI†). In order to study the origin of the loss of charge in the electrochemical cell in more detail, we use spectroelectrochemistry to monitor the changes of the absorption spectrum during the retention experiment. We observed the disappearance of the absorption features of the bipolaron on the n-type polymer when the cell is held at open circuit in the charged state. Based on this and the observation described above, we assign the charge retention limitation of the cell to the formation of hydrogen peroxide from the reaction of electrons on the n-type polymer and residues of oxygen left in the electrolyte (see reactions in Fig. 6b and data in Fig. S50, ESI†). As a result, we expect the p-type polymer to accumulate holes upon cycling of the cell, as confirmed by spectroelectrochemical measurements and by the drop in capacity illustrated in Fig. S51 (ESI†). This drop can be partially recovered by applying a negative bias (Fig. S52, ESI†). We finally note that the observed charge retention time is comparable to measurements carried out for other electrodes based on organic semiconductors in organic electrolytes and shows that oxygen free aqueous electrolytes can be an interesting electrolyte for the development of electrochemical energy storage devices.11
Conclusion
We demonstrated the development of solution processable, fast switchable p- and n-type electrode materials to operate in water, offering a route towards electrochemical energy storage in safe and sustainable electrolytes. Ethylene glycol based side chains attached to a p-type polymer backbone enabled reversible redox-reactions in aqueous electrolytes and fast charging of the polymer films on the second timescale for up to 8 μm thick films. For NDI-T2 n-type polymers we observed limited reversibility when using glycol side chains. We presented a chemical design strategy to prepare an n-type polymer with zwitterionic side chains via a post-functionalization reaction route and demonstrated improvement in both specific capacity and reversibility of the polymer electrode in aqueous electrolytes. When combined in a two-electrode cell, our p-type and n-type polymers showed reversible charging up to 1.4 V in a neutral aqueous sodium chloride salt solution. We used spectroelectrochemistry as an in operando measurement tool to monitor electrochemical charging/discharging of semitransparent electrodes and observed that both polymers formed reversible bipolaron states, approaching their theoretical capacities. Additionally, we used the technique to monitor the state of charge of the cell as well as to highlight the charge retention limitations of the n-type polymer. The study illustrates the potential and some of the limitations of using aqueous electrolytes as a safe and sustainable solution for energy storage applications. The materials reported here will also be applicable to novel devices for bio-sensing, electrochromic and memory applications, where control of mixed ionic-electronic conduction plays an important role.Conflicts of interest
DM, AG, PB, IM and JN have filed a patent application on the materials and results presented in this manuscript (PCT/GB2018/053329). All other authors declare no competing interests.Acknowledgements
We thank Peter R Haycock for the fruitful discussions about the NMR spectra. Funding: DM, PB and JN are grateful for funding from the EPSRC via the Supersolar Hub (grant EP/P02484X/1) and grant EP/P005543/1. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 742708) and EC H2020 Project SOLEDLIGHT (grant agreement No 643791), IMEC Synergy Grant SC2 (610115) and EPSRC EP/M005143/1. AG and IM are grateful for funding from EPSRC Project EP/G037515/1. AG and JN acknowledge funding form the EPSRC (EP/N509486/1) and from The Imperial College Faculty of Natural Sciences Strategic Research Fund. ER is grateful for funding from FRQNT.References
- J. Roncali, R. Garreau, D. Delabouglise, F. Garnier and M. Lemaireb, J. Chem. Soc., Chem. Commun., 1989, 679–681 RSC.
- I. F. Perepichka, M. Besbes, E. Levillain, M. Sallé and J. Roncali, Chem. Mater., 2002, 14, 449–457 CrossRef CAS.
- A. Giovannitti, D.-T. Sbircea, S. Inal, C. B. Nielsen, E. Bandiello, D. A. Hanifi, M. Sessolo, G. G. Malliaras, I. McCulloch and J. Rivnay, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12017–12022 CrossRef CAS PubMed.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef CAS PubMed.
- L. R. Savagian, A. M. Österholm, J. F. Ponder, K. J. Barth, J. Rivnay and J. R. Reynolds, Adv. Mater., 2018, 1–6 Search PubMed.
- S. Hellström, T. Cai, O. Inganäs and M. R. Andersson, Electrochim. Acta, 2011, 56, 3454–3459 CrossRef.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- A. M. Pappa, D. Ohayon, A. Giovannitti, I. P. Maria, A. Savva, I. Uguz, J. Rivnay, I. McCulloch, R. M. Owens and S. Inal, Sci. Adv., 2018, 4, eaat0911 CrossRef PubMed.
- D. J. MacInnes, M. A. Druy, P. J. Nigrey, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1981, 317–319 RSC.
- J. C. Carlberg and O. Inganäs, J. Electrochem. Soc., 1997, 144, L61–L64 CrossRef CAS.
