
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nature of the “Z”-phase in layered Na-ion battery cathodes†
James W.
Somerville
 a,
Adam
Sobkowiak‡
a,
Nuria
Tapia-Ruiz§
a,
Juliette
Billaud
b,
Juan G.
Lozano¶
a,
Robert A.
House
a,
Leighanne C.
Gallington
c,
Tore
Ericsson
d,
Lennart
Häggström
d,
Matthew R.
Roberts
a,
Urmimala
Maitra||
a and
Peter G.
Bruce
*ae
a,
Adam
Sobkowiak‡
a,
Nuria
Tapia-Ruiz§
a,
Juliette
Billaud
b,
Juan G.
Lozano¶
a,
Robert A.
House
a,
Leighanne C.
Gallington
c,
Tore
Ericsson
d,
Lennart
Häggström
d,
Matthew R.
Roberts
a,
Urmimala
Maitra||
a and
Peter G.
Bruce
*ae
aDept. of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: peter.bruce@materials.ox.ac.uk
bPaul Scherrer Institut, Electrochemical Laboratory, CH-5232 Villigen PSI, Switzerland
cX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Argonne, IL 60439, USA
dDept. of Chemistry, Ångstrom Lab., Uppsala University, Box 538, SE-751 21 Uppsala, Sweden
eThe Faraday Institution, Quad One, Becquerel Avenue, Harwell Campus, Didcot, OX11 0RA, UK
First published on 17th May 2019
Abstract
Layered sodium transition metal oxides with the P2 structure, e.g. Na2/3[Ni1/3Mn2/3]O2, are regarded as candidates for Na-ion battery cathodes. On charging, extraction of Na destabilizes the P2 phase (ABBA oxide ion stacking) in which Na+ is in trigonal prismatic coordination, resulting in layer gliding and formation of an O2 phase (ABAC stacking) with octahedral coordination. However, many related compounds do not exhibit such a simple P2 to O2 transition but rather form a so called “Z”-phase. Substituting Ni by Fe in Na2/3[Ni1/3Mn2/3]O2 is attractive as it reduces cost. The Fe containing compounds, such as Na2/3[Ni1/6Mn1/2Fe1/3]O2, form a “Z”-phase when charged above 4.1 V vs. Na+/Na. By combining ex situ and operando X-ray diffraction with scanning transmission electron microscopy and simulated diffraction patterns, we demonstrate that the “Z”-phase is most accurately described as a continuously changing intergrowth structure which evolves from P2 to O2 through the OP4 structure as an intermediate. On charging, Na+ removal results in O-type stacking faults within the P2 structure which increase in proportion. At 50% O-type stacking faults, the ordered OP4 phase forms and on further charging more O-type stacking faults are formed progressing towards a pure O2 structure. This gives the superficial appearance of a solid solution. Furthermore, in contrast to some previous studies, we did not detect Fe migration at any state-of-charge using 57Fe-Mössbauer spectroscopy. It was, however, found that the Fe-substitution serves to disrupt cation ordering in the material.
Broader contextThe Li-ion battery has revolutionised our lives. This cell chemistry, however, has a relatively high cost and suffers from sustainability issues, particularly surrounding the use of cobalt. The Na-ion battery represents a promising alternative as it avoids these problems and possesses several other advantageous characteristics (e.g. use of Al negative current collector). For high capacity layered oxide cathodes, the larger ionic radius of Na+ compared with Li+ grants a wider choice of transition metals that can be utilised and opens an avenue towards low cost, Co-free batteries. At the same time, the larger radius of Na+ enables a richer variety of stable layered configurations (O3, P2, P3 etc.), which can lead to multiple changes in crystal structure as the battery is charged and discharged. In this paper, we focus on the P2 layered oxide Na2/3[Ni1/6Mn1/2Fe1/3]O2 and unravel its complex structural changes at low Na contents (previously termed the “Z”-phase). Specifically, we identify this phase as a continuously evolving intergrowth structure between trigonal prismatic and octahedral stacking. This work can be reasonably extended to many other P2-type cathodes and will also have relevance to additional layered oxide cathode types (e.g. Li-ion Ni-rich cathodes) that exhibit high voltage structural transitions. |
Introduction
Layered sodium transition-metal oxides, NaMO2 (where M = Mn, Fe, Co, Ni, etc.), represent a promising class of materials for Na-ion battery cathodes due to their higher theoretical volumetric and gravimetric energy densities compared with other cathode candidates (e.g. Prussian blue analogues, polyanion, etc.).1,2 NaMO2 compounds crystallise in structures that vary in the coordination of Na (octahedral: O or trigonal prismatic: P) and the stacking of the oxygen layers (e.g. 2: ABBA ordering).3 To date, the full charge storage capacity of NaMO2 compounds cannot be utilized primarily because of phase transitions at low Na contents (high states of charge) which adversely affect capacity retention on cycling.4For example, P2-Na2/3[Ni1/3Mn2/3]O2 undergoes a phase transition to the O2 structure, shown in Fig. 1, when charged above 4.1 V vs. Na+/Na.5,6 This is characterised by the gliding of the transition metal oxide (TMO2) slabs resulting in a re-coordination of the Na sites from trigonal prisms to octahedra and a severe contraction of the unit cell along the c-axis.
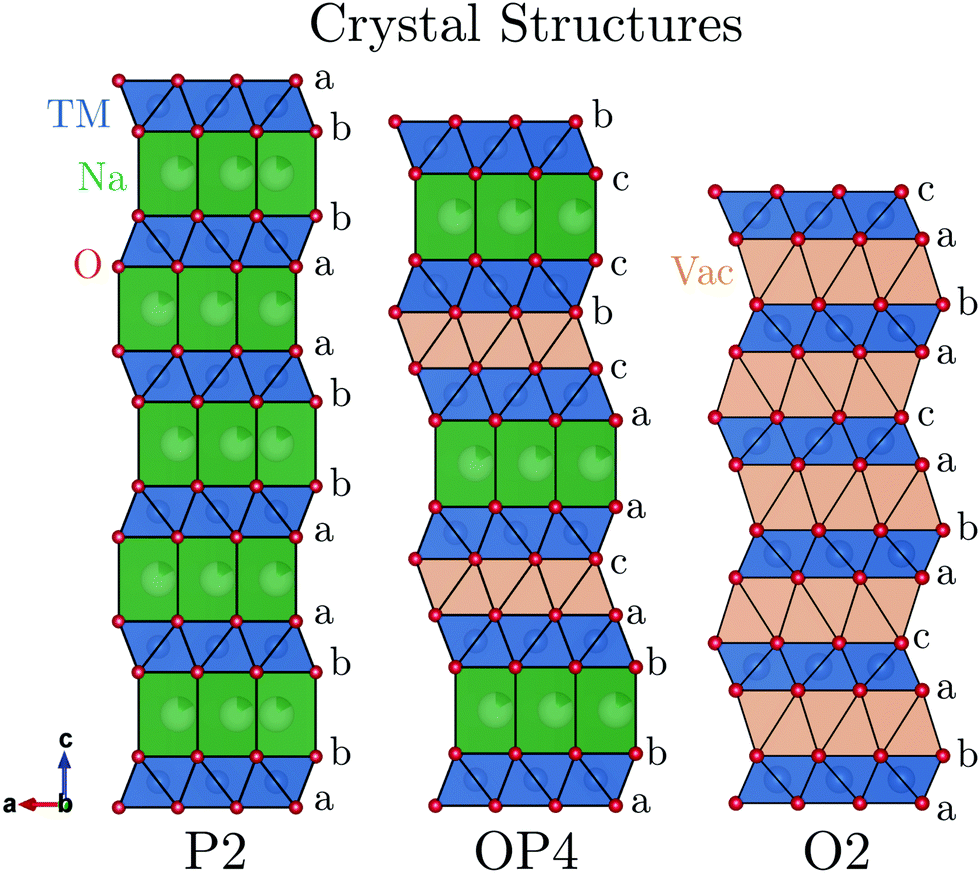 | ||
| Fig. 1 The P2, OP4, and O2 crystal structures are shown projected parallel to the layers showing the stacking sequence. | ||
P2-Na2/3[Fe1/2Mn1/2]O2 emerged as a promising cathode material because of its Earth-abundant elements and improved cycling stability compared with P2-Na2/3[Ni1/3Mn2/3]O2.7 This compound also undergoes a phase transition when charged above 4.1 V, but unlike the O2 structure, the new phase continues to evolve upon further charge and the overall volume contraction is less severe.8,9
The identification of this new high voltage phase and the mechanism by which it evolves has been a controversial subject in the literature. Originally, Yabuuchi et al. described the high voltage phase of P2-Na2/3[Fe1/2Mn1/2]O2 as an OP4-type structure (alternating layers of O and P-type stacking, shown in Fig. 1) based on ex situ synchrotron powder X-ray diffraction (PXRD) results.7 After carrying out in situ PXRD work on the same compound, Mortemard de Boisse et al. concluded that many model structures could be used to fit the small number of Bragg reflections observed and therefore referred to this high voltage phase more generally as the “Z”-phase.8 Work by Talaie et al. later postulated that this “Z”-phase was not OP4 but instead an O2 phase where some Fe (and/or other transition metal cations) had migrated into tetrahedral sites of the Na layer.9 Discussion of the high voltage phase has since expanded to a range of P2 compounds containing elements such as Mg, Co, Ni, and Cu.9–18 Many descriptors have been used to identify the high voltage phase in these compounds including “Z”-phase, O2, OP4, and P2–O2 intergrowth structures. A common observation however is the sudden decrease of long-range order and a continuous contraction (002) peak to higher 2θ value in in situ XRD. From here on, structures producing this signature will be referred to only as “Z”-phase for simplicity.
Despite the large amount of interest, questions remain over the dynamic nature of this “Z”-phase (seemingly a solid solution) and whether transition metal migration plays a critical role.
Two new sodium intercalation cathodes targeted for high specific energy and elemental abundance, P2-Na2/3[Ni1/3−y/2Mn2/3−y/2Fey]O2 (y = 1/6, 1/3), were recently reported by us.19 It was found that the cycling stability of these Fe-substituted compositions was superior to Fe-free P2-Na2/3[Ni1/3Mn2/3]O2. In the current paper, we focus on comparing the structural evolution of the Fe-free compound (y = 0) with one of these Fe-substituted compositions, Na2/3[Ni1/3−y/2Mn2/3−y/2Fey]O2 (y = 1/3), as they are representative examples of Na-ion cathodes transitioning to a well-defined O2-structure and the “Z”-phase, respectively, when charged above 4.1 V vs. Na+/Na. It is worth mentioning that we also analysed the composition Na2/3[Ni1/3−y/2Mn2/3−y/2Fey]O2 (y = 1/6), and that the results from this compound are included in the ESI† for comparison and are largely the same as the y = 1/3 composition. We show that the “Z”-phase is most accurately described as a continuously changing intergrowth structure which evolves between P2 and O2 with OP4 as an intermediary. This is in contrast to the Fe-free compound, which is confirmed to transform directly to the O2 structure via a biphasic reaction. Finally, 57Fe-Mössbauer spectroscopy did not show any evidence of Fe migration to the Na-layers for Na2/3[Ni1/6Mn1/2Fe1/3]O2 at high voltages or after cycling.
Experimental section
Stoichiometric amounts of NiO (Sigma Aldrich 99.8%), MnO2 (Sigma Aldrich ≥ 99%), and Fe2O3 (Sigma Aldrich ≥ 99%) were ball milled with 5% excess of Na2CO3 (Sigma Aldrich ≥ 99.5%) for 1 h at 400 rpm in acetone. The powders were subsequently pressed into approximately 250 mg pellets of 13 mm in diameter using a pressure of 5 tons cm−2 and heated in a tube furnace under flowing oxygen gas at a temperature of 950 °C for Na2/3[Ni1/3Mn2/3]O2 and 1030 °C for Na2/3[Ni1/6Mn1/2Fe1/3]O2. The employed heating and cooling rate was 10 °C min−1 and the pellets were immediately transferred to an Ar-filled glovebox for storage and further characterisation.Synchrotron XRD data was measured at the 11 ID-B beamline of the Advanced Photon Source. The wavelength (λ) used was 0.2113 Å. Data was collected on a 2D area detector and subseqently reduced using the GSAS-II software.20 Rietveld refinements were also carried out using GSAS-II. Operando XRD measurements were performed at room temperature on a PANanalytical Empyrean diffractometer equipped with a Cu Kα source.
Powder neutron diffraction (ND) measurements were carried out at ISIS, Rutherford Appleton Laboratories, at the POLARIS medium resolution instrument in the time of flight (TOF) mode.
Annular-dark field scanning transmission electron microscopy (ADF-STEM) images were collected on an aberration corrected JEOL ARM 200F which was operated at a voltage of 200 kV. 22 mrad and 69.6–164.8 mrad were used for the convergence semi-angle and the collection semi-angle, respectively. Each image is composed of multiple frames which were recorded and subsequently aligned correcting for drift and scan distortions using SmartAlign.21
Electrodes were prepared in a glovebox under Ar atmosphere and had a composite weight ratio of 75% active material, 15% Carbon black (Super S, Timcal), and 10% PVDF (Kynar) using NMP (Sigma Aldrich) as a casting solvent. These electrodes were then electrochemically tested in 2325-type coin cells using Whatman glass fibre separators and an electrolyte composed of 1 M NaPF6 in EC/DEC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio). Metallic Na pressed into a flat sheet was used as the counter electrode. For ex situ measurements, the coin cells were disassembled, the electrodes were washed in DMC to remove any remaining electrolyte salt and solvent and then dried under vacuum. Self-standing electrodes for operando XRD measurements were prepared using a weight ratio of 75% active material, 10% Carbon black (Imerys) and 15% Kynar Flex (Solvay) dissolved in a suspension of acetone and ethanol. The solution was cast using a doctor blade onto a glass window. After evaporation of the solvent, the electrode sheet was peeled off and electrodes were punched out.
:1 volumetric ratio). Metallic Na pressed into a flat sheet was used as the counter electrode. For ex situ measurements, the coin cells were disassembled, the electrodes were washed in DMC to remove any remaining electrolyte salt and solvent and then dried under vacuum. Self-standing electrodes for operando XRD measurements were prepared using a weight ratio of 75% active material, 10% Carbon black (Imerys) and 15% Kynar Flex (Solvay) dissolved in a suspension of acetone and ethanol. The solution was cast using a doctor blade onto a glass window. After evaporation of the solvent, the electrode sheet was peeled off and electrodes were punched out.
The operando XRD cell is a custom-made cell and has been described elsewhere.22 The measurements were performed at a rate of 5 mA g−1 between 1.5 and 4.5 V vs. Na+/Na. Analysis of the diffraction patterns was performed with the GSAS-II software and colour maps were generated using MATLAB. Operando XRD patterns were corrected for sample displacement and the background was subtracted before being plotted.
Simulation of intergrowth structures was carried out with FAULTS profile refinement program (based on the DIFFaX simulation program).23,24 Structural parameters were taken from the operando XRD data. P-type layers were defined to have a Na content of 1/3 Na per formula unit and an interlayer distance equal to that of P2-Na2/3[Ni1/6Mn1/2Fe1/3]O2 immediately before transitioning to the “Z”-phase (dP-type = 5.69 Å). O-type layers were defined to be fully de-sodiated with an interlayer spacing matching that of the O2 structure, [Ni1/3Mn2/3]O2 (dO-type = 4.44 Å). Both possible glide vectors, [1/3 2/3 z] and [2/3 1/3 z], were assumed to have equal probability to achieve O-type stacking. Crystal structure models were drawn with the freely available software, VESTA.25
57Fe-Mössbauer spectra were collected in transmission mode using a 57CoRh source of constant acceleration covering a velocity span of ±4 mm s−1. The samples were prepared in an Ar-filled glovebox by mixing 10–20 mg of the composite electrode with a suitable amount of boron nitride as an inert filler. This powder mixture was then evenly spread over a 13 mm in diameter absorber disc. Due to the moisture sensitivity of the active material, the samples were kept under a constant flow of N2 throughout the measurements. The spectra were Lorentzian line least-squares fitted, as applied by Lagarec and Rancourt, using the software Recoil.26 The centre shift, CS, being the sum of the true isomer shift and the second order Doppler shift, is given relative to metallic iron (α-Fe) at room temperature. The magnitude of the quadrupole splitting, QS, is given as the peak separation of the doublets, and the line width, W, is the experimental full-width half maximum (FWHM) of the spectral signals.
Results and discussion
Pristine characterisation
Na2/3[Ni1/3Mn2/3]O2 and Na2/3[Ni1/6Mn1/2Fe1/3]O2 were synthesised via solid state methods. Powder X-ray and neutron diffraction (PXRD and ND) patterns are presented in Fig. 2a and b. Rietveld refinements of the PXRD patterns confirmed crystallisation in a P2-type structure for both compositions (refinement details are presented in Fig. S1, S2 and Tables S2, S3, ESI†). The in-plane a-parameter increases with Fe content, from 2.897 to 2.925 Å, corresponding well with the slightly larger radius of high spin Fe3+ (0.65 Å) compared to the average radii of Mn4+ (0.53 Å) and Ni2+ (0.69 Å) ions, for which the Fe is substituted. The c-parameter, reflecting the interlayer distances, showed only small variations with no clear trend as a function of Fe content.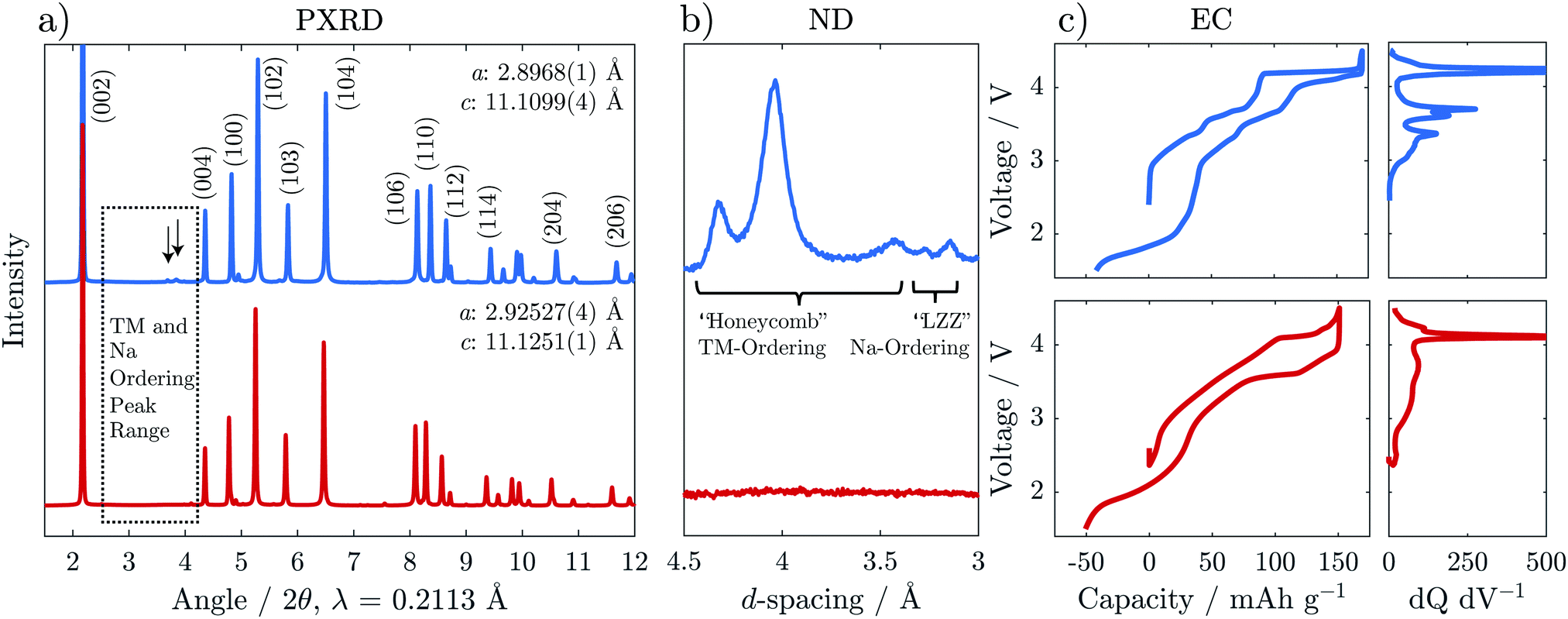 | ||
| Fig. 2 (a) Synchrotron powder X-ray diffraction (PXRD), (b) a section of neutron diffraction (ND) highlighting peaks due to ordering, and (c) electrochemical load curves of the first cycle for Na2/3[Ni1/3Mn2/3]O2 (blue, top) and Na2/3[Ni1/6Mn1/2Fe1/3]O2 (red, bottom) are shown. Na-ordering is indicated by the arrows in (a) and also seen in the ND in (b). To emphasise the subtle steps in the electrochemical load curves in (c), the differential capacity with respect to voltage (dq dV−1) is presented in the right panel for the charge curve only. | ||
The PXRD pattern of Fe-free Na2/3[Ni1/3Mn2/3]O2 in Fig. 2a shows weak reflections in the 2θ range 26–30° indicated by the arrows, which are not accounted for by the structural model. These reflections have been attributed previously to a “large zig zag” (LZZ) Na ordering.6 Extra reflections stemming from honeycomb ordering of the Ni and Mn are also evident in the highlighted region (3.6–4.6 Å) of the ND pattern in Fig. 2b. The honeycomb-ordering between Ni and Mn is only discernible in ND because of the greater difference in neutron scattering lengths vs. X-ray scattering factors.27,28 With the introduction of Fe in Na2/3[Ni1/6Mn1/2Fe1/3]O2, both the Na-ordering and the transition metal ordering reflections disappear. A similar effect was reported for Co-substitution into Na2/3[Ni1/3Mn2/3]O2.29
The interruption of the transition metal ordering with Fe substitution was also found to have a strong effect on the electrochemical behaviour. The steps in the galvanostatic voltage profile become much less pronounced for the Fe-substituted compound which is also reflected in broadened features of the dQ dV−1 trace (Fig. 2c). This effect is similar to that observed through Mg-doping, as reported recently by us and by others,30,31 and suggests a disruption in the Na and charge ordering.
Operando X-ray diffraction
Operando XRD was performed on the first cycle of both Na2/3[Ni1/3Mn2/3]O2 and Na2/3[Ni1/6Mn1/2Fe1/3]O2 to gain a deeper understanding of the structural evolution. The results are presented in Fig. 3 and Fig. S5 (ESI†) for Na2/3[Ni1/4Mn7/12Fe1/6]O2. On initial charge to 4.1 V, both compositions show shifts of diffraction peaks which are consistent with an increase in interlayer distance (i.e. 0.5 c-parameter) and decrease in the in-plane TM to TM distance (a-parameter). The interlayer distance increases due to reduced screening between adjacent negatively charged oxygen layers when Na is removed. The in-plane TM–TM distance decreases due to the shortening of the TM–O bond as the transition metals are oxidized.32,33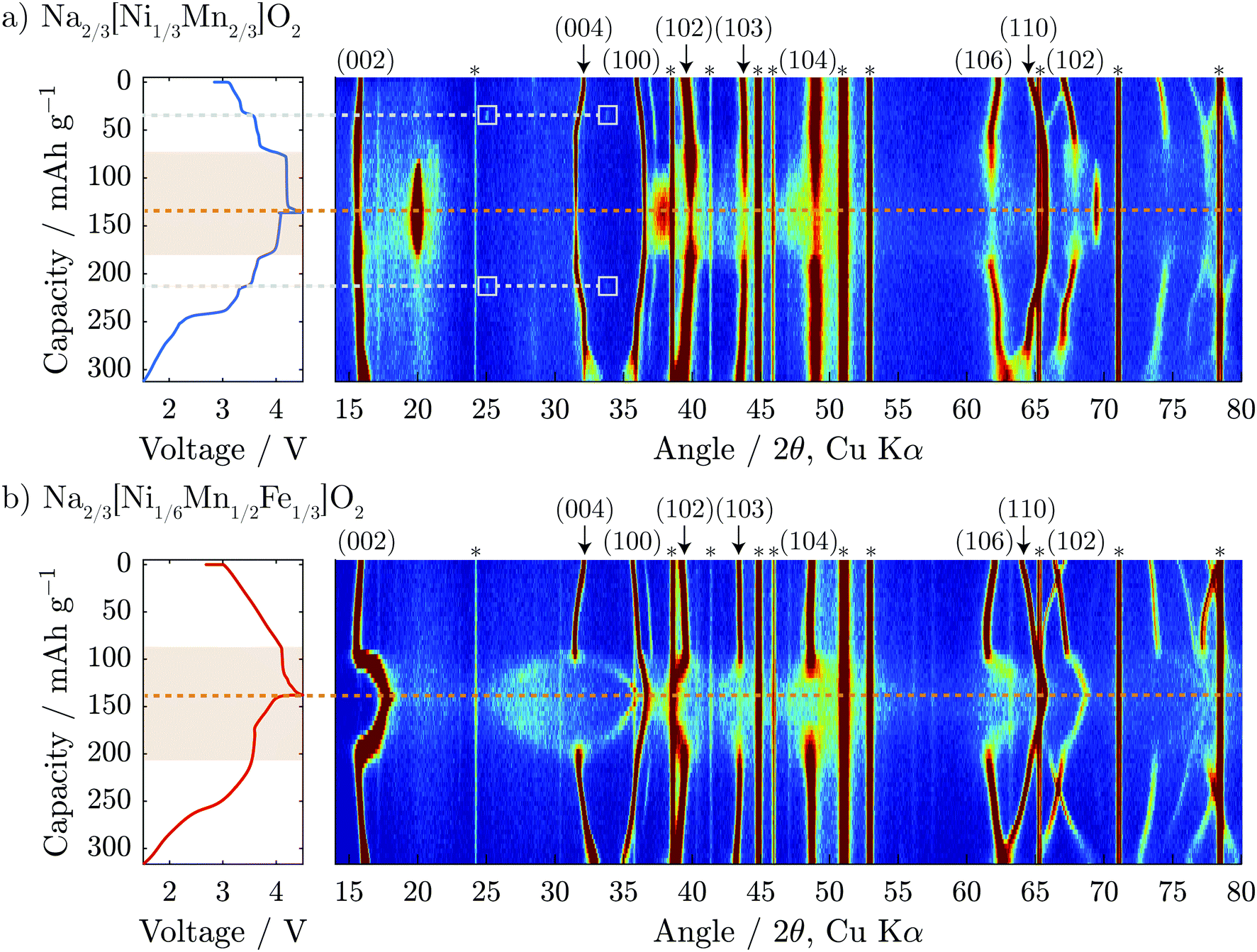 | ||
| Fig. 3 Operando XRD colour maps of the first complete electrochemical cycle are shown for (a) Na2/3[Ni1/3Mn2/3]O2 and (b) Na2/3[Ni1/6Mn1/2Fe1/3]O2. The corresponding electrochemical load curves are shown on the left side of the figure with an orange dashed line denoting the top of charge and light grey dashed lines denoting the Na-ordering event at Na1/2[Ni1/3Mn2/3]O2. The regions of the electrochemistry which correspond to the high voltage structural changes are highlighted in orange. Asterisks (*) denote reflections associated with the operando cell window and inactive electrode constituents (e.g. carbon). | ||
A closer look at the Na2/3[Ni1/3Mn2/3]O2 data presented in Fig. 3a shows two small reflections that appear in the diffraction pattern at 25° and 34° 2θ at 35 mA h g−1 on the cumulative capacity scale (approx. Na1/2[Ni1/3Mn2/3]O2). The reflections reappear at the same 2θ-position and the same state-of-charge on discharge (at 215 mA h g−1) demonstrating that this is a reversible phenomenon. Upon further inspection of the structure using higher intensity synchrotron XRD (Fig. S4, ESI†), several superstructure peaks could be detected and were indexed to a 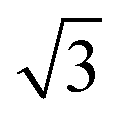 a × 2a × c superstructure (space group: Pnmn). The extra reflections are apparent due to Na-ordering and the identified superstructure matches that which was previously found for Na1/2CoO2 by Huang et al.34 In contrast, no evidence of Na-ion ordering was seen for Na2/3[Ni1/6Mn1/2Fe1/3]O2.
a × 2a × c superstructure (space group: Pnmn). The extra reflections are apparent due to Na-ordering and the identified superstructure matches that which was previously found for Na1/2CoO2 by Huang et al.34 In contrast, no evidence of Na-ion ordering was seen for Na2/3[Ni1/6Mn1/2Fe1/3]O2.
When charged above 4.1 V, both compositions show a plateau in the electrochemical load curve indicating a two-phase transition, which decreases in length with Fe-substitution. In the case of the Fe-free composition, Na2/3[Ni1/3Mn2/3]O2, it has been reported previously that the end member in this two-phase region is a fully desodiated structure of O2-type with octahedral vacancies in the alkali-ion layer,5 as shown in Fig. 1. From our operando diffraction results, we confirm that the decrease of the P2 (002) reflection intensity at 2θ = 15.6° is concurrent with the appearance and increase of the O2 (002) reflection intensity at 2θ = 20.0° during charging. This results in a change of the interlayer distance from approximately 5.68 to 4.44 Å and a total unit cell volume contraction (from pristine to fully charged) of 23%. This biphasic transition along the voltage plateau continues until a sudden increase in potential reaches the upper cut-off at 4.5 V vs. Na+/Na.
For the Fe-containing composition, Na2/3[Ni1/6Mn1/2Fe1/3]O2, shown in Fig. 3b, the structural evolution when charged above 4.1 V differs significantly from the Fe-free composition. The initial P2 structure reaches an interlayer spacing of 5.69 Å before entering a biphasic region, identified by the disappearance of the P2 (002) reflection during charge at ≈95 mA h g−1 (approx. Na1/3[Ni1/6Mn1/2Fe1/3]O2), and a new reflection appearing at only slightly higher 2θ, characteristic of the so-called “Z”-phase. As soon as this “Z”-phase is formed, further Na-extraction appears to proceed through a solid solution process, as indicated by a continuous shift of this new reflection towards higher 2θ angles, initially sharpening in peak shape and then broadening. This can be seen clearly in Fig. 6a. At the top of charge, the angle of the new reflection reaches 2θ = 17.8° corresponding to an interlayer spacing of 4.99 Å and a total volume change of 15%.
A new feature of the high voltage “Z”-phase not, to our knowledge, observed previously, is a faint splitting of the P2 (004) reflection to both higher and lower angles of 2θ. This is in contrast to Na2/3[Ni1/3Mn2/3]O2 for which the evolution of the (004) reflection on charging mirrors that of the (002) reflection. This splitting is not consistent with the “Z”-phase being a solid solution based purely on P2, O2, or OP4-type structures. Instead, we show below that the “Z”-phase can be described as an evolving intergrowth involving the P2, O2, and OP4 structures varying in ratios from P2 through OP4 to O2 on charging (Na extraction).
Scanning transmission electron microscopy
Annular-dark field scanning transmission electron microscopy (ADF-STEM) was carried out to provide direct structural visualization of the “Z”-phase. Fig. 4 compares pristine Na2/3[Ni1/6Mn1/2Fe1/3]O2 with an ex situ sample (charged to 4.5 V) along the [100] projection in order to identify the stacking of the TMO2 slabs. ADF mode images are dominated by the contribution of heavier elements, thus the atomic columns seen in the image are a result of the transition metals in the structure.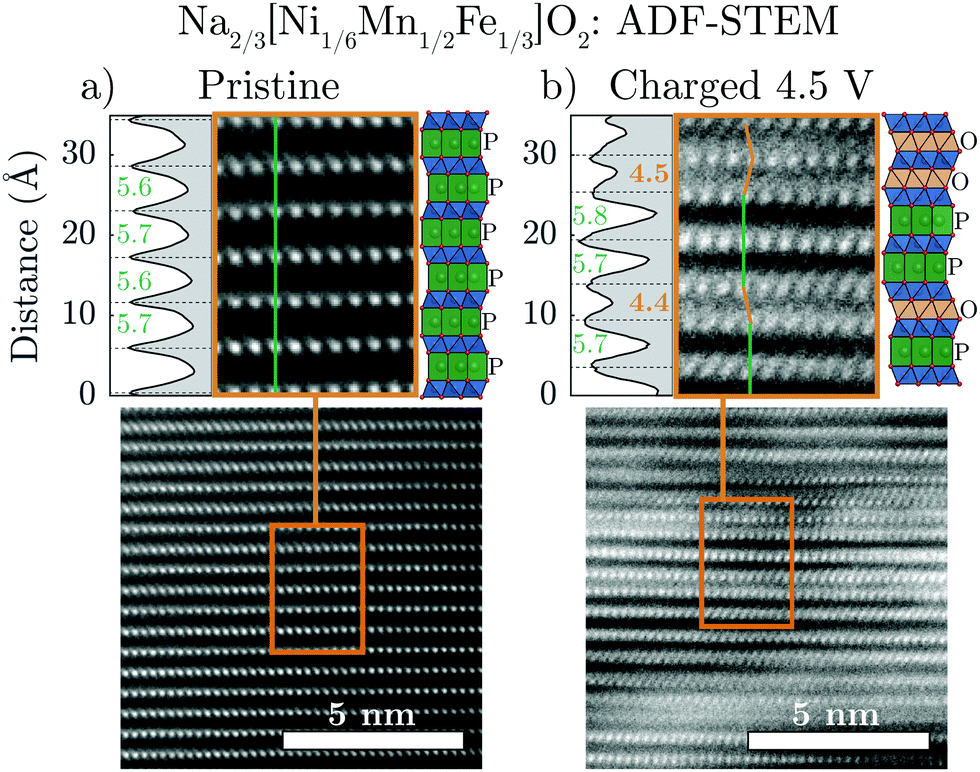 | ||
| Fig. 4 Scanning transmission electron microscopy (STEM) images of (a) pristine and (b) charged to 4.5 V Na2/3[Ni1/6Mn1/2Fe1/3]O2 taken along the [100] projection are shown in annular-dark field (ADF) mode. The highlighted region of each image clearly shows the stacking of the layers which are identified as either P-type (no TM offset: green line) or O-type (TM offset: orange line). The intensity variation is plotted to the left of each highlighted image in order to measure the interlayer distances between adjacent TMO2 slabs. Structural illustrations are also shown to the right of each of the highlighted images. | ||
In the pristine material (Fig. 4a), the TM columns in adjacent layers are aligned along the vertical direction (indicated by the green line). This is consistent with trigonal prismatic Na sites in the interslab space (i.e. P-type layers). In contrast, the image of the sample charged to 4.5 V (Fig. 4b) shows some adjacent layers which are aligned (green line) and others which are offset from one another (orange line). This observation is consistent with the simultaneous presence of P-type and O-type layers in the “Z”-phase and once again discounts the hypothesis that the “Z”-phase is purely of O2-type.
Interestingly, the interlayer distances between the observed P-type and O-type layers in the charged sample differ from one another. The P-type layers have a relatively large interlayer spacing of approximately 5.7 Å, approximately equal to the interlayer spacing of the P2 structure before transformation to the “Z”-phase. On the other hand, the O-type layers have a much smaller interlayer spacing of approximately 4.4 Å matching that of the O2 structure of fully charged Na2/3[Ni1/3Mn2/3]O2. As the Na content in charged O2 phase of Na2/3[Ni1/3Mn2/3]O2 is close to zero, the STEM results of the charged Na2/3[Ni1/6Mn1/2Fe1/3]O2 “Z”-phase strongly suggest that the Na-ions remaining in the structure should be predominately located in the P-type layers. These new insights are important for creating more accurate models of the “Z”-phase.
Ex situ synchrotron XRD
Ex situ synchrotron XRD was carried out on Na2/3[Ni1/3Mn2/3]O2 and Na2/3[Ni1/6Mn1/2Fe1/3]O2 after the materials were charged to 4.5 V, shown in Fig. 5a and b, respectively. For Na2/3[Ni1/3Mn2/3]O2, the majority phase is seen to be the O2 structure although some reflections associated with the P2 phase remain in the diffraction pattern at the end of charge. The O2 reflections, in particular, show dramatic hkl-dependent broadening as a result of the equal probability that the TMO2 slabs glide along two distinct vectors, [1/3 2/3 z] and [2/3 1/3 z], as the structure transforms Na layers containing trigonal prismatic sites (P-type Na layers) to octahedral sites (O-type Na layers).5,17 For comparison to the experimental data, the P2 and O2 diffraction patterns were simulated with the program FAULTS (an extension of DIFFAX), which can adequately model the interlayer disorder caused by such a transition. It is clear that the experimental data is well represented by the two simulated patterns.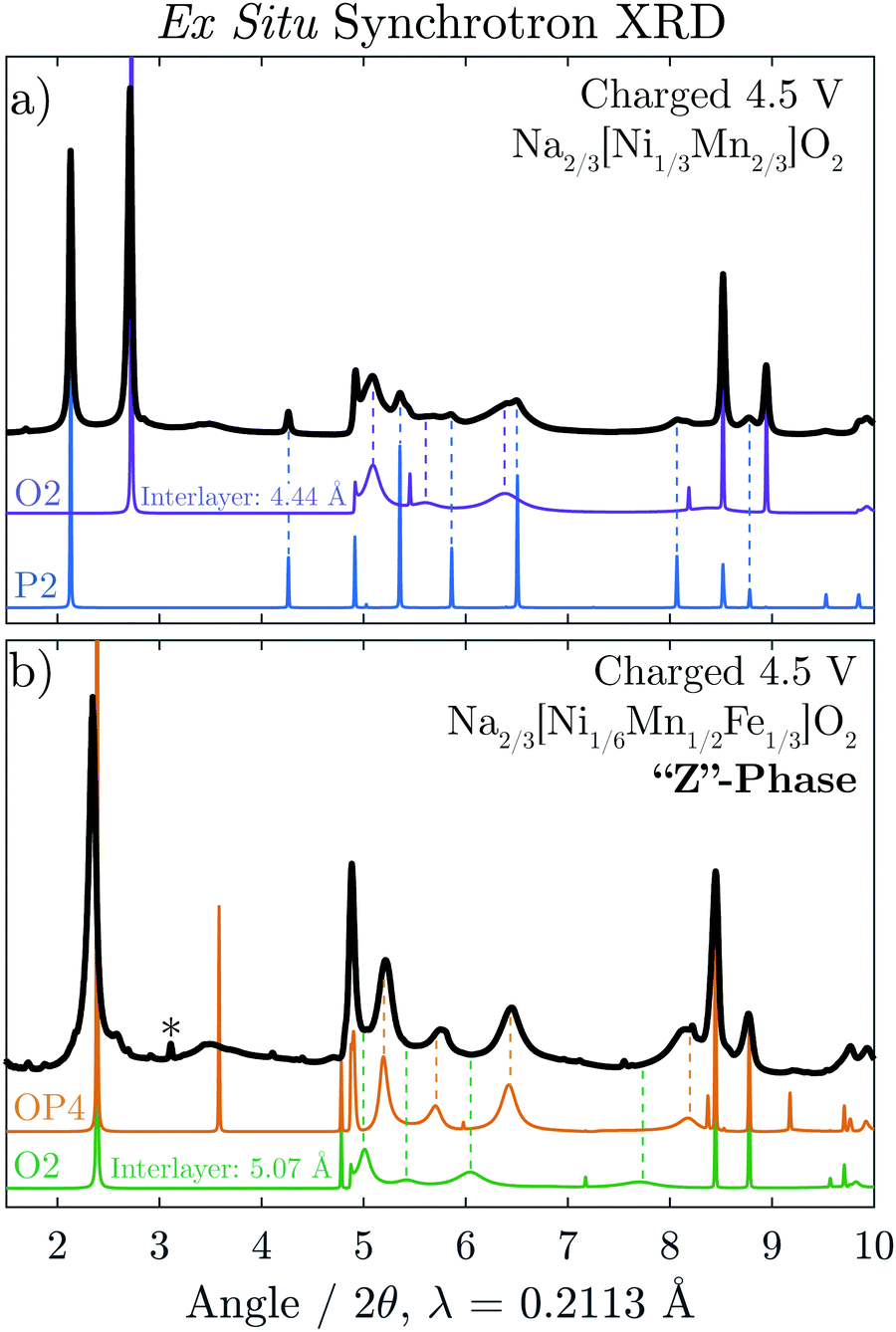 | ||
| Fig. 5 Ex situ synchrotron XRD is shown for (a) charged Na2/3[Ni1/3Mn2/3]O2 and (b) charged Na2/3[Ni1/6Mn1/2Fe1/3]O2 shown in black. In (a), a diffraction pattern for the O2 structure with an interlayer distance of dO-type = 4.44 Å is simulated using FAULTS alongside a simulated diffraction pattern for the P2 structure. For comparison in (b), a diffraction pattern for the OP4 structure is simulated using FAULTS with differing interlayers spacing (dP-type = 5.69 Å and dO-type = 4.44 Å) alongside a simulated diffraction pattern for an O2 structure with uniform interlayer spacing (dO-type = 5.07 Å). | ||
The diffraction pattern of charged Na2/3[Ni1/6Mn1/2Fe1/3]O2 (Fig. 5b) also shows severe hkl-dependent broadening. The average interlayer distance of this phase, indicated by the position of the first intense reflection, is significantly larger than that of the fully de-sodiated O2 phase (≈5.1 Å). As the STEM results indicated the simultaneous presence of P and O-type layers, a diffraction pattern for the OP4 structure (as seen in Fig. 1) was simulated. Our methodology is described in detail in the Experimental Section, but it is important to point out that the assumption was made that the O-type layers are completely vacant of Na+ and for this reason have a smaller interlayer spacing equal to that of the O2 structure observed for O2-[Ni1/3Mn2/3]O2, 4.44 Å. As the octahedral sites in the O2-type layers share a face with high-valence transition metals, it is indeed very likely that all Na is removed from these layers. The P-type layers are modeled with partial Na occupancy of 1/3 Na per formula unit and an interlayer distance equal to that of P2-Na2/3[Ni1/6Mn1/2Fe1/3]O2 immediately before the “Z”-phase transition, 5.69 Å. The OP4 simulated pattern matches the broadened peaks of the “Z”-phase rather well with the exception of the sharp peak at 2θ = 3.6°. Fig. S7 (ESI†), however, shows that the sharpness of this peak reduces dramatically with minimal deviation from the ideal P–O–P–O alternation.
A diffraction pattern for an O2 structure with an interlayer distance of 5.07 Å, equivalent to the average interlayer spacing of the simulated OP4 structure, was also simulated (green). In contrast to the OP4 simulated pattern, the broaden peaks of the O2 pattern clearly do not match that of the “Z”-phase. Together, the ex situ STEM and synchrotron XRD results provide strong evidence that the “Z”-phase (after charge to 4.5 V) is an intergrowth containing both P and O-type layers.
Modelling the “Z”-phase as an evolving intergrowth
Attention can now be turned to the interesting evolution of the “Z”-phase which would appear to move through a solid solution-like regime from the operando XRD. To study this evolution, diffraction patterns were simulated for two intergrowth regimes; from 0–100% P2–OP4 and from 0–100% O2–OP4 and plotted in Fig. 6b and d. Among the simulated patterns in Fig. 6b, the sharpest reflections are associated with the ordered P2, OP4, and O2 structures, as would be expected as these have long range order. The more disordered intergrowth structures between the P2, OP4, and O2 are relatively broader. Fig. 6d shows these same simulated patterns over a wider 2θ range in colour map format. In this plot, it can be seen that the P2 (004) peak splits into the OP4 (006) and (008) peaks moving down along the y-axis. Moreover, the P2 (102) reflection decreases in 2θ as it morphs into the OP4 (103) peak.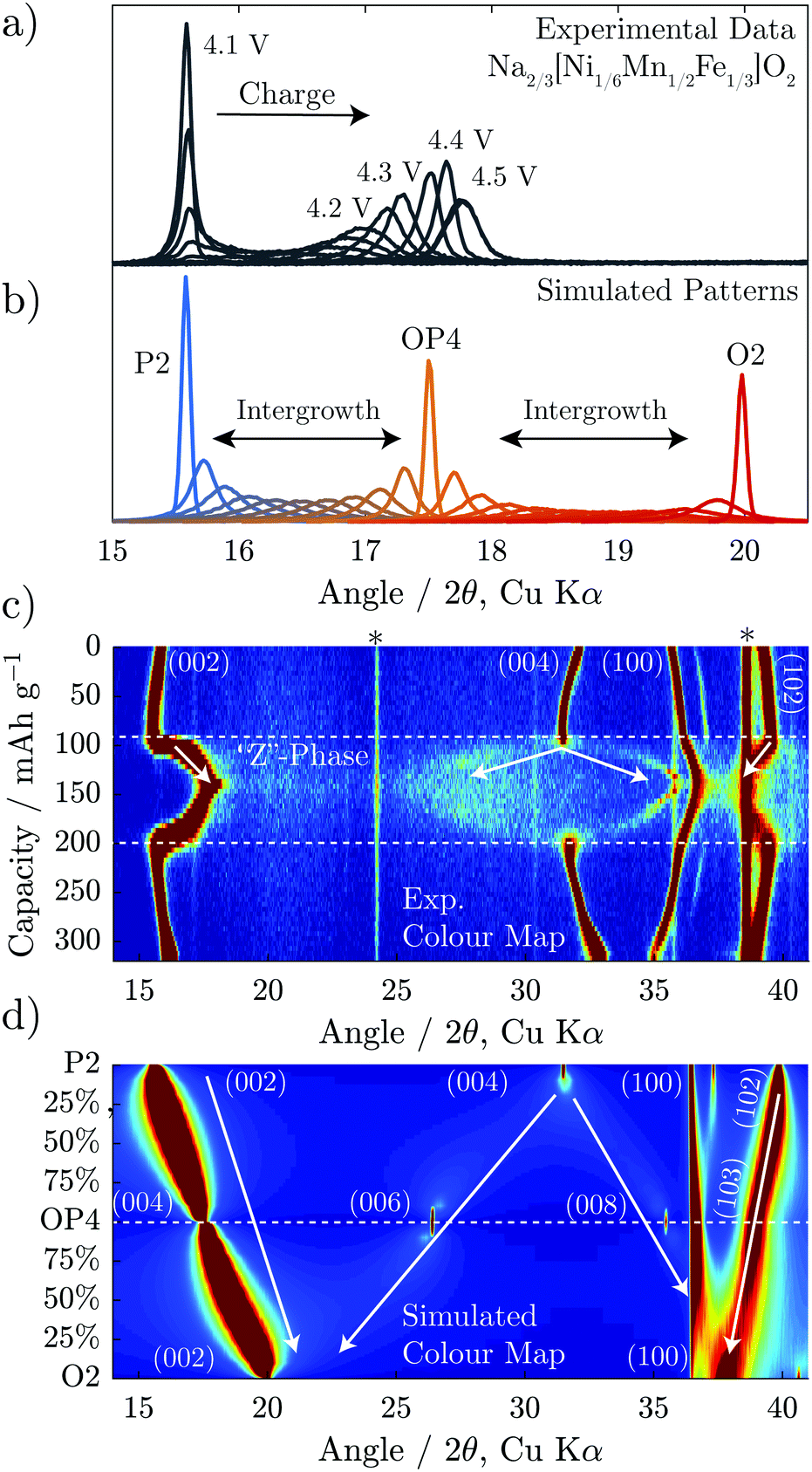 | ||
| Fig. 6 Diffraction patterns are simulated using FAULTS for intergrowth structures composed of P2 and OP4, and OP4 and O2. These are plotted overlaid in (b) and over a wider 2θ range in (d) as a colour map. Experimental data for Na2/3[Ni1/6Mn1/2Fe1/3]O2 (also presented in Fig. 3b) is replotted in (a and c) for comparison to the simulations. | ||
The simulated patterns for the intergrowth structure varying ratios from P2 through OP4 to O2 match well with the evolution of the operando XRD of Na2/3[Ni1/6Mn1/2Fe1/3]O2 shown in Fig. 6a. The first peak of the experimental data sharpens at almost exactly the same 2θ value (≈17.5°) as that of the simulated OP4 phase. Furthermore, at higher and lower voltages, the first peak of experimental patterns is much broader, closely resembling the intergrowth model patterns (Fig. 6a and b). The simulated patterns also explain the splitting of the P2 (004) peak, as it is shown to split into the OP4 (006) and (008) peaks in Fig. 6d. The (002) peak movement to higher 2θ and the (004) peak splitting are both consequences of continuously increasing quantity of O-type layers in the intergrowth structure.
From the above, we see that the “Z”-phase in Na2/3[Ni1/6Mn1/2Fe1/3]O2 is not a solid solution upon Na extraction but instead an evolving intergrowth structure. The evolution of the “Z”-phase on charging progresses through a staging process where Na+ is removed from P2-type layers to form vacant O-type Na layers randomly distributed within the P2 structure. The proportion of such O-type stacking faults increases on charging until it reaches 50% at which point an ordered OP4 phase forms (alternating P and O-stacking). On further charging, more P-type layers glide to form O-type layers resulting in random O-type stacking faults within an otherwise OP4 structure. A choice of two equally probable glide vectors which the TMO2 slabs can traverse to transform from P-type to O-type stacking, causes most of the long-range interlayer order of the structure to be lost. Meanwhile, the average interlayer distance (i.e. 0.5 c-parameter) has a steep and continual decrease as Na+ is removed due to the smaller spacing of vacant O-type layers compared with partially sodiated P-type layers. This gives the misleading appearance of a solid solution region on charging.
57Fe-Mössbauer spectroscopy
In order to achieve a more complete picture regarding the role of Fe in the structural evolution, 57Fe-Mössbauer Spectroscopy (Fe-MS) was employed. Fe-MS measurements were conducted on the Na2/3[Ni1/6Mn1/2Fe1/3]O2 composition at several states of charge. Ex situ samples charged to 4.0 V, 4.2 V, and 4.5 V, respectively, were compared to the pristine material as well as fully discharged samples (1.5 V) after 1 and 5 full charge–discharge cycles. The spectrum of the pristine sample (Fig. 7a) shows a single sharp doublet with hyperfine parameters (Table 1) consistent with high-spin Fe3+ (S = 5/2) in an octahedral environment. | ||
| Fig. 7 57Fe-Mössbauer spectroscopy spectra at various states of charge with blue lines representing a Fe3+ doublet fit, orange lines representing a Fe4+ doublet fit, and the black line representing the total fit. | ||
| CS (mm s−1) | QS (mm s−1) | FWHM (mm s−1) | Relative intensity | |
|---|---|---|---|---|
| * = fixed value. | ||||
| Pristine | 0.34(1) | 0.68(1) | 0.25(1) | 100% (Fe3+) |
| Charged 4.0 V | 0.33(1) | 0.85(1) | 0.24(1) | 90% (Fe3+) |
| 0.14(1) | 0.68(1) | 0.24* | 10% (Fe4+) | |
| Charged 4.2 V | 0.30(1) | 0.75(1) | 0.31(1) | 84% (Fe3+) |
| −0.09(1) | 0.54(1) | 0.31* | 16% (Fe4+) | |
| Charged 4.5 V | 0.30(1) | 0.78(1) | 0.35(1) | 80% (Fe3+) |
| −0.14(1) | 0.58(1) | 0.35* | 20% (Fe4+) | |
| Discharged 1.5 V (1 cycle) | 0.34(1) | 0.69(1) | 0.25(1) | 100% (Fe3+) |
| Discharged 1.5 V (5 cycles) | 0.35(1) | 0.64(1) | 0.25(1) | 100% (Fe3+) |
As the material is charged to 4.0 V, however, an asymmetry weighted toward the left-hand side of the main doublet becomes apparent (Fig. 7b) and is attributed to oxidation of Fe within the transition metal layer to a tetravalent state. This agrees with changes in the XANES Fe K-edge that we reported previously.19 As the material is charged further across the high-voltage plateau, the Fe4+ signal increases in intensity, as seen in the spectra of the samples charged to 4.2 V (Fig. 7d) and 4.5 V (Fig. 7f), respectively, and reaches a spectral contribution of 20% at the end of charge. Additionally, it should be noted that during charging, both the Fe3+ and Fe4+ signals show clear trends of continuously increasing peak widths (FWHM) and decreasing centre shifts (CS), as presented in Table 1. The peak broadening is attributed to a decrease in crystallinity (increased distribution of Fe environments) as a result of the phase transition from the P2 to the “Z”-phase, which is supported by the general peak broadening observed in the operando XRD results, shown above. The change in the CS parameter to more negative values on charging is consistent with an increase in electron density at the Fe nuclei as the Fe–O bonds contract. This agrees with the contraction in the in-plane TM–TM distance (a-parameter) seen in the operando XRD. Moreover, the quadrupole splitting (QS) for the Fe3+ signal is generally larger for the charged samples as compared to the pristine/discharged, indicating an increased distortion of the octahedral environment within the transition metal layers. It should be mentioned that we also attempted to fit an extra signal to assess for the presence of tetrahedrally coordinated Fe that may have formed due to migration of Fe to the interlayer space upon charge, as has been suggested in several recent publications.9,35,36 Such a model did not, however, improve the fit in our case and did not result in logical trends of the hyperfine parameters.
The spectra (Fig. 7c and e) and the corresponding hyperfine parameters (Table 1) for the samples discharged to 1.5 V after 1 and 5 cycles are nearly identical to that of the pristine material, showing a single sharp doublet corresponding to octahedrally coordinated Fe3+. This further highlights the facile reversibility between the P2 and “Z”-phases.
Discussion
It has been demonstrated that the “Z”-phase of Na2/3[Ni1/6Mn1/2Fe1/3]O2, which forms on charging above 4.1 V, is in fact an intergrowth between the P2 and O2 structures with OP4 as an intermediate. Removal of Na+ from the P2 structure occurs by staging, i.e. introducing stacking faults of highly depleted Na+ layers with O-type stacking while neighbouring layers still contain Na+ and P-type stacking. The ratio of random O-type stacking faults within the P2 structure increases on charging passing through an ordered OP4 structure at 50% O-type stacking faults and continuing towards a pure O2 structure, as shown in Fig. 8. This constantly evolving intergrowth on charge gives the superficial appearance of a solid solution upon first inspection of the operando XRD. It was previously hypothesized that the continuous decrease of the average interlayer spacing was a result of progressive Fe migration;9 however, we found no evidence of any tetrahedral Fe species through 57Fe-Mössbauer spectroscopy at the top of charge or after cycling. Moreover, other layered oxide cathodes undergo a similar staging mechanism when they are severely depleted of alkali ions. For example, when LiCoO2 is charged beyond 4.5 V vs. Li+/Li it too contracts along the c-axis as it transforms from O3 to the H1–3 phase, which contains lithiated O3-type layers alternating with vacant O1-type layers.37,38 When more Li+ is removed from this compound, the average interlayer spacing further decreases until the pure O1 phase is formed.39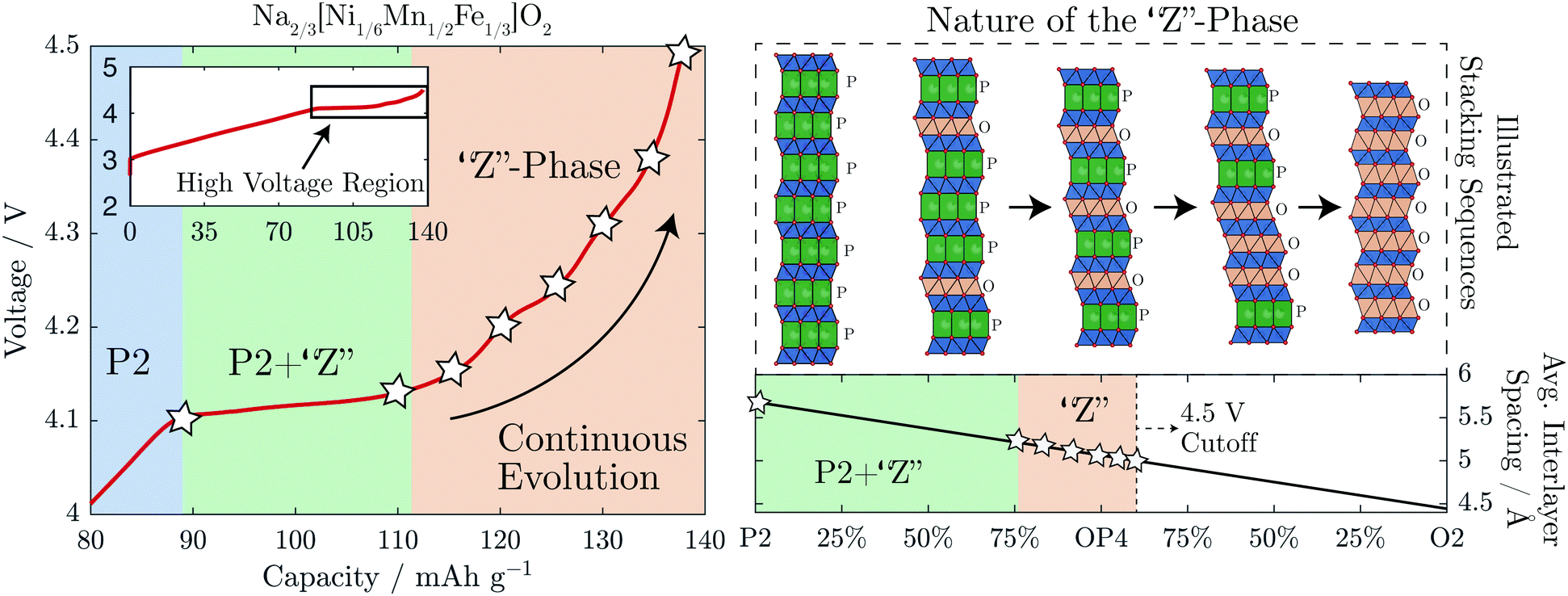 | ||
| Fig. 8 A summary of the proposed phase transformation mechanism for the “Z”-phase is shown. Above 4.1 V, Na is extracted via the introduction of O-type stacking faults into the P2 structure decreasing the average interlayer spacing of the structure. The inset on the top left shows the whole charging curve, highlighting the high voltage region to which the main plot relates. | ||
It is still not clear why some P2-NaMO2 compounds transform directly to the O2-phase on charge while others evolve via the intergrowth mechanism described here. In the case of Na2/3[Ni1/3Mn2/3]O2, perhaps the transition metal and/or Na-ordering play a role in changing the energetics. “Z”-phase formation is beneficial compared to O2, as the overall volume changes are smaller in magnitude and less abrupt, leading to better capacity retention.19
Conclusion
The nature of the “Z”-phase has been elusive for some time due to minimal long-range interlayer order and its complicated evolution on charge. We have been able to show here that the “Z”-phase is an evolving intergrowth of P2 and O2 with OP4 as an intermediate which varies its Na content by a staging phenomenon; Na removal is compensated by the introduction of vacant O-type stacking faults into the P2 structure. Much of the long-range order is lost because these O-type stacking faults can occur by the layers gliding in two directions with equal probability.Many P2 compounds have been shown to transform into the “Z”-phase, especially those containing Fe, when charged to high voltages. The exploitation of low-cost Fe, which is active in NaMO2 cathodes, is a major advantage of the Na-ion chemistry over analogous Li-ion compounds. Therefore, correctly discerning the structural identity of the “Z”-phase is a key step forward in the development of Earth-abundant, high capacity P2-NaMO2 materials as cathodes for Na-ion batteries.
Author contributions
JWS synthesized the compounds in the study. Experimental data was collected by JWS (synchrotron XRD, electrochemistry, ICP-OES), AS (Fe-MS), TE (Fe-MS), LH (Fe-MS), JB (operando XRD), JGL (STEM) and LCG (ex situ synchrotron XRD). JWS analysed the data with vital contributions from AS, NTR, RAH, MRR, UM, and PGB. JWS, AS, and PGB wrote the paper with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Claire Villevieille for experimental assistance and discussions and for providing access to the operando XRD cell. PGB is indebted to the Engineering and Physical Sciences Research Council (EPSRC), including the SUPERGEN Energy Storage Hub [EP/L019469/1], Enabling Next Generation Lithium Batteries [EP/M009521/1], Henry Royce Institute for capital equipment [EP/R010145/1] and the Faraday Institution All-Solid-State Batteries with Li and Na Anodes [FIRG007, FIRG008] for financial support. We gratefully acknowledge the Science and Technology Facilities Council (STFC) for access to neutron beamtime at ISIS, and also for the provision of sample preparation, POLARIS facilities. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors acknowledge use of characterisation facilities within the David Cockayne Centre for Electron Microscopy, Department of Materials, University of Oxford, alongside financial support provided by the Henry Royce Institute.References
- K. Kubota, N. Yabuuchi, H. Yoshida, M. Dahbi and S. Komaba, MRS Bull., 2014, 39, 416–422 CrossRef CAS.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS.
- Y. Yoda, K. Kubota, H. Isozumi, T. Horiba and S. Komaba, ACS Appl. Mater. Interfaces, 2018, 10, 10986–10997 CrossRef CAS PubMed.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef CAS.
- D. H. Lee, J. Xu and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- B. Mortemard de Boisse, D. Carlier, M. Guignard, L. Bourgeois and C. Delmas, Inorg. Chem., 2014, 53, 11197–11205 CrossRef CAS PubMed.
- E. Talaie, V. Duffort, H. L. Smith, B. Fultz and L. F. Nazar, Energy Environ. Sci., 2015, 8, 2512–2523 RSC.
- N. Tapia-Ruiz, W. M. Dose, N. Sharma, H. Chen, J. Heath, J. W. Somerville, U. Maitra, M. S. Islam and P. G. Bruce, Energy Environ. Sci., 2018, 11, 1470–1479 RSC.
- U. Maitra, R. A. House, J. W. Somerville, N. Tapia-Ruiz, J. G. Lozano, N. Guerrini, R. Hao, K. Luo, L. Jin, M. A. Pérez-Osorio, F. Massel, D. M. Pickup, S. Ramos, X. Lu, D. E. McNally, A. V. Chadwick, F. Giustino, T. Schmitt, L. C. Duda, M. R. Roberts and P. G. Bruce, Nat. Chem., 2018, 10, 288–295 CrossRef CAS PubMed.
- S. Kumakura, Y. Tahara, S. Sato, K. Kubota and S. Komaba, Chem. Mater., 2017, 29, 8958–8962 CrossRef CAS.
- J. Yoshida, E. Guerin, M. Arnault, C. Constantin, B. Mortemard de Boisse, D. Carlier, M. Guignard and C. Delmas, J. Electrochem. Soc., 2014, 161, A1987–A1991 CrossRef CAS.
- E. Talaie, S. Y. Kim, N. Chen and L. F. Nazar, Chem. Mater., 2017, 29, 6684–6697 CrossRef CAS.
- C. Marino, E. Marelli and C. Villevieille, RSC Adv., 2017, 7, 13851–13857 RSC.
- L. Liu, X. Li, S.-H. Bo, Y. Wang, H. Chen, N. Twu, D. Wu and G. Ceder, Adv. Energy Mater., 2015, 5, 1500944 CrossRef.
- K. Kubota, S. Kumakura, Y. Yoda, K. Kuroki and S. Komaba, Adv. Energy Mater., 2018, 8, 1703415 CrossRef.
- K. Kubota, Y. Yoda and S. Komaba, J. Electrochem. Soc., 2017, 164, A2368–A2373 CrossRef CAS.
- J. W. Somerville, R. A. House, N. Tapia-Ruiz, A. Sobkowiak, S. Ramos, A. V. Chadwick, M. R. Roberts, U. Maitra and P. G. Bruce, J. Mater. Chem. A, 2018, 6, 5271–5275 RSC.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- L. Jones, H. Yang, T. J. Pennycook, M. S. J. Marshall, S. Van Aert, N. D. Browning, M. R. Castell and P. D. Nellist, Adv. Struct. Chem. Imaging, 2015, 1, 8 CrossRef.
- P. Bleith, H. Kaiser, P. Novák and C. Villevieille, Electrochim. Acta, 2015, 176, 18–21 CrossRef CAS.
- M. Casas-Cabanas, M. Reynaud, J. Rikarte, P. Horbach and J. Rodríguez-Carvajal, J. Appl. Crystallogr., 2016, 49, 2259–2269 CrossRef CAS.
- M. M. J. Treacy, J. M. Newsam and M. W. Deem, Proc. R. Soc. A, 1991, 433, 499–520 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K. Lagarec and D. Rancourt, Nucl. Instrum. Methods Phys. Res., Sect. B, 1997, 129, 266–280 CrossRef CAS.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- B. L. Henke, E. M. Gullikson and J. C. Davis, At. Data Nucl. Data Tables, 1993, 54, 181–342 CrossRef CAS.
- Z. Lu, R. a. Donaberger and J. R. Dahn, Chem. Mater., 2000, 12, 3583–3590 CrossRef CAS.
- G. Singh, N. Tapia-Ruiz, J. M. Lopez del Amo, U. Maitra, J. W. Somerville, A. R. Armstrong, J. Martinez de Ilarduya, T. Rojo and P. G. Bruce, Chem. Mater., 2016, 28, 5087–5094 CrossRef.
- P. F. Wang, Y. You, Y. X. Yin, Y. S. Wang, L. J. Wan, L. Gu and Y. G. Guo, Angew. Chem., Int. Ed., 2016, 55, 7445–7449 CrossRef CAS PubMed.
- W. K. Pang, S. Kalluri, V. K. Peterson, N. Sharma, J. Kimpton, B. Johannessen, H. K. Liu, S. X. Dou and Z. Guo, Chem. Mater., 2015, 27, 3150–3158 CrossRef CAS.
- K. Kubota, T. Asari, H. Yoshida, N. Yaabuuchi, H. Shiiba, M. Nakayama and S. Komaba, Adv. Funct. Mater., 2016, 26, 6047–6059 CrossRef CAS.
- Q. Huang, M. L. Foo, J. W. Lynn, H. W. Zandbergen, G. Lawes, Y. Wang, B. H. Toby, A. P. Ramirez, N. P. Ong and R. J. Cava, J. Phys.: Condens. Matter, 2004, 16, 5803–5814 CrossRef CAS.
- N. Yabuuchi, H. Yoshida and S. Komaba, Electrochemistry, 2012, 80, 716–719 CrossRef CAS.
- R. Kataoka, K. Kuratani, M. Kitta, N. Takeichi, T. Kiyobayashi and M. Tabuchi, Electrochim. Acta, 2015, 182, 871–877 CrossRef CAS.
- A. Van der Ven, M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1998, 145, 2149–2155 CrossRef CAS.
- Z. Chen, Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1604–A1609 CrossRef CAS.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114–1123 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Refinement of Synchtrotron PXRD. Operando XRD of Na ordering in Na1/2[Ni1/3Mn2/3]O2. Operando XRD and analysis for Na2/3[Ni1/4Mn7/12Fe1/6]O2. See DOI: 10.1039/c8ee02991a. Supporting research data has been deposited in the Oxford Research Archive and is available under this DOI: http://10.5287/bodleian:9Rajw9NDY. |
| ‡ Present address: Etteplan Sweden AB, SE-171 54 Solna, Sweden. |
| § Present address: Dept. of Chemistry, University of Lancaster, Lancaster, UK. |
| ¶ Present address: Dept. de Ingeniería y Ciencia de los Materiales y del Transporte, Universidad de Sevilla (EPS-US), Sevilla, Spain. |
| || Present address: Inst. of Physical Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |