
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergy of the catalytic activation on Ni and the CeO2–TiO2/Ce2Ti2O7 stoichiometric redox cycle for dramatically enhanced solar fuel production†
Chongyan
Ruan‡
ab,
Zheng-Qing
Huang‡
 c,
Jian
Lin
a,
Lin
Li
a,
Xiaoyan
Liu
a,
Ming
Tian
a,
Chuande
Huang
a,
Chun-Ran
Chang
*c,
Jun
Li
d and
Xiaodong
Wang
*a
c,
Jian
Lin
a,
Lin
Li
a,
Xiaoyan
Liu
a,
Ming
Tian
a,
Chuande
Huang
a,
Chun-Ran
Chang
*c,
Jun
Li
d and
Xiaodong
Wang
*a
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xdwang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
cShaanxi Key Laboratory of Energy Chemical Process Intensification, School of Chemical Engineering and Technology, Xi’an Jiaotong University, Xi’an 710049, China. E-mail: changcr@mail.xjtu.edu.cn
dDepartment of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing, 100084, China
First published on 31st January 2019
Abstract
Solar thermochemical approaches to CO2 and H2O splitting have emerged as an attractive pathway to solar fuel production. However, efficiently producing solar fuel with high redox kinetics and yields at lower temperature remains a major challenge. In this study, Ni promoted ceria–titanium oxide (CeO2–TiO2) redox catalysts were developed for highly effective thermochemical CO2 and H2O splitting as well as partial oxidation of CH4 at 900 °C. Unprecedented CO and H2 production rates and productivities of about 10–140 and 5–50 times higher than the current state-of-the-art solar thermochemical carbon dioxide splitting and water splitting processes were achieved with simultaneous close to complete CH4 conversions and high selectivities towards syngas. The underlying mechanism for the exceptional reaction performance was investigated by combined experimental characterization and density functional theory (DFT) calculations. It is revealed that the metallic Ni and the Ni/oxide interface manifest catalytic activity for both CH4 activation and CO2 or H2O dissociation, whereas CeO2–TiO2 enhances the lattice oxygen transport via the CeO2–TiO2/Ce2Ti2O7 stoichiometric redox cycle for CH4 partial oxidation and the subsequent CO2 or H2O splitting promoted by catalytically active Ni. Such findings substantiate the significance of the synergy between the reactant activation by catalytic sites and the stoichiometric redox chemistry governing oxygen ion transport, paving the way for designing prospective materials for sustainable solar fuel production.
Broader contextSolar energy, with tremendous potential as an environmentally sound and sustainable energy source, dwarfs all the derivative sources by a wide margin. The challenge, however, is to take full advantage of the abundant and infinite solar energy and to convert it into readily utilisable and storable forms. Solar thermochemical CO2 and H2O splitting, tapping sunlight directly and storing solar energy in renewable fuel, are emerging technologies towards meeting this goal. Successful adoption of solar-to-fuel technologies is predicated upon identifying advanced materials with higher efficiency. The present work demonstrates the application of novel Ni promoted ceria–titanium oxide redox catalysts for solar thermochemical CO2 and H2O splitting coupled with CH4 partial oxidation, which exhibit considerably higher CO and H2 production rates and productivities than the conventional solar thermochemical CO2 and H2O splitting processes with ∼100% CH4 utilization. Supported by detailed experimental characterization and DFT calculations, a synergism between the catalytic activation on Ni and the CeO2–TiO2/Ce2Ti2O7 stoichiometric redox cycle is shown to be responsible for the high efficiency of CO2 and H2O splitting as well as CH4 conversion. These findings provide fundamental insights into the mechanisms underlying the remarkable reactivity and constitute a basis for engineering efficient materials for enhanced solar energy conversion. |
1. Introduction
Access to carbon-neutral, affordable and sustainable energy sources is widely recognized as the cornerstone of sustained economic growth and increasing prosperity of modern society.1,2 Solar-based routes hold great promise given sunlight's infinite abundance and accessibility, since the annual sunlight striking the earth (∼120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TW) is far exceeding the world energy consumption rate (estimated at ∼43 TW by 2100).2–5 Solar fuel production via two-step solar thermochemical carbon dioxide splitting (STCDS) and water splitting (STWS) is an attractive alternative to artificial photosynthesis, biomass production and conversion, and photovoltaic-driven electrolysis in particular as it can be potentially more energy efficient and require less land and water to implement.6,7 In the two-step redox cycle, the metal oxide is first reduced at low oxygen partial pressures and elevated temperatures (above 1400 °C) utilizing concentrated solar energy. Following thermal reduction, the oxygen deficient metal oxide is re-oxidized at lower temperatures (below 1100 °C) with CO2 and/or H2O, yielding CO and/or H2. Although extensive research efforts have been devoted to two-step STCDS and STWS processes,8–12 challenges associated with rather high operating temperatures,8,9 limited fuel yields restricted by the thermodynamics,10 and thermal shocks induced by temperature swing8 greatly hamper their practical implementation.
000 TW) is far exceeding the world energy consumption rate (estimated at ∼43 TW by 2100).2–5 Solar fuel production via two-step solar thermochemical carbon dioxide splitting (STCDS) and water splitting (STWS) is an attractive alternative to artificial photosynthesis, biomass production and conversion, and photovoltaic-driven electrolysis in particular as it can be potentially more energy efficient and require less land and water to implement.6,7 In the two-step redox cycle, the metal oxide is first reduced at low oxygen partial pressures and elevated temperatures (above 1400 °C) utilizing concentrated solar energy. Following thermal reduction, the oxygen deficient metal oxide is re-oxidized at lower temperatures (below 1100 °C) with CO2 and/or H2O, yielding CO and/or H2. Although extensive research efforts have been devoted to two-step STCDS and STWS processes,8–12 challenges associated with rather high operating temperatures,8,9 limited fuel yields restricted by the thermodynamics,10 and thermal shocks induced by temperature swing8 greatly hamper their practical implementation.
Alternatively, introducing reducing agents such as methane,13,14 syngas15 or hydrogen16–19 notably lowers the external oxygen partial pressure, thereby facilitating lattice oxygen extraction from metal oxides. Among them, methane (the main constituent of natural gas) is particularly attractive as an economically accessible primary energy source, albeit it has shown poor reactivity with metal oxides compared to syngas or hydrogen. Integrating partial oxidation of methane into the reduction step allows for operating the redox cycle isothermally at significantly lower temperatures (800–1100 °C)13,14,20,21 with higher fuel yields and a theoretically predicted solar-to-fuel efficiency greater than 45%,22–24 hence increasing the practicality of these processes. A schematic diagram of two-step STCDS and STWS processes coupled with methane driven reduction (MDR) is illustrated in Fig. 1. In the endothermic MDR step, the lattice oxygen of the redox metal oxide is well exploited in partial oxidation of CH4, generating syngas with a suitable H2:CO ratio for downstream methanol or Fischer–Tropsch synthesis. In the subsequent exothermic oxidation step, the lattice oxygen abstracted by CH4 is replenished by CO2 and/or H2O splitting, producing concentrated CO and/or H2. The overall reaction is highly endothermic, thus permitting efficient storage of solar energy in chemical form (CO, H2).22
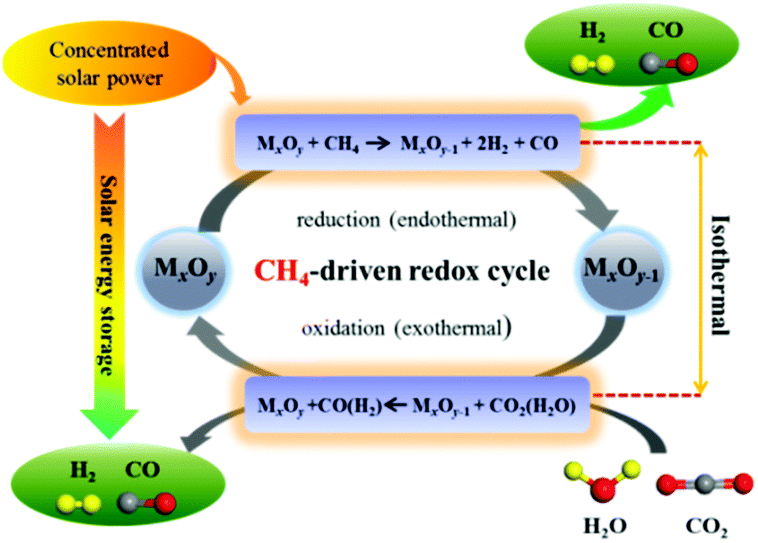 | ||
| Fig. 1 Schematic of two-step solar thermochemical carbon dioxide splitting and water splitting processes coupled with methane driven reduction (MDR-STCDS, MDR-STWS). | ||
The metal oxides function as the redox intermediates to store and deliver oxygen ions in between the MDR and the following CO2 (H2O) splitting step. Therefore, the thermodynamic and kinetic properties of the metal oxides dictate the techno-economic feasibility of two-step MDR-STCDS and MDR-STWS technologies. Among the various redox metal oxides developed, iron- and ceria-based oxides have attracted significant attention.13,14,21,23–26 Iron oxide represents a very promising candidate by virtue of its earth abundance, low cost and minimal environmental impact.26 Nevertheless, the poor reactivity with CH4, low selectivity toward syngas, slow re-oxidation kinetics with CO2 and H2O and the susceptibility to sintering with successive redox cycles constitute significant drawbacks.25,26 Recent studies indicated that the activity, selectivity and stability of iron-based oxides can be significantly enhanced promoted by tailored supports, such as lanthanum strontium ferrite (LSF)13,14 and calcium manganese oxide (Ca0.5Mn0.5O).21 Unfortunately, the CO2 or H2O splitting kinetics was still far from satisfactory, which may render these processes less efficient.
In comparison to iron oxide, ceria is particularly attractive owing to its (i) rapid redox kinetics, (ii) excellent syngas selectivity, and (iii) robust structural and crystallographic stability.23,24,27–29 Nonetheless, the solar fuel yield is limited by the low ceria reduction extent (Ce4+ → Ce3+: 0.2–1.6 mol%) via the nonstoichiometric redox cycle (CeO2 ↔ CeO2−δ without phase transitions) and the inferior CH4 reactivity (CH4 conversion: 2–20%).23 This is primarily due to the high activation energy required for lattice oxygen removal and the low surface activity for CH4 activation. To address the aforementioned challenges, two potential strategies have been proposed. The first one is through doping or lattice site substitution (CexZr1−xO2, CexFe1−xO2, CexHf1−xO2, etc.), in an attempt to introduce crystallographic defects that facilitate oxygen ion diffusion.30–32 However, such metal doping approaches have achieved limited success in enhancing the ceria reduction extent and CH4 reactivity based on nonstoichiometric chemistries. For example, Ce0.8Zr0.2O2 solid solutions were evaluated for CH4 partial oxidation and H2O splitting at 800 °C, which exhibited restricted CH4 conversions of 36–55% with Ce4+ reduction degrees ranging between 50% and 70%.33 The second strategy is to promote CeO2 with noble metal catalysts such as Pt and Pd.34 While this remarkably decreased the activation energy for CH4 partial oxidation at the gas–solid interface and enhanced the lattice oxygen consumption from the bulk, the cost-effectiveness and cycling stability of incorporating noble metals are debatable.
In the current work, we explore and demonstrate a new approach operating by stoichiometric redox chemistry. This novel chemistry enables a complete reduction of Ce4+ to Ce3+ with a reversible phase change (CeO2–TiO2 ↔ Ce2Ti2O7), dramatically different from current metal oxide cycles utilizing oxygen non-stoichiometry in ceria. Note that stoichiometric redox chemistry has recently drawn significant attention in enhancing solar fuel production for two-step STCDS and STWS processes,10,12,35 and in enhancing the robustness of catalysts via the unique phase transition for sorption enhanced steam reforming of bio-glycerol,36 yet it is still rarely exploited in two-step MDR-STCDS and MDR-STWS processes. Furthermore, non-noble metal Ni was introduced to accelerated CH4 partial oxidation, as it has been proposed in heterogeneous catalysis that metallic nickel has sufficient catalytic activity for CH4 activation by lowering the dissociation barrier of C–H bond cleavage.37,38 Encouragingly, the Ni promoted ceria–titanium oxide nanocomposite (CeO2–TiO2) developed here exhibited exceptional efficacy for CO2 and H2O splitting as well as partial oxidation of CH4. One to two orders of magnitude higher CO/H2 production rates and productivities were achieved as compared to the state-of-the-art STCDS/STWS and MDR-STCDS/MDR-STWS processes regarding CO2 and H2O splitting, and nearly complete CH4 conversions with excellent syngas selectivities were achieved in the MDR step. By combining detailed experimental characterization of the physicochemical properties during the redox cycles with density functional theory (DFT) calculations, a synergistic effect between reduced Ni species and ceria–titanium oxide is suggested to be responsible for the superior CH4 reactivity and CO2 and H2O splitting performance. Specifically, the metallic Ni and Ni/oxide interface act as active sites to lower the reaction barrier for CH4 activation during reduction and accelerate CO2 and H2O dissociation kinetics during oxidation. On the other hand, the CeO2–TiO2 oxide solid compound serves as the reactive intermediates to transport active lattice oxygen for CH4 partial oxidation and the subsequent CO2 and H2O splitting via the CeO2–TiO2/Ce2Ti2O7 stoichiometric chemistry with a complete Ce4+ ↔ Ce3+ redox cycle readily accessible. The lower formation energies of oxygen vacancies on Ti-doped CeO2 indicate that the incorporation of TiO2 into CeO2 weakens the Ce–O bonds and thus enhances the lattice oxygen transport. The combination of experimental investigation and theoretical calculations allows for a step forward in the understanding of the structure–activity relationship in designing highly promising candidates for solar fuel production applications.
2. Experimental section
2.1 Catalyst synthesis
The nickel-promoted ceria–titanium oxide catalysts were fabricated by a two-step synthesis approach. First, the ceria–titanium oxide solid compound (CeO2–TiO2) was synthesized, followed by deposition of nickel via a simple incipient wetness technique. The CeO2–TiO2 solid compound was prepared via a co-precipitation method. Briefly, stoichiometric amounts of Ce(NO3)3·6H2O and (C4H9O)4Ti were dissolved in 200 mL deionized water and 200 mL ethanol, respectively. The molar ratio of Ce4+ to Ti4+ was 1:1. Subsequently, the transparent solution was mixed under continuous stirring. Afterwards, a precipitating agent (NH3·H2O, 25%) was added dropwise to the mixed solution to adjust the pH to 9, keeping it at 80 °C for 3 h with stirring. The resulting precipitate was aged for another 1 h, filtered and washed thoroughly with deionized water. The washed precipitate was dried in an oven at 110 °C overnight, and subsequently calcined under stagnant air at 900 °C for 4 h (a heating rate of 5 °C min−1). Finally, the calcined sample was ground into a fine powder using a mortar and pestle.
The supported nickel catalysts were synthesized by wet impregnation of the as-synthesized CeO2–TiO2 solid compound with an appropriate amount of Ni(NO3)2·6H2O dissolved in deionized water. The obtained slurry was then dried in a vacuum at room temperature to evaporate excess moisture. Finally, the impregnated catalysts were dried at 110 °C overnight and annealed at 900 °C in air for 8 hours (a heating rate of 5 °C min−1) to stabilize the structure properties. Catalysts with nominal Ni loadings of 5, 2, 1, and 0.5 wt% were prepared and are referred to as 5Ni/CeO2–TiO2, 2Ni/CeO2–TiO2, 1Ni/CeO2–TiO2, and 0.5Ni/CeO2–TiO2, respectively.
2.2 Reactivity investigation
Catalytic activity tests were carried out at atmospheric pressure in a differential quartz tube microreactor (10 mm i.d.) positioned in an infrared image furnace (VTH-E44, ULVAC-RIKO). Typically, 200 mg of the catalyst sample was sandwiched between two layers of quartz wool, and the temperature was measured with an alumina-shielded R-type thermocouple (±3.8 °C) in direct contact with the catalyst bed. The cyclic redox reactions were performed isothermally at 900 °C. Inlet gas flow rates were regulated by electronic mass flow controllers. In the MDR step, the catalysts were reduced using methane (1.5 mL min−1) diluted in argon (148.5 mL min−1) for 6 min. The following CO2 or H2O splitting reaction was initiated by introducing CO2 (99.999%) or water vapor balanced with Ar (H2O/Ar at yH2O = 4.2 mol%) into the reactor at a total flow rate of 700 mL min−1 for 10 min. The line for the steam to the reactor was heated to 150 °C to prevent H2O condensation. In between each half cycle, the reactor was purged with Ar (500 mL min−1) for 5–10 min to avoid poorly defined mixtures, thereby allowing clear determination of products from each segment. To initiate each experiment, several reduction–oxidation cycles were conducted until syngas production stabilized. Besides, preliminary experiments were also conducted to identify reduction durations to avoid coke formation. The outlet gas concentrations were constantly analyzed and recorded after condensation of H2O via a calibrated quadrupole mass spectrometer (MS, GAM200 InProcess Instruments), and the effective volumetric flow rates (Vi) of observed species were determined. The MS signals were calibrated before each experiment using standard calibration gases. CH4 was monitored at m/z 16, CO2 at m/z 44 and 28, CO again at m/z 28, H2 at m/z 2 and Ar at m/z 40. The fraction originating from CO2 was subtracted from the signal at m/z 28 before the CO quantification. The CH4 conversion (XCH4), CO selectivity (SCO) and H2/CO molar ratio (RH2/CO) during the reduction half-cycle were calculated using:

A carbon balance was performed to check the accuracy of the measurement and a maximum deviation of 5% was obtained for the reported experiments.
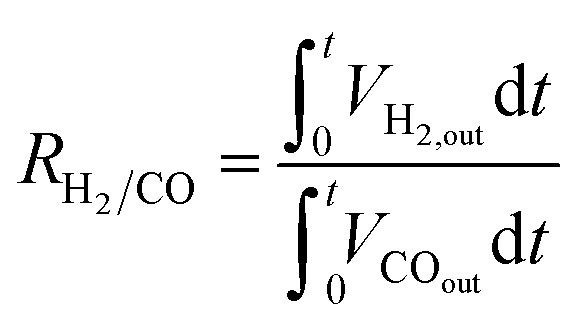

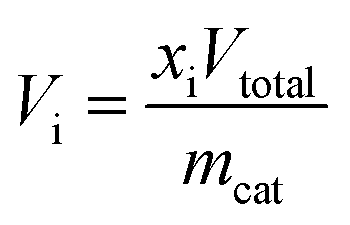 where Vi denotes the volumetric rates of CO or H2 produced per unit of mass of the catalyst; xi denotes the mole fraction of CO or H2 monitored by the mass spectrometer; Vtotal is the total volumetric flow rate regulated by the digital mass flow controller; and mcat is the mass of the catalyst. The corresponding CO or H2 yield was calculated by integrating the transient CO or H2 evolution rate with respect to time.
where Vi denotes the volumetric rates of CO or H2 produced per unit of mass of the catalyst; xi denotes the mole fraction of CO or H2 monitored by the mass spectrometer; Vtotal is the total volumetric flow rate regulated by the digital mass flow controller; and mcat is the mass of the catalyst. The corresponding CO or H2 yield was calculated by integrating the transient CO or H2 evolution rate with respect to time.
2.3 Catalyst characterization
The catalysts were characterized at various stages (after synthesis, after CH4 reduction, and after multicycle reactive tests) via thorough physical and chemical techniques. Powder X-ray diffraction (XRD) was performed to investigate the crystallographic phase evolution of the catalysts using a PANalytical diffractometer (40 kV, 40 mA), with Cu Kα radiation (λ = 1.5418 Å). The diffraction patterns were collected at ambient conditions between 2θ values of 20 and 70° with a step size of 0.02° and 30 s counting time per angle. The crystal phases were identified using the JCPDS database and the lattice spacing derived from the peak position was determined based on Bragg's diffraction law.X-ray photoelectron spectroscopy (XPS) measurements were conducted on an ESCALAB 250 photoelectron spectrometer (Thermo Fisher Scientific, Al Kα, hν = 1486.6 eV) with a chamber pressure of 3 × 10−8 Pa to probe the near-surface element states. All binding energies were calibrated with respect to the C 1s peak centered at 284.8 eV. Deconvolution of the peaks was processed with the XPSPEAK program using Shirley background subtraction and a mix of Gaussian–Lorentzian functions.
The redox behavior of the catalysts was assessed via temperature-programmed reduction using H2 (H2-TPR) instead of CH4 to avoid complications from possible carbon deposition. H2-TPR measurements were carried out using Micromeritics Auto Chem II 2920 apparatus equipped with a thermal conductivity detector (TCD). In a typical experiment, approximately 0.1 g of the catalyst sample was loaded in a U-shape quartz tube. Pretreatment was carried out in flowing Ar (30 mL min−1) at 350 °C for 30 min to eliminate water and gas adsorbed on the surface of the sample, followed by cooling down to 50 °C. Thereafter, TPR analysis was performed under 10% H2/Ar (30 mL min−1) up to 900 °C at a ramp rate of 10 °C min−1.
High-resolution scanning electron microscopy (HRSEM; JSM-7800F, 1 kV accelerating voltage) was applied to observe the microstructure and morphology of the catalyst samples before and after redox cycles. High-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy combined with energy-dispersive X-ray spectroscopy (STEM-EDX) were employed to identify the morphology, crystallinity, and elemental distribution and composition of samples after various process steps. These techniques were implemented on a JEOL JEM-2200F microscope operated at 200 kV and with a linear resolution of 0.10 nm. Prior to the measurement, the samples were crushed into fine powders and dispersed ultrasonically in ethanol. After dispersion, a few droplets of each sample were deposited on a carbon coated Cu grid and allowed to dry.
X-ray absorption near edge structure (XANES) experiments were carried out at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) with a ring current of 140–210 mA at 3.5 GeV. Samples (∼10 mg) diluted with BN (∼90 mg) were ground finely and pressed into self-supporting wafers for XANES measurements. Spectra were collected at the Ce LIII and Ti K edges in transmission mode with a Si(111) double-crystal monochromator. Reagent grade CeO2 and Ce(NO3)3·6H2O were used as Ce4+ and Ce3+ standards respectively, whereas TiO2 (Degussa-P25) and Ti2O3 were used as Ti4+ and Ti3+ standards respectively. Data processing and analysis were performed with the Athena software program in a standard curve-fitting procedure.
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements were performed on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation) to quantify the actual Ni loadings in the as-prepared catalysts.
2.4 Computational details
3. Results and discussion
3.1 Redox performance and stability
Since the feasibility of two-step MDR-STCDS and MDR-STWS processes is contingent upon satisfactory performance for CO2 and H2O splitting as well as CH4 conversion, consecutive redox cycles were carried out to investigate the activity and stability of Ni/CeO2–TiO2 catalysts. Fig. 2a and b show the transient CO and H2 formation rates during the CO2 and H2O splitting steps (indicated by the red curves) for 5Ni/CeO2–TiO2 after the isothermal MDR step, respectively. As can be seen, both reactions proceeded rapidly with extraordinary peak CO and H2 evolution rates followed by a quick decay, indicating very fast CO2 and H2O splitting kinetics. The peak CO/H2 production rates and CO/H2 productivities reported here exceed virtually all of those reported in the STCDS/STWS, HDR-STCDS (H2 driven reduction solar thermochemical CO2 splitting)/HDR-STWS (H2 driven reduction solar thermochemical H2O splitting) and MDR-STCDS/MDR-STWS processes, as summarized in Tables S4 and S5 (ESI†). Specifically, 5Ni/CeO2–TiO2 showed unprecedented CO/H2 production rates and total CO/H2 yields of about 10–140 and 5–50 times higher than the state-of-the-art ceria-based materials and perovskites in conventional STCDS and STWS processes performed at significantly higher temperatures (above 1250 °C). Moreover, 5Ni/CeO2–TiO2 also far outperformed most redox materials in the MDR-STCDS/MDR-STWS processes and even in HDR-STCDS/HDR-STWS redox schemes. Note that no detectable amount of CO2 or CO was observed during the H2O splitting half-cycle (Fig. 2b), indicating that almost no carbon deposition occurred after the MDR step. To further disclose the impact of Ni species on the CO2 and H2O splitting performance, CeO2–TiO2 in the absence of Ni was chemically reduced with 1% H2/Ar at 900 °C to ensure complete reduction. Subsequently, the completely reduced CeO2–TiO2 was subjected to CO2 and H2O splitting reactions at identical operating conditions as in the oxidation half cycle of two-step MDR-STCDS and MDR-STWS processes. As shown in Fig. 2a and b, it is evident that the reduced CeO2–TiO2 (indicated by the black curves) behaved significantly differently. The peak CO and H2 production rates were less than 19% and 21% of the reduced 5Ni/CeO2–TiO2, with CO and H2 evolution profiles being lower and broader, and it can be attributed to the considerably slower CO2 and H2O splitting kinetics over the reduced CeO2–TiO2. These results strongly suggest that the reduced Ni species substantially accelerated the catalytic splitting of CO2 and H2O, which will be discussed in detail in Section 3.3. In heterogeneous catalysis,53–55 Heine et al.55 also showed that CO2 molecules could be activated on Ni(111) and dissociated into CO and atomic oxygen in the methanation reaction. Carrasco et al.54 demonstrated that the strong electronic perturbations induced by nickel/ceria interactions could lead to an unexpectedly low H2O dissociation activation barrier and thus faster dissociation of H2O in the water-gas shift reaction as compared to bare CeO2. Consecutive CO2/H2O splitting cycles verified the stability of the 5Ni/CeO2–TiO2 redox catalyst (see Fig. 2c and d), which showed reproducible kinetic curves of CO and H2 evolution rates in the second and subsequent oxidation. The CO (Fig. 2c) or H2 (Fig. 2d) productivity, estimated by integrating the corresponding transient CO or H2 evolution rate with respect to time, stayed relatively constant over the course of 10 repetitive cycles. Interestingly, the CO or H2 productivity was comparable to the theoretically expected value (44.5 mL CO/H2 g−1 assuming a full reduction to Ce3+) stoichiometrically available for complete oxidation of Ce3+ to Ce4+. These results might imply that the 5Ni/CeO2–TiO2 redox catalyst was completely reoxidized after the CO2 or H2O splitting step. The slightly excess amount of CO or H2 above the theoretical maximum might result from the partial reoxidation of the reduced Ni species on the surface of 5Ni/CeO2–TiO2 with CO2 or H2O as no carbon was deposited.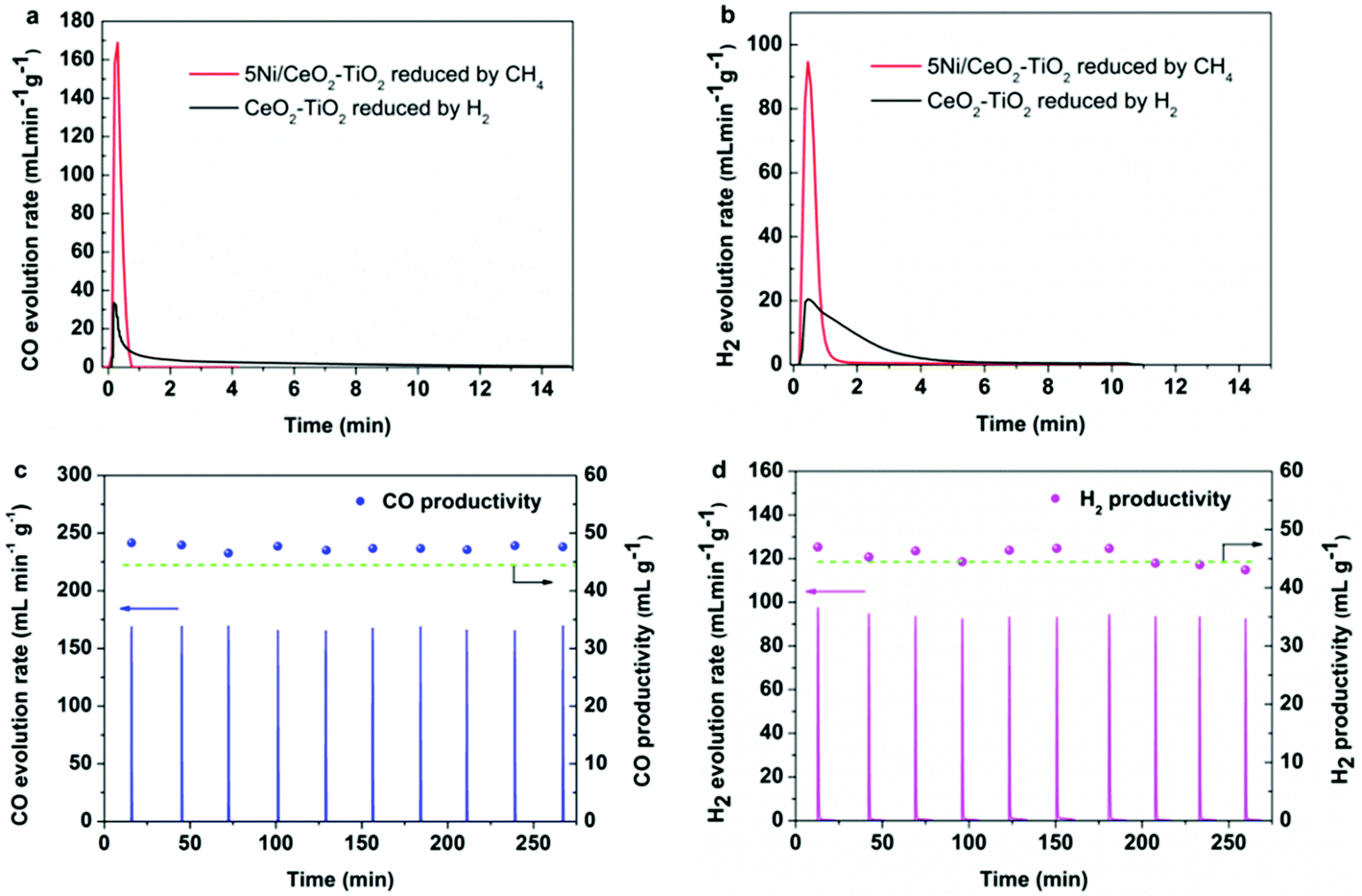 | ||
| Fig. 2 (a) Transient CO evolution rate during the CO2 splitting half cycle for 5Ni/CeO2–TiO2 of the isothermal MDR-STCDS process at 900 °C, the CO evolution rate for CeO2–TiO2 reduced by H2 is also shown. (b) Transient H2 evolution rate during the H2O splitting half cycle for 5Ni/CeO2–TiO2 of the isothermal MDR-STWS process at 900 °C, the H2 evolution rate for CeO2–TiO2 reduced by H2 is also shown. Transient CO and H2 evolution rates for 5Ni/CeO2–TiO2 during the CO2 and H2O splitting half cycles of the isothermal (c) MDR-STCDS and (d) MDR-STWS processes over 10 redox cycles. The dashed horizontal lines give the theoretically expected quantities of CO and H2, i.e., 44.5 mL CO/H2 g−1 for 5Ni/CeO2–TiO2 assuming a full reduction to Ce3+. | ||
In regard to the MDR step, representative CH4 isothermal reduction results are illustrated in Fig. 3a and b. For both processes, the as-prepared 5Ni/CeO2–TiO2 catalyst exhibited promising CH4 conversions (>99%) with negligible CH4 detected. As can be seen, the reaction between CH4 and 5Ni/CeO2–TiO2 followed a similar pattern characterized by two distinct regions. The initial stage (region I) was dominated by complete oxidation of CH4 to CO2, which was typically attributed to oxygen derived from NiO and/or loosely bonded surface oxygen56,57 on CeO2–TiO2. The second stage (region II) was dominated by partial oxidation of CH4 to CO and H2 by the strongly bonded bulk oxygen of CeO2–TiO2 that was sufficiently favored over the metallic nickel.58 The amount of oxygen converted to CO for 5Ni/CeO2–TiO2 reached 36.3 mL g−1 and 40.5 mL g−1 in MDR-STCDS and MDR-STWS processes, respectively, which was close to the theoretical removable oxygen (44.5 mL g−1) assuming a complete reduction from Ce4+ to Ce3+. Notably, the reaction of CeO2–TiO2 with CH4 in the absence of Ni shows drastically different results. As evidenced in Fig. S3a (ESI†), the CH4 concentration rapidly reached the nominal value during the reduction step in the MDR-STWS process with inferior CH4 conversion (5.9%), and the amount of oxygen converted to CO was only 1.0 mL g−1, indicating the bulk oxygen of CeO2–TiO2 remained largely unconsumed without Ni promotion. Additionally, the conversion of CH4 using CeO2–TiO2 samples with different Ni loadings (0, 0.5, 1, 2 and 5 wt%) is illustrated in Fig. S3b (ESI†). It is evident that CH4 conversion increased concurrently with increasing Ni content. The above observations reveal that the presence of Ni species is vital for catalyzing CH4 activation, thereby enhancing the bulk oxygen consumption originating from CeO2–TiO2 and hence the partial oxidation of CH4. The instantaneous gaseous product concentrations during the reduction of 5Ni/CeO2–TiO2 varied slightly among 10 isothermal MDR-STCDS and MDR-STWS cycles (Fig. S4a and b, ESI†), confirming that 5Ni/CeO2–TiO2 could be reduced with CH4 repeatedly with high reproducibility. The corresponding catalytic performance of 5Ni/CeO2–TiO2 during CH4 reduction is summarized in Fig. 3c and d, in terms of the CH4 conversion (CH4 Con), H2/CO ratio, and CO selectivity (CO Sele). Remarkably, essentially complete conversions of CH4 were achieved for both processes over the entire 10 redox cycles, with H2:CO ratios stabilized at ∼1.8 (Fig. 3c) and ∼1.9 (Fig. 3d), i.e., close to the typically desired ratio for downstream Fischer–Tropsch or methanol synthesis. The CO selectivity for 5Ni/CeO2–TiO2 in the isothermal MDR-STCDS and MDR-STWS processes amounted to ∼76 and ∼85%, respectively, which is most likely due to the different surface oxygen species as will be discussed later. To summarize, the extraordinary CO2 and H2O splitting performance and the superior CH4 partial oxidation activity render 5Ni/CeO2–TiO2 a highly promising redox catalyst in the MDR-STCDS and MDR-STWS processes.
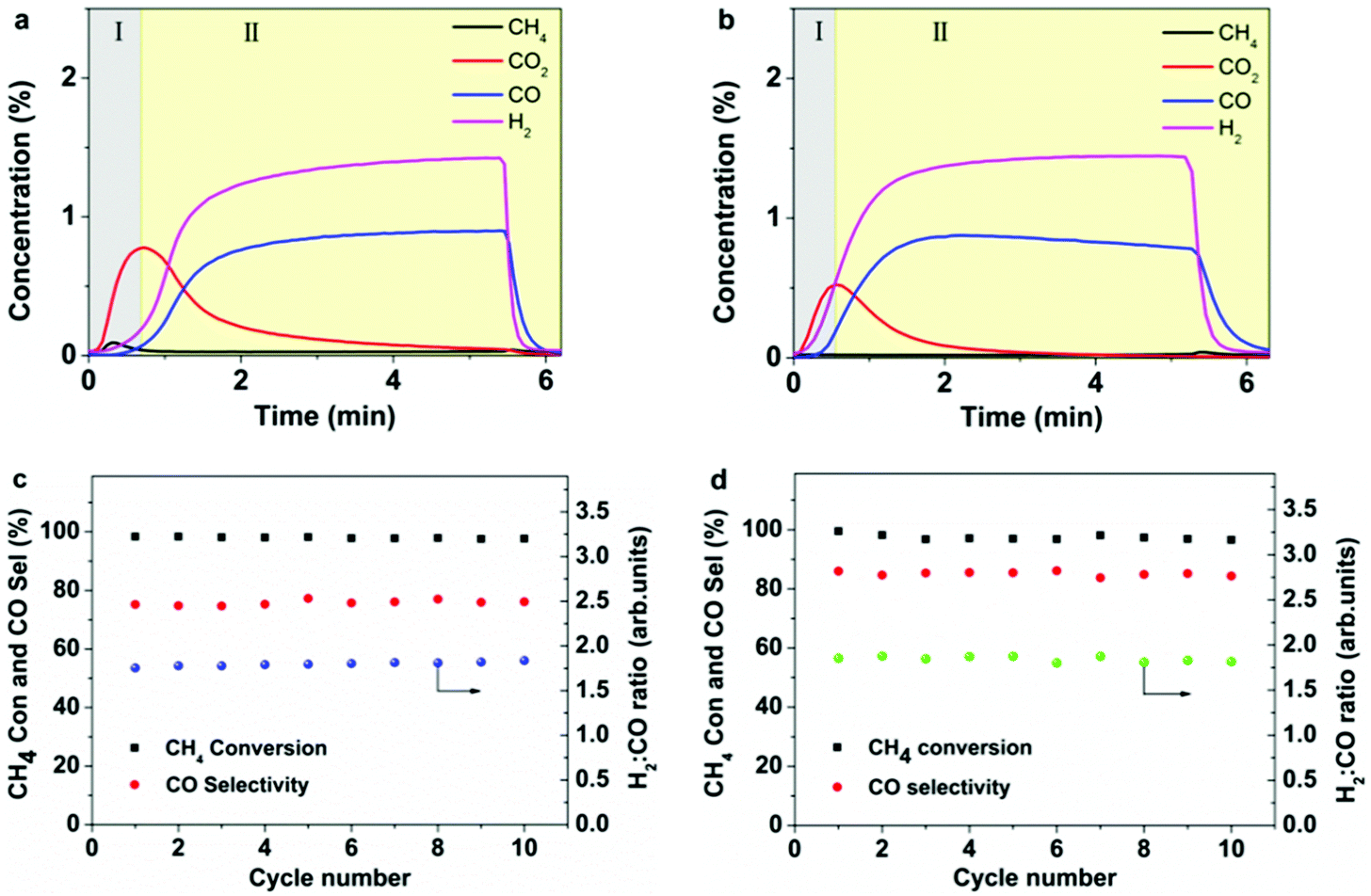 | ||
| Fig. 3 Temporal gaseous product concentrations for 5Ni/CeO2–TiO2 during the CH4 reduction half cycle of the isothermal (a) MDR-STCDS and (b) MDR-STWS processes at 900 °C. Average CH4 conversion, CO selectivity and H2/CO ratio for 5Ni/CeO2–TiO2 during the CH4 reduction half cycle of the isothermal (c) MDR-STCDS and (d) MDR-STWS processes over 10 redox cycles. | ||
3.2 Redox catalyst characterization
In order to elucidate the underlying reasons for the exceptional efficacy of the 5Ni/CeO2–TiO2 redox catalyst, various physicochemical characterization techniques complemented by detailed theoretical studies were applied to probe the redox chemistry and reaction pathways. Fig. 4 illustrates the XRD patterns of the 5Ni/CeO2–TiO2 redox catalyst at various stages (after synthesis, after the MDR step, and after multicycle reactive tests). As observed in Fig. 4a, the as synthesized sample exhibited distinct reflections characteristic of NiO, CeO2, and TiO2 (rutile, JCPDS 01-083-2242; brookite, JCPDS 01-076-1934), evidencing the solid state reactions between individual compounds remained negligible after the initial calcination process. Intriguingly, reflection peaks attributable to CeO2 and TiO2 completely disappeared for the reduced sample after the MDR step, accompanied by the emergence of new reflection peaks indexable to Ce2Ti2O7 pyrochlore (JCPDS 00-047-0667) with decreased crystallinity (Fig. 4b). Additional peaks attributed to metallic Ni (JCPDS 00-001-1272) were also observed at about 2θ = 46.5° and 67.9° (Fig. 4b) with a crystallite size of 17.4 ± 0.8 nm determined by the Scherrer equation, which contributed essentially to CH4 activation. On the contrary, no apparent structure change was identified for the CeO2–TiO2 sample in the absence of NiO, illustrating the negligible phase transformation of CeO2–TiO2 after the MDR step (Fig. S7, ESI†). Hence, it can be concluded that a stoichiometric reaction between CeO2 and TiO2 occurred for the 5Ni/CeO2–TiO2 catalyst after reducing with CH4, forming Ce2Ti2O7 pyrochlore promoted by the presence of Ni species. Meanwhile, catalytically active metallic Ni dispersed on Ce2Ti2O7 pyrochlore was obtained after the reduction treatment. Furthermore, regeneration of CeO2 and TiO2 was clearly indicated by the XRD spectra of 5Ni/CeO2–TiO2 catalysts after isothermal MDR-STCDS and MDR-STWS redox cycling, where the peaks corresponding to Ce2Ti2O7 pyrochlore totally vanished (Fig. 4c). Thus, it is evidenced that the transformation between Ce2Ti2O7 pyrochlore and CeO2 and TiO2 mixed oxides was entirely reversible during the redox cycles, substantiating the excellent recyclability of the 5Ni/CeO2–TiO2 redox catalyst. Additionally, weak NiO peaks as well as the reduced metallic Ni were identified after the final CO2 and H2O reoxidation half cycle (Fig. 4c), confirming the surface oxidation of the reduced Ni species despite the bulk oxidation not being thermodynamically favorable.59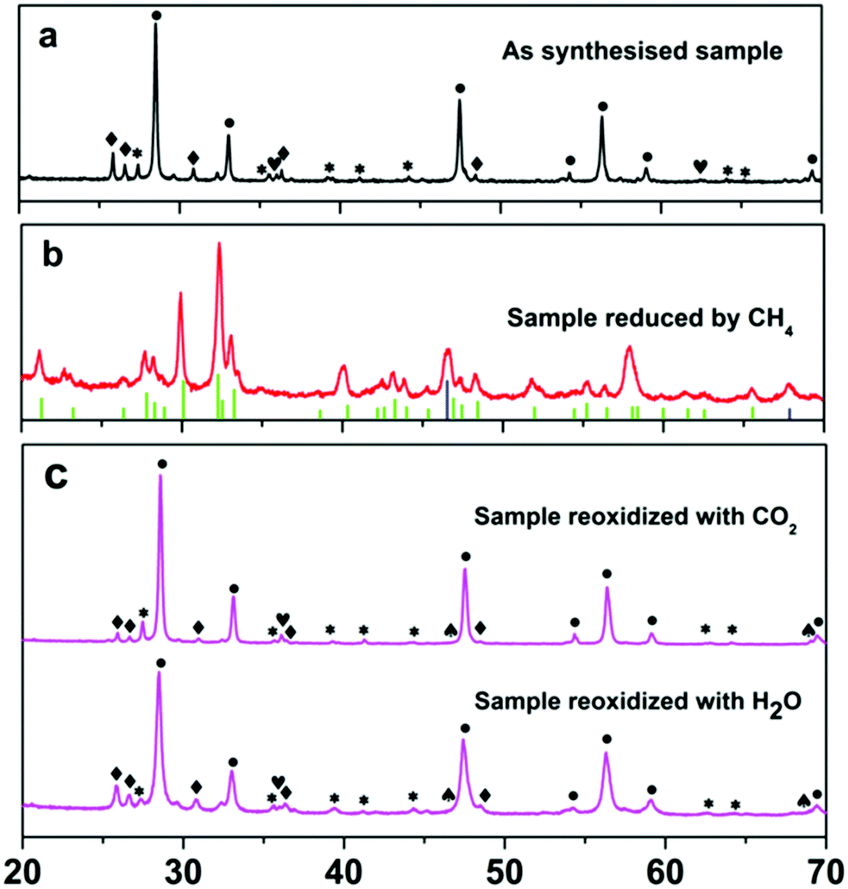 | ||
Fig. 4 X-ray powder diffraction patterns of (a) as synthesized 5Ni/CeO2–TiO2, (b) 5Ni/CeO2–TiO2 after the CH4 reduction half cycle and (c) 5Ni/CeO2–TiO2 after isothermal MDR-STCDS (up) and MDR-STWS (below) redox cycling at 900 °C. The following compounds were identified: (●) CeO2, (*) TiO2 (rutile), (✦) TiO2 (brookite), (♥) NiO, (♠) Ni, ( ) Ce2Ti2O7 (JCPDS 00-047-0667) and ( ) Ce2Ti2O7 (JCPDS 00-047-0667) and ( ) Ni (JCPDS 00-001-1272). ) Ni (JCPDS 00-001-1272). | ||
H2-TPR experiments were performed to investigate the redox properties of the as synthesized 5Ni/CeO2–TiO2 and CeO2–TiO2 samples, together with those of TiO2 and CeO2 as references, and the results are presented in Fig. 5. TiO2 exhibited no appreciable reduction in the temperature range of 100–900 °C. For CeO2 two weak reduction peaks were observed. The first peak located at about 480 °C was generally ascribed to the reduction of surface capping oxygen,60,61 and the second peak centered at about 800 °C was related to the removal of bulk lattice oxygen,60,61 which still proceeded significantly at above 900 °C, revealing that bulk lattice oxygen of CeO2 was extremely stable. Notably, in the case of CeO2–TiO2 mixed oxides, the H2-TPR profile featured a single broad peak ranging from 400 to 700 °C with intensity much greater than pure CeO2, indicative of relatively high lattice oxygen mobility. The enhanced reducibility can potentially be explained by the reduction of the energy barrier for oxygen anion migration via the stoichiometric reaction between CeO2 and TiO2 during reduction, resulting in Ce2Ti2O7 pyrochlore as indicated by the XRD pattern of CeO2–TiO2 after the H2-TPR measurement (see Fig. S8, ESI† for further details). The anion-deficient Ce2Ti2O7 pyrochlore may enable efficient oxygen anion transport and facilitate the accessibility of bulk lattice oxygen. These results correspond well with the earlier work of Luo et al.,62 whereby the reduction degree of CeO2–TiO2 mixed oxides was greatly enhanced owing to incorporation of TiO2 into the CeO2 lattice. It is noteworthy that the presence of NiO can further improve the oxygen anion diffusivity within CeO2–TiO2. This is validated by the fact that the reduction peak of 5Ni/CeO2–TiO2 shifted down to ∼350 °C, overlapping with the reduction band of NiO,63,64 while the H2-TPR profile was characterized by a narrower intense reduction peak at 300–400 °C, compared to CeO2–TiO2 without NiO. Such superior lattice oxygen mobility is highly beneficial for CH4 partial oxidation and the subsequent CO2 and H2O splitting reactions.
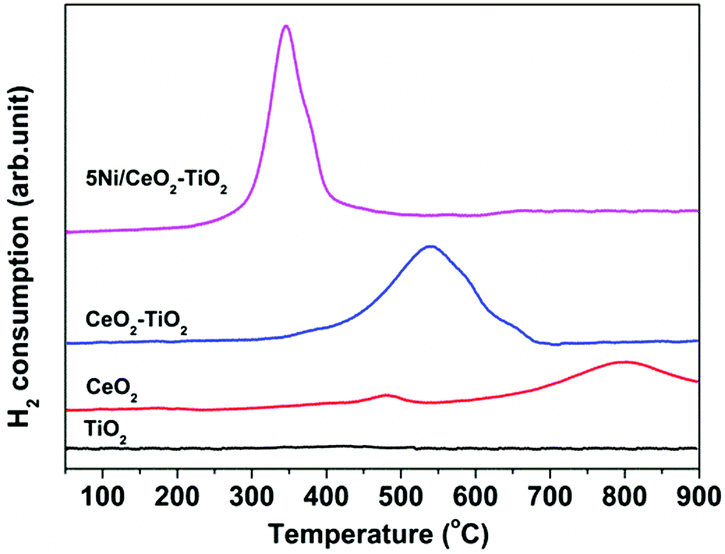 | ||
| Fig. 5 H2-TPR profiles of as synthesized 5Ni/CeO2–TiO2 and CeO2–TiO2. For comparison, the data for TiO2 and CeO2 are also shown. | ||
X-ray photoelectron spectroscopy (XPS) was employed to shed light on the evolution of the surface Ni and O species of the 5Ni/CeO2–TiO2 catalyst. The Ni 2p3/2 spectra of the catalyst at different stages are compared in Fig. 6a. The as synthesized catalyst showed a strong peak at 855.4 eV (red) along with a broad satellite peak at 861.5 eV (blue). These features could be assigned to the Ni2+ in NiO,65,66 evidencing that the surface Ni species were present as NiO. After the MDR step, a peak at 852.2 eV (yellow) associated with zero-valent metallic Ni0 was clearly identified,66,67 indicating that metallic nickel was evolved after CH4 isothermal reduction. In addition, a NiO contribution resulting from air exposure was observed due to the ex situ and surface sensitivity of XPS measurements as described elsewhere.66,67 Apparently, for 5Ni/CeO2–TiO2 catalysts after isothermal MDR-STCDS and MDR-STWS redox cycling, the contributions of metallic Ni0 decreased remarkably, pointing toward the reoxidation of the surface reduced Ni species by CO2 and H2O combined with the post-reaction air exposure. Furthermore, the O 1s spectra of the catalysts after isothermal MDR-STCDS and MDR-STWS redox cycling are displayed in Fig. 6b. Deconvolution of the O 1s band showed two distinct components. The predominant component at about 529.5 eV (pink) agrees well with the signature of lattice oxygen,55,65 while the minor components centered at 531.5 eV (26.3%) and 532.0 eV (20.4%) may originate from low coordination surface oxygen in hydroxyl and carbonate species of the catalysts after the MDR-STCDS and MDR-STWS cyclic experiments, respectively.55,68 The electrophilic surface oxygen species have been associated with deep oxidation of CH4 to CO2,68,69 and this is consistent with the fact that a higher CO selectivity was obtained on the 5Ni/CeO2–TiO2 catalyst in MDR-STWS redox cycling as the surface oxygen species were relatively fewer.
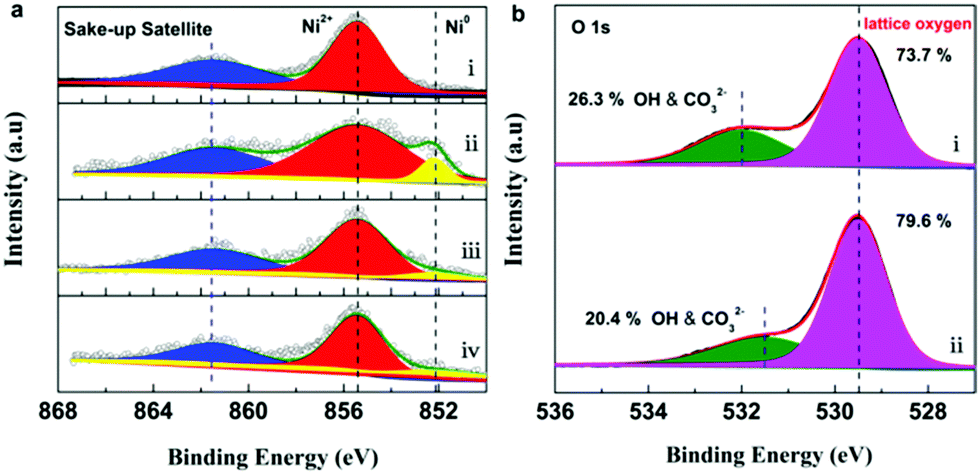 | ||
| Fig. 6 (a) Ni 2p3/2 XPS spectra of the 5Ni/CeO2–TiO2 sample: (i) as synthesized, (ii) after the CH4 reduction half cycle, (iii) after isothermal MDR-STCDS redox cycling at 900 °C and (iv) after isothermal MDR-STWS redox cycling at 900 °C. (b) O 1s XPS spectra of the 5Ni/CeO2–TiO2 sample: (i) after isothermal MDR-STCDS redox cycling at 900 °C, and (ii) after isothermal MDR-STWS redox cycling at 900 °C. | ||
To gain further insight into the redox chemistry during solar fuel production, the oxidation states of cerium and titanium were probed using XANES spectroscopy. Fig. 7 shows the normalized Ce LIII and Ti K edge XANES spectra of the 5Ni/CeO2–TiO2 redox catalyst after three different stages in isothermal MDR-STCDS and MDR-STWS redox cycling. For comparison, standard reference spectra of Ce4+ (CeO2), Ce3+ (Ce(NO3)3·6H2O), Ti4+ (TiO2), and Ti3+ (Ti2O3) are also included. The spectrum of Ce4+ is characterized by two absorption peaks at around 5732.2 eV and 5739.0 eV, whereas the spectrum of Ce3+ is dominant with one intense absorption peak at around 5727.9 eV.70,71 As observed in Fig. 7a, the Ce LIII-edge XANES spectrum of the as synthesized catalyst resembled that of Ce4+ in CeO2. Remarkably, the spectrum of the catalyst after the MDR step exhibited exclusively features characteristic of cerium in the completely reduced state (Ce3+), indicating the complete reduction of Ce4+ after reacting with CH4. Contrary to 5Ni/CeO2–TiO2, only minute changes are observed in the Ce LIII-edge spectra of CeO2–TiO2 after the MDR step (Fig. S9, ESI†), suggesting no significant change in the oxidation state of cerium in CeO2–TiO2 without Ni promotion. The reduced 5Ni/CeO2–TiO2 after the final CO2 or H2O splitting step recovered the initial doublet feature indicative of Ce4+. Therefore, the valence state of cerium in 5Ni/CeO2–TiO2 changed reversibly during the redox reactions. On the other hand, Ti K-edge XANES spectra of 5Ni/CeO2–TiO2 were in close resemblance to TiO2 irrespective of the different processing steps, i.e., after initial synthesis, after reduction with CH4 or after CO2 or H2O splitting redox cycling (Fig. 7b). The absence of shifts in the Ti K-edge position reveals that unlike cerium, all titanium remained in the formal +4 oxidation state upon reduction and oxidation. In conclusion, XANES analysis establishes that a complete Ce4+ ↔ Ce3+ redox cycle was attainable for the 5Ni/CeO2–TiO2 catalyst promoted by Ni species, while negligible change in the cerium oxidation state was observed for CeO2–TiO2 without Ni promotion, and this can account for the superior CH4 partial oxidation activity and outstanding CO and H2 productivities of 5Ni/CeO2–TiO2 in MDR-STCDS and MDR-STWS processes.
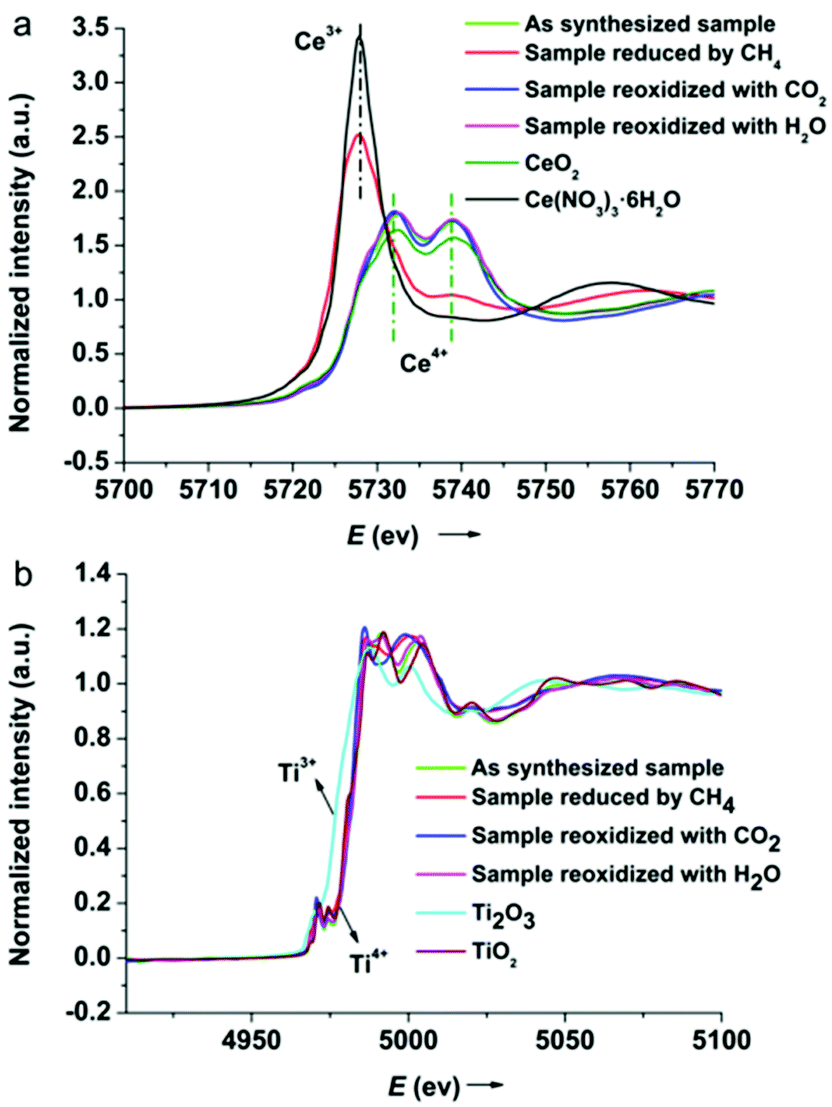 | ||
| Fig. 7 Normalized XANES spectra for the 5Ni/CeO2–TiO2 catalyst after various steps in isothermal MDR-STCDS and MDR-STWS redox cycling at (a) the Ce LIII-edge and (b) the Ti K-edge, together with the spectra of standard reference compounds for different oxidation states. | ||
The morphological and the structural changes of the catalyst were confirmed via HRSEM analysis. Representative HRSEM images of the as synthesized 5Ni/CeO2–TiO2 along with the ones after two-step MDR-STCDS and MDR-STWS redox cycling are presented in Fig. 8a–c. Fig. 8a shows that the fresh catalyst was composed of small nanosized grains with relatively uniform diameters (50–100 nm). No significant grain size growth was observed over multiple redox cycles (Fig. 8b and c), confirming the stability and robustness of the 5Ni/CeO2–TiO2 nanocomposite and its potential application in MDR-STCDS and MDR-STWS processes for solar fuel production.
 | ||
| Fig. 8 Representative SEM images of the 5Ni/CeO2–TiO2 catalyst: (a) as synthesized, (b) after isothermal MDR-STCDS redox cycling at 900 °C and (c) after isothermal MDR-STWS redox cycling at 900 °C. | ||
The elemental distribution and structure of the 5Ni/CeO2–TiO2 catalyst were further characterized by STEM-EDX and HRTEM techniques and the results are indicated in Fig. 9. For the initial catalyst, STEM-based EDX mapping (Fig. 9b1–b4) showed an inhomogeneous distribution of the elements with distinct Ce, Ti, and Ni rich regions (see Fig. S10, ESI† for more details). The corresponding HRTEM images exhibited well-resolved lattice fringes of 0.31, 0.35, and 0.24 nm, which can be ascribed to CeO2(111), TiO2(210), and NiO(111) facets, respectively (Fig. 9c). The above results further confirm the intimately mixed oxides of CeO2, TiO2, and NiO in the as-prepared catalyst. In contrast, elemental mapping for 5Ni/CeO2–TiO2 after reduction with CH4 (Fig. 9e1–e4) demonstrates clearly that the distributions of Ce, Ti, and O overlap with each other, whereas Ni was at oxidation state Ni0 as no clear O element was identified within the same region. Besides, clear lattice fringes of 0.29 and 0.20 nm corresponding to (112) Ce2Ti2O7 (JCPDS 00-047-0667) and (111) Ni (JCPDS 00-001-1272) structural domains, respectively, were revealed (Fig. 9f). These results thus provide unambiguous evidence that TiO2 was incorporated into the CeO2 lattice, resulting in Ce2Ti2O7 pyrochlore, while NiO was reduced to metallic nickel after CH4 reduction, in accordance with the XRD analysis. However, the distribution of Ce and Ti remained inhomogeneous for CeO2–TiO2 before and after the MRD step (Fig. S11, ESI†), indicating the incorporation of TiO2 into CeO2 appeared to be negligible. Additionally, STEM-EDX mapping of 5Ni/CeO2–TiO2 catalysts after MDR-STCDS and MDR-STWS redox cycling verified a homogeneous distribution of Ce, Ti, and O elements (Fig. S12, ESI†), implying that CeO2 and TiO2 were regenerated with a homogeneous dispersion, with Ni species dispersed on the CeO2–TiO2 matrix. In essence, the STEM-EDX mapping and HRTEM analysis visualized the crystalline phase evolution of the 5Ni/CeO2–TiO2 catalyst, i.e., the as-prepared catalyst existed in the form of mixed oxides, which transformed into Ce2Ti2O7 pyrochlore and metallic Ni after the MDR step and regenerated with uniformly distributed CeO2 and TiO2 upon CO2 or H2O splitting.
 | ||
| Fig. 9 (a) STEM image, (b1–b4) the corresponding element-mapping images and (c) HRTEM images of as synthesized 5Ni/CeO2–TiO2; (d) STEM image, (e1–e4) the corresponding element-mapping images and (f) HRTEM images of 5Ni/CeO2–TiO2 after the CH4 reduction half cycle. | ||
3.3 DFT studies
To provide a more fundamental understanding of the observed catalytic performance, DFT calculations were undertaken to probe the reaction landscape associated with CH4 dissociation and CO2/H2O splitting, as well as the performance of oxygen anion migration within the CeO2–TiO2 support. For the methane driven reduction step, the breaking of the first C–H bond in CH4 was studied on the metal, oxide and metal–oxide interface, respectively, as shown in Fig. 10a. The CH4 dissociation on metal oxide surfaces, CeO2(111) and TiO2(110), is endothermic with a reaction energy larger than 1.00 eV and possesses high reaction barriers. In contrast, on the Ni(111) surface the reaction is slightly endothermic (0.17 eV) and the reaction barrier decreases to 0.73 eV, much lower than that on metal oxide surfaces with 1.44 eV for CeO2(111) and 1.27 eV for TiO2(110). Furthermore, at the interface of Ni/CeO2(111) and Ni/TiO2(110), despite the reaction barriers being slightly higher than that on Ni(111), the reaction energies change to significantly exothermic, indicating that the Ni/CeO2–TiO2 interfaces are more preferential for CH4 dissociation. Therefore, our calculation results suggest that both Ni and the Ni/CeO2–TiO2 interface are more active than the pure metal oxide, which echoes the constructive role of Ni in metal oxides for methane driven reduction. | ||
| Fig. 10 DFT calculation results. Potential energy diagrams for (a) methane dissociation (CH4(g) → CH3* + H*), (b) carbon dioxide splitting (CO2(g) → CO* + O*) and (c) water splitting (H2O(g) → H2* + O*) on the metal, oxide and metal–oxide interface. (d) The formation energies of oxygen vacancies (E(VO)) at different sites of Ti-doped CeO2(111), and E(VO) for CeO2(111) and TiO2(110) are also shown as references. The zero energy reference corresponds to the sum of energies of gas-phase molecules and a clean surface. The reaction energies and activation energies for the elementary steps involved in the three reactions are displayed in Tables S6–S8 (ESI†). The geometric structures of states labelled in (a–c) are displayed in Fig. S13–S15 (ESI†). On the Ti-doped CeO2(111) in (d), the oxygen atoms in the first atomic layer are labelled with “u” and the oxygen atoms in the third atomic layer are labelled with “d”. The surface structures with oxygen vacancies are displayed in Fig. S16 (ESI†). The red, yellow and gray spheres represent O, Ce and Ti atoms, respectively. | ||
In the process of CO2 or H2O splitting, the reduced Ce2Ti2O7(211), Ni(111) and Ni/Ce2Ti2O7(211) interfaces were considered as active sites. As shown in Fig. 10b, the CO2 dissociation on Ce2Ti2O7(211) is an endothermic reaction with a reaction energy of 1.08 eV and a large reaction barrier of 1.45 eV. On Ni(111) the reaction energy becomes exothermic with a value of −1.14 eV and the reaction barrier is as low as 0.46 eV. However, the adsorption energy of CO2 on Ni(111) is insignificant (only −0.01 eV), which can lead to a low coverage of adsorbed CO2 and thus is detrimental to the total rate of CO2 dissociation. At the interface between Ni and Ce2Ti2O7(211), though the reaction barrier is increased by 0.36 eV compared to that on Ni(111), the reaction is more exothermic and the adsorption of CO2 is dramatically enhanced with an adsorption energy of −0.82 eV, suggesting the Ni/Ce2Ti2O7(211) interface is more favorable for CO2 splitting. For the water dissociation reaction on the Ce2Ti2O7(211) surface (Fig. 10c), the breakage of the first O–H bond only needs to overcome a small reaction barrier of 0.34 eV, resulting in the OH group binding with metal atoms and the H atom binding with surface oxygen atoms (state A-w1). However, the subsequent hydrogen formation on the Ce2Ti2O7(211) surface is extremely difficult as the reaction barrier and the reaction energy are up to 3.53 eV and 2.99 eV, respectively. On the Ni(111) surface, water first experiences two steps of O–H bond breaking, then followed by one step of hydrogen generation. The breaking of the second O–H bond (from state A-w2 to B-w2) is the rate-determining step with a reaction barrier of 1.09 eV. At the interface of Ni/Ce2Ti2O7(211), water needs to go through three steps similar to that on Ce2Ti2O7(211) and two steps of hydrogen atom diffusion (state A-w3 to A′-w3 and state B-w3 to B′-w3). The reaction barrier of the rate-determining step (the breaking of the first O–H bond) is 1.13 eV, which is only 0.04 eV higher than that on the Ni(111) surface. However, the adsorption of water at interface sites (−1.07 eV) is stronger than that on Ni(111) (−0.50 eV). Therefore, Ni and the Ni/Ce2Ti2O7 interface show comparable activity in water splitting, but are much more active than the pure Ce2Ti2O7 surface.
Additionally, to explore the effect of TiO2 on the oxygen anion diffusivity within CeO2, the formation energies of oxygen vacancies E(VO) of Ti-doped CeO2 were calculated with CeO2 and TiO2 as references (Fig. 10d). The calculated results indicate that the E(VO) of Ti-doped CeO2(111) are lower than those of CeO2(111) and TiO2(110). Moreover, the oxygen atoms (u2, d2 in Fig. 10d) with lower coordination numbers after replacing one Ce atom with a Ti atom are more likely to be removed from the surface with the lowest E(VO) of 1.57 eV and 1.70 eV. Overall, the lower formation energies of oxygen vacancies approaching the Ti atom on Ti-doped CeO2(111) reflect the weakening of the Ce–O bond induced by the Ti atom, which indicates the promotional effect of TiO2 on oxygen anion diffusion within CeO2.
3.4 Reaction mechanism
On the basis of the experimental evidence and DFT calculations presented above, the potential reaction mechanism for the 5Ni/CeO2–TiO2 redox catalyst in MDR-STCDS and MDR-STWS processes is depicted in Fig. 11. We propose that in the MDR step, oxygen originating from NiO and loosely bonded surface oxygen were responsible for the total oxidation of CH4 to CO2 and H2O with simultaneous reduction of NiO to metallic Ni. Furthermore, a stoichiometric reaction between CeO2 and TiO2 was promoted by the presence of Ni, providing lattice oxygen necessary for partial oxidation of CH4 and resulting in Ce2Ti2O7 pyrochlore. In the subsequent CO2 or H2O splitting step, the anion-deficient Ce2Ti2O7 pyrochlore was reoxidized back to CeO2 and TiO2 after incorporation of O anions, producing CO or H2. Meanwhile, the surface metallic Ni was partially reoxidized by CO2 or H2O. The reduced Ni species in 5Ni/CeO2–TiO2 are identified as the active sites for CH4 activation and accelerate CO2 and H2O dissociation in the reduction and oxidation steps, as strongly indicated by the drastically enhanced CH4 reactivity (Fig. 3 and Fig. S3, ESI†) and CO2 and H2O splitting kinetics (Fig. 2a and b) over those of CeO2–TiO2. The phase transformations between CeO2–TiO2 and Ce2Ti2O7 pyrochlore proceeded reversibly with the promotion of Ni species for enhanced lattice oxygen transport, accompanied by the complete Ce4+ ↔ Ce3+ redox cycle through reduction with CH4 and oxidation with CO2 or H2O, and it was backed up by XRD, XANES and electron microscopy (STEM-EDX, HRTEM) observations. DFT calculations also indicate that the metallic Ni and Ni/CeO2–TiO2 interface sites enhance the CH4 activation and the dissociation of CO2 and H2O. The calculated formation energies of oxygen vacancies suggest that the incorporation of TiO2 into CeO2 can weaken the Ce–O bond and thus promote the oxygen anion diffusion within CeO2. Overall, the synergistic effect between the catalytic activation on Ni and the CeO2–TiO2/Ce2Ti2O7 stoichiometric redox cycle enabled the highly effective solar fuel production in MDR-STCDS and MDR-STWS processes.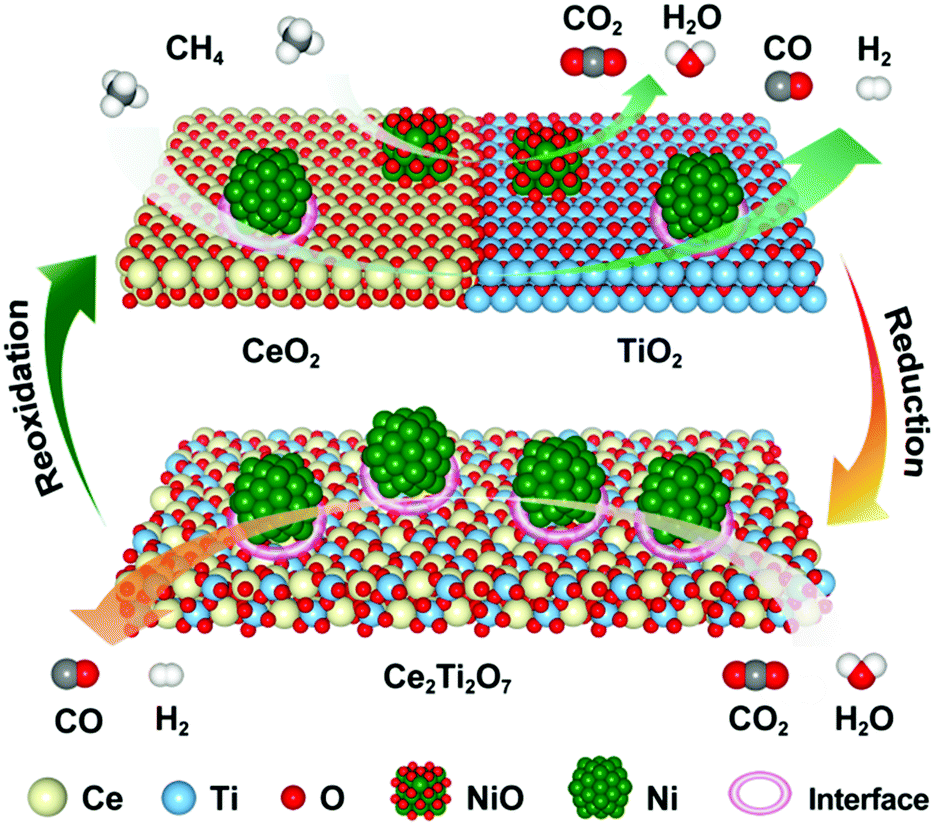 | ||
| Fig. 11 Schematic of the proposed reaction mechanism for MDR-STCDS and MDR-STWS processes over the 5Ni/CeO2–TiO2 redox catalyst. | ||
4. Conclusions
In summary, 5Ni/CeO2–TiO2 has emerged as an exceptional catalytic system for robust operation in MDR-STCDS and MDR-STWS processes. The redox catalyst featured ∼100% CH4 conversions and syngas selectivities up to ∼76 and ∼85% in the MDR step. In regard to the CO2 and H2O splitting steps, one to two orders of magnitude higher CO/H2 production rates and productivities were achieved compared to the state-of-the-art STCDS/STWS and MDR-STCDS/MDR-STWS studies. In addition, the catalyst developed here possesses excellent stability over multiple redox cycles. As revealed by the experimental investigations and DFT calculations, the superior performance of 5Ni/CeO2–TiO2 is driven by the synergy between the catalytic activation on Ni and the CeO2–TiO2/Ce2Ti2O7 stoichiometric redox cycle as they provide active sites and reactive intermediates to transport lattice oxygen for CH4 partial oxidation and CO2 or H2O splitting in a complementary fashion. We anticipate the fundamental understanding on the crucial roles of the catalytic sites for reactant activation and the stoichiometric redox chemistry for enhanced lattice oxygen availability can provide important guidance for the rational design of advanced materials toward solar fuel production.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (21476232, 21590792, 21776271, 21676269, 21603170, 91645203), National Key Projects for Fundamental Research and Development of China (2016YFA0202801), the “Strategic Priority Research Program” of the Chinese Academy of Sciences (XDB17020100) and Department of Science and Technology of Liaoning province under contract 2015020086-101. C. R. C. gratefully acknowledges the China Postdoctoral Science Foundation (2018T111034), the Fundamental Research Funds for the Central Universities (xtr0218016 and cxtd2017004) and the support of K. C. Wong Education Foundation. The calculations were performed by using the HPC Platform at Xi’an Jiaotong University and National Supercomputing Center in Shenzhen. This paper is dedicated to the 70th anniversary of the Dalian Institute of Chemical Physics, Chinese Academy of Sciences.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- N. S. Lewis, Science, 2016, 351, 1920–1929 CrossRef PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- E. M. Nichols, J. J. Gallagher, C. Liu, Y. Su, J. Resasco, Y. Yu, Y. Sun, P. Yang, M. C. Chang and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11461–11466 CrossRef CAS PubMed.
- D. S. Mallapragada, N. R. Singh, V. Curteanu and R. Agrawal, Ind. Eng. Chem. Res., 2013, 52, 5136–5144 CrossRef CAS.
- J. A. Herron, J. Kim, A. A. Upadhye, G. W. Huber and C. T. Maravelias, Energy Environ. Sci., 2015, 8, 126–157 RSC.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- M. Tou, R. Michalsky and A. Steinfeld, Joule, 2017, 146–154, DOI:10.1016/j.joule.2017.07.015.
- C. L. Muhich, B. W. Evanko, K. C. Weston, P. Lichty, X. Liang, J. Martinek, C. B. Musgrave and A. W. Weimer, Science, 2013, 341, 540–542 CrossRef CAS PubMed.
- A. H. Bork, M. Kubicek, M. Struzik and J. L. M. Rupp, J. Mater. Chem. A, 2015, 3, 15546–15557 RSC.
- C. Ruan, Y. Tan, L. Li, J. Wang, X. Liu and X. Wang, AIChE J., 2017, 63, 3450–3462 CrossRef CAS.
- F. He, J. Trainham, G. Parsons, J. S. Newman and F. Li, Energy Environ. Sci., 2014, 7, 2033–2042 RSC.
- F. He and F. Li, Energy Environ. Sci., 2015, 8, 535–539 RSC.
- J. R. Scheffe, M. D. Allendorf, E. N. Coker, B. W. Jacobs, A. H. McDaniel and A. W. Weimer, Chem. Mater., 2011, 23, 2030–2038 CrossRef.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, J. Phys. Chem. C, 2017, 121, 11055–11068 CrossRef CAS.
- Z. Zhao, M. Uddi, N. Tsvetkov, B. Yildiz and A. F. Ghoniem, Phys. Chem. Chem. Phys., 2017, 19, 25774–25785 RSC.
- D. Maiti, B. J. Hare, Y. A. Daza, A. E. Ramos, J. N. Kuhn and V. R. Bhethanabotla, Energy Environ. Sci., 2018, 11, 648–659 RSC.
- B. J. Hare, D. Maiti, Y. A. Daza, V. R. Bhethanabotla and J. N. Kuhn, ACS Catal., 2018, 8, 3021–3029 CrossRef CAS.
- R. Michalsky, D. Neuhaus and A. Steinfeld, Energy Technol., 2015, 3, 784–789 CrossRef CAS.
- J. Zhang, V. Haribal and F. Li, Sci. Adv., 2017, 3, e1701184 CrossRef PubMed.
- P. T. Krenzke and J. H. Davidson, Energy Fuels, 2014, 28, 4088–4095 CrossRef CAS.
- P. T. Krenzke, J. R. Fosheim, J. Zheng and J. H. Davidson, Int. J. Hydrogen Energy, 2016, 41, 12799–12811 CrossRef CAS.
- K. J. Warren, J. Reim, K. Randhir, B. Greek, R. Carrillo, D. W. Hahn and J. Scheffe, Energy Technol., 2017, 5, 2138–2149 CrossRef CAS.
- A. Steinfeld, P. Kuhn and J. Karni, Energy, 1993, 18, 239–249 CrossRef CAS.
- Q. Imtiaz, N. S. Yüzbasi, P. M. Abdala, A. M. Kierzkowska, W. van Beek, M. Broda and C. R. Müller, J. Mater. Chem. A, 2016, 4, 113–123 RSC.
- M. M. Nair and S. Abanades, Energy Fuels, 2016, 30, 6050–6058 CrossRef.
- X. Gao, A. Vidal, A. Bayon, R. Bader, J. Hinkley, W. Lipiński and A. Tricoli, J. Mater. Chem. A, 2016, 4, 9614–9624 RSC.
- M. Welte, K. Warren, J. R. Scheffe and A. Steinfeld, Ind. Eng. Chem. Res., 2017, 56, 10300–10308 CrossRef CAS PubMed.
- Y. Zheng, Y. Wei, K. Li, X. Zhu, H. Wang and Y. Wang, Int. J. Hydrogen Energy, 2014, 39, 13361–13368 CrossRef CAS.
- X. Zhu, Y. Wei, H. Wang and K. Li, Int. J. Hydrogen Energy, 2013, 38, 4492–4501 CrossRef CAS.
- J. R. Scheffe, R. Jacot, G. R. Patzke and A. Steinfeld, J. Phys. Chem. C, 2013, 117, 24104–24114 CrossRef CAS.
- Y. Zheng, K. Li, H. Wang, X. Zhu, Y. Wei, M. Zheng and Y. Wang, Energy Fuels, 2016, 30, 638–647 CrossRef CAS.
- K. Otsuka, Y. Wang, E. Sunada and I. Yamanaka, J. Catal., 1998, 175, 152–160 CrossRef CAS.
- D. Arifin, V. J. Aston, X. Liang, A. H. McDaniel and A. W. Weimer, Energy Environ. Sci., 2012, 5, 9438 RSC.
- C. Dang, Y. Li, S. M. Yusuf, Y. Cao, H. Wang, H. Yu, F. Peng and F. Li, Energy Environ. Sci., 2018, 11, 660–668 RSC.
- D. R. Killelea, V. L. Campbell, N. S. Shuman and A. L. Utz, Science, 2008, 319, 790–793 CrossRef CAS PubMed.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T. D. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- S. Fabris, G. Vicario, G. Balducci, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2005, 109, 22860–22867 CrossRef CAS PubMed.
- J. B. Park, J. Graciani, J. Evans, D. Stacchiola, S. D. Senanayake, L. Barrio, P. Liu, J. F. Sanz, J. Hrbek and J. A. Rodriguez, J. Am. Chem. Soc., 2010, 132, 356–363 CrossRef CAS PubMed.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- H. Jónsson, G. Mills and K. W. Jacobsen, in Classical and Quantum Dynamics inCondensed Phase Simulations, ed. B. J. Berne, G. Ciccotti and D. F. Coker, World Scientific, Singapore, 1998, pp. 385–404 Search PubMed.
- A. Preuss and R. Gruehn, J. Solid State Chem., 1994, 110, 363–369 CrossRef CAS.
- L. Foppa, T. Margossian, S. M. Kim, C. Müller, C. Copéret, K. Larmier and A. Comas-Vives, J. Am. Chem. Soc., 2017, 139, 17128–17139 CrossRef CAS PubMed.
- S. Takenaka, N. Hanaizumi, V. T. D. Son and K. Otsuka, J. Catal., 2004, 228, 405–416 CrossRef CAS.
- J. Carrasco, D. Lopez-Duran, Z. Liu, T. Duchon, J. Evans, S. D. Senanayake, E. J. Crumlin, V. Matolin, J. A. Rodriguez and M. V. Ganduglia-Pirovano, Angew. Chem., Int. Ed., 2015, 54, 3917–3921 CrossRef CAS PubMed.
- C. Heine, B. A. Lechner, H. Bluhm and M. Salmeron, J. Am. Chem. Soc., 2016, 138, 13246–13252 CrossRef CAS PubMed.
- Q. G. Yan, W. Z. Weng, H. L. Wan, H. Toghiani, R. K. Toghiani and C. U. Pittman, Appl. Catal., A, 2003, 239, 43–58 CrossRef CAS.
- S. Chen, L. Zeng, H. Tian, X. Li and J. Gong, ACS Catal., 2017, 7, 3548–3559 CrossRef CAS.
- M. Rydén, M. Johansson, A. Lyngfelt and T. Mattisson, Energy Environ. Sci., 2009, 2, 970 RSC.
- A. Löfberg, J. Guerrero-Caballero, T. Kane, A. Rubbens and L. Jalowiecki-Duhamel, Appl. Catal., B, 2017, 212, 159–174 CrossRef.
- I. Atribak, A. Buenolopez and A. Garciagarcia, J. Catal., 2008, 259, 123–132 CrossRef CAS.
- X. Wang, D. Liu, J. Li, J. Zhen and H. Zhang, NPG Asia Mater., 2015, 7, e158 CrossRef CAS.
- M. F. Luo, J. Chen, L. S. Chen, J. Q. Lu, Z. Feng and C. Li, Chem. Mater., 2001, 13, 197–202 CrossRef CAS.
- T. J. Lin, H. Xie, X. Meng and L. Shi, Catal. Commun., 2015, 68, 88–92 CrossRef CAS.
- H. Zhu, D. C. Rosenfeld, D. H. Anjum, S. S. Sangaru, Y. Saih, S. Ould-Chikh and J.-M. Basset, J. Catal., 2015, 329, 291–306 CrossRef CAS.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- A. Antzara, E. Heracleous, L. Silvester, D. B. Bukur and A. A. Lemonidou, Catal. Today, 2016, 272, 32–41 CrossRef CAS.
- V. V. Galvita, H. Poelman and G. B. Marin, J. Power Sources, 2015, 286, 362–370 CrossRef CAS.
- Y. Gao, L. M. Neal and F. Li, ACS Catal., 2016, 6, 7293–7302 CrossRef CAS.
- O. Mihai, D. Chen and A. Holmen, J. Catal., 2012, 293, 175–185 CrossRef CAS.
- T. Yamamoto, A. Suzuki, Y. Nagai, T. Tanabe, F. Dong, Y. Inada, M. Nomura, M. Tada and Y. Iwasawa, Angew. Chem., Int. Ed., 2007, 119, 9413–9416 CrossRef.
- S. Zhang, S. Muratsugu, N. Ishiguro, S.-I. Ohkoshi and M. Tada, ChemCatChem, 2012, 4, 1783–1790 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee03069c |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2019 |