- C. Karlsson, J. Nicholas, D. Evans, M. Forsyth, M. Strømme, M. Sjödin, P. C. Howlett and C. Pozo-Gonzalo, ChemSusChem, 2016, 9, 2112–2121 CrossRef CAS PubMed.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- P. Poizot, F. Dolhem and J. Gaubicher, Curr. Opin. Electrochem., 2018, 9, 70–80 CrossRef CAS.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed.
- L. Q. Flagg, R. Giridharagopal, J. Guo and D. S. Ginger, Chem. Mater., 2018, 30, 5380–5389 CrossRef CAS.
- D. M. de Leeuw, M. M. J. Simenon, A. R. Brown and R. E. F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- B. M. Savoie, M. A. Webb and T. F. Miller, J. Phys. Chem. Lett., 2017, 8, 641–646 CrossRef CAS PubMed.
- Y. Liu, Z. A. Page, T. P. Russell and T. Emrick, Angew. Chem., Int. Ed., 2015, 54, 11485–11489 CrossRef CAS PubMed.
- C. G. Tang, M. C. Y. Ang, K. K. Choo, V. Keerthi, J. K. Tan, M. N. Syafiqah, T. Kugler, J. H. Burroughes, R. Q. Png, L. L. Chua and P. K. H. Ho, Nature, 2016, 539, 536–540 CrossRef CAS PubMed.
- I. S. Chae, M. Koyano, K. Oyaizu and H. Nishide, J. Mater. Chem. A, 2013, 1, 1326–1333 RSC.
- C. K. Song, B. J. Eckstein, T. Lip, D. Tam, L. Trahey and T. J. Marks, ACS Appl. Mater. Interfaces, 2014, 6, 19347–19354 CrossRef CAS PubMed.
- F. S. Kim, X. Guo, M. D. Watson and S. A. Jenekhe, Adv. Mater., 2010, 22, 478–482 CrossRef CAS PubMed.
- A. Giovannitti, C. B. Nielsen, D.-T. Sbircea, S. Inal, M. Donahue, M. R. Niazi, D. A. Hanifi, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Nat. Commun., 2016, 7, 13066 CrossRef CAS PubMed.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS PubMed.
- B. W. D’Andrade, S. Datta, S. R. Forrest, P. Djurovich, E. Polikarpov and M. E. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef.
- C. Enengl, S. Enengl, S. Pluczyk and M. Havlicek, ChemPhysChem, 2016, 17, 3836–3844 CrossRef CAS PubMed.
- K. Koumoto, H. Ochiai and N. Sugimoto, Tetrahedron, 2008, 64, 168–174 CrossRef CAS.
- D. Trefz, A. Ruff, R. Tkachov, M. Wieland, M. Goll, A. Kiriy and S. Ludwigs, J. Phys. Chem. C, 2015, 119, 22760–22771 CrossRef CAS.
- J. C. Carlberg, Solid State Ionics, 1996, 85, 73–78 CrossRef.
- A. Giovannitti, K. J. Thorley, C. B. Nielsen, J. Li, M. J. Donahue, G. G. Malliaras, J. Rivnay and I. McCulloch, Adv. Funct. Mater., 2018, 28, 1706325 CrossRef.
- K. Koshika, N. Sano, K. Oyaizu and H. Nishide, Chem. Commun., 2009, 836–838 RSC.
- N. Sano, W. Tomita, S. Hara, C. M. Min, J. S. Lee, K. Oyaizu and H. Nishide, ACS Appl. Mater. Interfaces, 2013, 5, 1355–1361 CrossRef CAS PubMed.
- A. W. Lang, J. F. Ponder, A. M. Österholm, N. J. Kennard, R. H. Bulloch and J. R. Reynolds, J. Mater. Chem. A, 2017, 5, 23887–23897 RSC.
- J. F. Ponder, A. M. Österholm and J. R. Reynolds, Chem. Mater., 2017, 29, 4385–4392 CrossRef CAS.
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1–6 CrossRef CAS PubMed.
- C. Cendra, A. Giovannitti, A. Savva, V. Venkatraman, I. McCulloch, A. Salleo, S. Inal and J. Rivnay, Adv. Funct. Mater., 2018, 0, 1807034 Search PubMed.
- A. Savva, C. Cendra, A. Giugni, B. Torre, J. Surgailis, D. Ohayon, A. Giovannitti, I. McCulloch, E. Di Fabrizio, A. Salleo, J. Rivnay and S. Inal, Chem. Mater., 2019, 31, 927–937 CrossRef CAS.
- Y. Liang, Y. Jing, S. Gheytani, K. Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- E. Mitraka, M. Gryszel, M. Vagin, M. J. Jafari, A. Singh, M. Warczak, M. Mitrakas, M. Berggren, T. Ederth, I. Zozoulenko, X. Crispin and E. D. Głowacki, Adv. Sustainable Syst., 2018, 1800110 Search PubMed.
- R. Kerr, C. Pozo-Gonzalo, M. Forsyth and B. Winther-Jensen, ECS Electrochem. Lett., 2013, 2, F29–F31 CrossRef CAS.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee03518k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |