
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly stable, Au/Ru heterobimetallic photoredox catalyst with a [2.2]paracyclophane backbone†
Daniel M.
Knoll
 a,
Christoph
Zippel
a,
Zahid
Hassan
a,
Martin
Nieger
b,
Patrick
Weis
d,
Manfred M.
Kappes
cd and
Stefan
Bräse
*ae
a,
Christoph
Zippel
a,
Zahid
Hassan
a,
Martin
Nieger
b,
Patrick
Weis
d,
Manfred M.
Kappes
cd and
Stefan
Bräse
*ae
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
bDepartment of Chemistry, University of Helsinki, P.O. Box 55 A.I. Virtasen aukio 1, 00014 University of Helsinki, Finland
cInstitute of Nanotechnology, Karlsruhe Institute of Technology, Herman-vonHelmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-HaberWeg 2, 76131 Karlsruhe, Germany
eInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Herman-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 15th November 2019
Abstract
We report the synthesis and catalytic application of a highly stable distance-defined Au/Ru heterobimetallic complex. [2.2]Paracyclophane serves as a backbone, holding the two metal centers in a spatial orientation and metal–metal fixed distance. The Au/Ru heterobimetallic complex is highly stable, easily accessible and exhibits promising catalytic activity in a visible-light mediated dual Au/Ru Meyer–Schuster rearrangement.
Catalysis with mononuclear complexes employs sophisticated ligands to fine-tune the coordination sphere of the metal centre to achieve the desired reactivity.1–5 An elegant way to introduce electronic flexibility into the catalytic cycle and additionally enable cooperative or tandem catalysis is found in heterobimetallic complexes.6–8 Here, two possibilities arise. In one mode, one metal fulfils the classic activation of the substrate, whereas its catalytic activity is enhanced by the second metal centre. Alternatively, both metal centers can work together in a truly cooperative fashion, as has been demonstrated for metallosalens.9,10 This latter concept is widely found in enzyme bio-catalysis, which rationalizes their outstanding activity and selectivity.11–13 Despite the vast number of examples for enzymatic catalysis and even their diverse applications in industry, only a few examples for defined binuclear organometallic complexes which exhibit cooperative effects are known.14–16
Enabling chemical synthesis with low-energy visible light can be used to promote photoredox reactions. One of the highest achievements of chemistry as a field is the use of sustainable practices employing low-cost and nature abundant visible-light energy to drive reactions instead of thermal, electrical or mechanical energy. Visible-light mediated photoredox catalysis has seen a rapid growth over the past 20 years.17–22 Similarly, gold catalysis has gained new prominence for driving efficient and selective processes under mild conditions,23–25 especially in combination with photoredox catalysis.26
However, to date, heterobimetallic complexes with molecular architectures of spatial orientation and metal–metal fixed bond distance have seldom been explored. Exploring distance-defined visible light absorbing coordination complexes based on PCP as a new platform might allow to study the effect of metal–metal distance and their electronic communication on cooperativity. The unique “bent and battered” structure of [2.2]paracyclophane (PCP) features planar chirality, high rigidity and transannular electronic communication which makes this scaffold an ideal playground for the synthesis of distance-defined bimetallic complexes.38 Disubstituted PCPs with two substituents located on different decks display four different regioisomers that are named pseudo-geminal, -ortho, -meta and -para shown in Fig. 1. The short distance between the two decks of the [2.2]paracyclophane causes an overlap of the π-system and leads to a transannular electronic communication.27–29 The through-space conjugation effect is especially interesting in the case of the pseudo-para and -meta isomers. In these isomers, no direct metal–metal interaction is possible and the electronic communication via PCP between the two metal centres can be studied selectively.
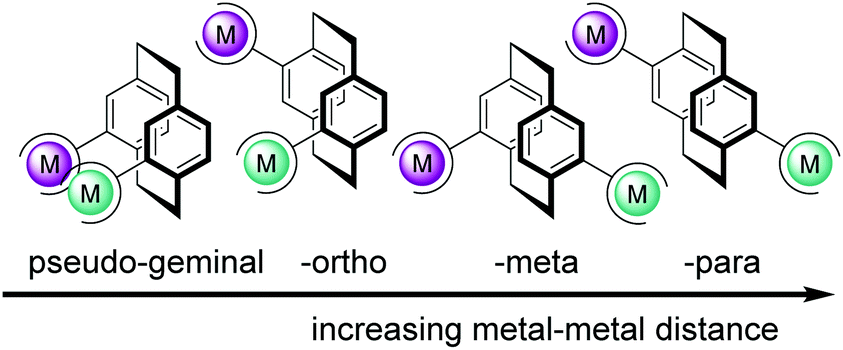 | ||
| Fig. 1 Design of heterobimetallic PCPs with increasing M–M distance. | ||
To evaluate the catalytic performance of the newly synthesized heterobimetallic PCP Au/Ru complexes, skeletal rearrangements are good probes and the visible-light mediated arylative Meyer–Schuster rearrangement also is known to be a good “acid test” to examine stability and catalytic potential. We investigated a well-studied reaction in which both metals participate in the mechanism instead of one metal only being the “fine-tuning” for the other.
Results and discussion
Inspired by recent investigations on the visible-light mediated arylative Meyer–Schuster rearrangement by the groups of Glorius, Luna and Shin,30–32 we focused on a new heterobimetallic version of the photoredox catalyst system employed. The gold(I) catalyst, PPh3AuCl can be incorporated by replacing one of the phenyl rings with PCP. We changed the Ru(bpy)32+ cation for Ru(bpy)2ppy+ because of easier synthetic access,5 a wider spectral excitation window and its suitability as a photosensitizer.33 Furthermore, the reaction studied in detail entails oxidative quenching of the excited state of the photocatalyst, which is facilitated for cyclometalated ruthenium complexes when compared to their non-cyclometalated counterparts.20To be able to compare the influence of PCP on the outcome of catalysis, we first set out to prepare the respective PCP-Au and PCP-Ru, resembling the coordination environments found in the later heterobimetallic complex (Scheme 1).
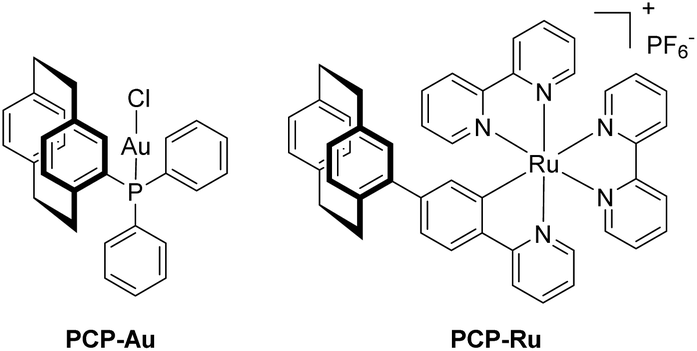 | ||
| Scheme 1 Monometallic complexes PCP-Au and PCP-Ru. | ||
Starting from PCP dibromide 1, selective monolithiation5 and reaction with chlorodiphenylphosphine gave 4-bromo-16-diphenylphosphoryl[2.2]paracyclophane (2). The high selectivity of the monolithiation allows for a further functionalization at the 4-position via a cross-coupling reaction. Under optimized Suzuki–Miyaura reaction conditions for PCP,5 a new C–C bond to 2-phenylpyridine was formed. After two steps, compound 3 was obtained which bears the (oxidized) phosphine as a selective binding site for Au(I) and the 2-phenylpyridine moiety for the incorporation of Ru(II). Reduction of the phosphoryl moiety with trichlorosilane to the free phosphine 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 directly followed by auration provided us with the Au(I) complex 5. Initial efforts to introduce ruthenium into the 2-phenylpyridine moiety were met with failure, presumably due to the use of commonly employed AgBF4.33 However, a milder and silver-free method35 was successfully applied to obtain the cationic heterobimetallic complex pAuRu in 27% yield over 5 steps (Scheme 2). Due to the chiral-at-metal centre and the inherent planar chirality of heterodisubstituted PCP, pAuRu was obtained as a mixture of diastereomers. Despite our efforts, we did not succeed in separation of the diastereomeric mixture by HPLC.
34 directly followed by auration provided us with the Au(I) complex 5. Initial efforts to introduce ruthenium into the 2-phenylpyridine moiety were met with failure, presumably due to the use of commonly employed AgBF4.33 However, a milder and silver-free method35 was successfully applied to obtain the cationic heterobimetallic complex pAuRu in 27% yield over 5 steps (Scheme 2). Due to the chiral-at-metal centre and the inherent planar chirality of heterodisubstituted PCP, pAuRu was obtained as a mixture of diastereomers. Despite our efforts, we did not succeed in separation of the diastereomeric mixture by HPLC.
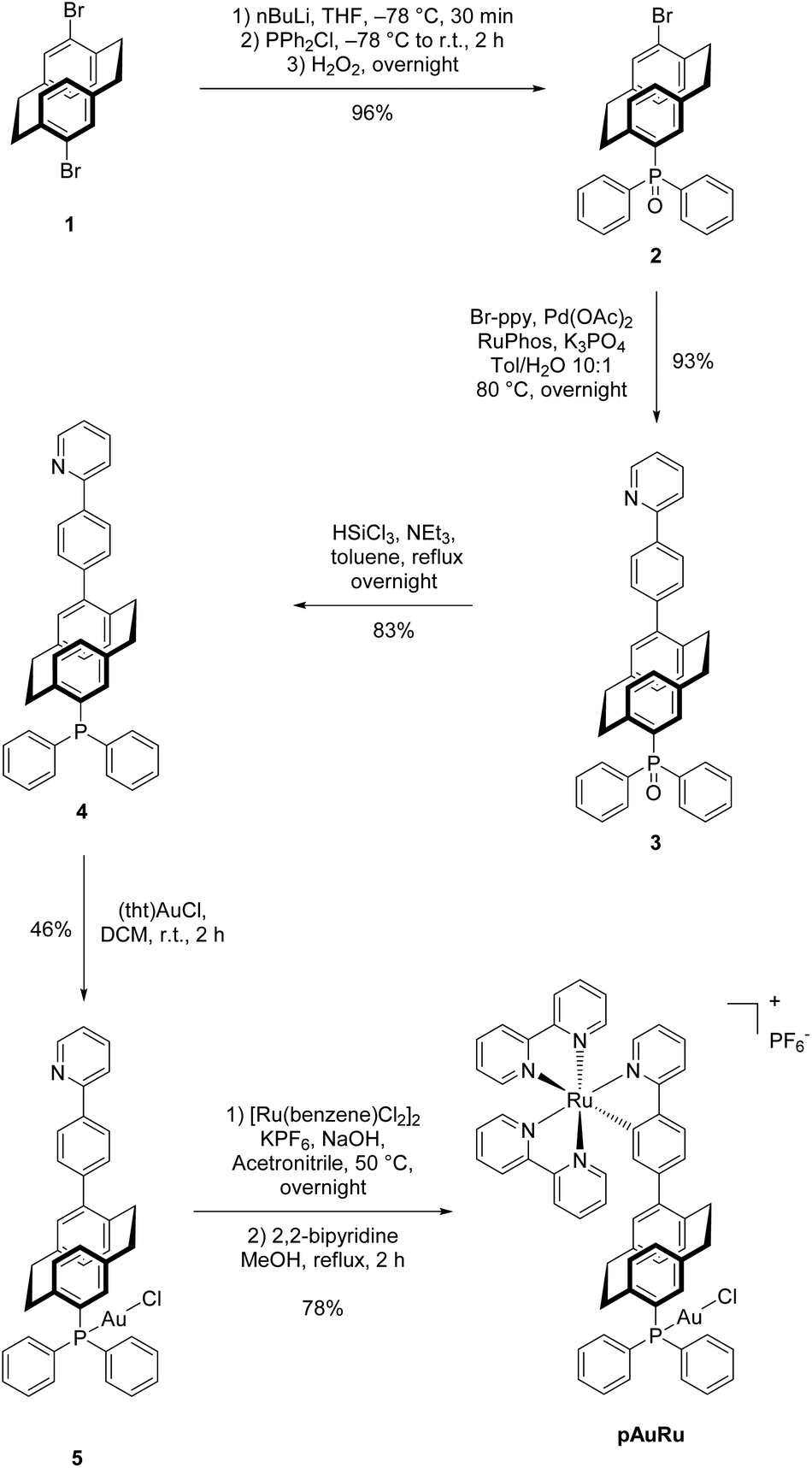 | ||
| Scheme 2 Synthetic access to target complex pAuRu. | ||
Absorption spectra of the complexes PPh3AuCl (red), [Ru(bpy)2(ppy)]PF6 (blue), PCP-Au (black), PCP-Ru (orange) and pAuRu (green) were obtained in MeOH/MeCN, 3:1 at 298 K (Fig. 2). Absorption maxima in the UV-vis spectra and molar extinction coefficients are summarized in Table 1.
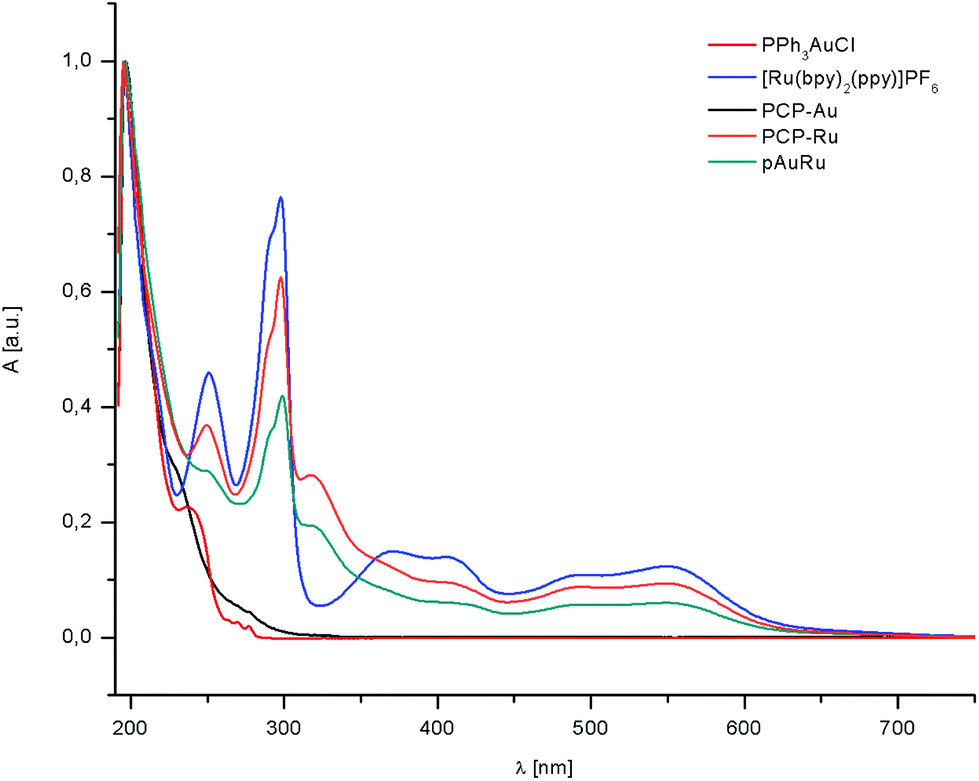 | ||
| Fig. 2 Absorption spectra in MeOH/MeCN, 3:1 at 298 K. | ||
| Entry | Compound | λ absmax [nm] (ε [104 M−1 cm−1]) |
|---|---|---|
|
a MeOH/MeCN, 3:1, 298 K.
|
||
| 1 | [Ru(bpy)2(ppy)]PF6 | 371 (0.99) |
| 410 (0.91) | ||
| 490 (0.71) | ||
| 552 (0.81) | ||
| 2 | PCP-Ru | 366 (1.21) |
| 414 (0.89) | ||
| 490 (0.83) | ||
| 552 (0.89) | ||
| 3 | pAuRu | 366 (0.87) |
| 414 (0.62) | ||
| 490 (0.60) | ||
| 552 (0.63) | ||
The absorption bands in the ultraviolet region below 350 nm arise from spin allowed ligand centered (LC) π → π* transition. As the only feature, PCP–Ru and pAuRu exhibit an additional LC band at 320 nm which arises from the PCP backbone. Examination of the lower energy absorption bands reveals a large spectral envelope which arises from the cyclometalated phenylpyridine ligand. This also causes additional features below 300 nm which can be assigned to intraligand π → π* transitions. In the spectra, two bands which are consistent with metal-to-ligand charge transfer (MLCT) are found in the region between 350–450 nm and 450–600 nm.
The first band can be assigned to an MLCT from the ruthenium center to the cyclometalating phenylpyridyl ligand, whereas the band at higher wavelengths is primarily localized on the polypyridyl ligands.
The absorption of PCP-Ru and pAuRu is slightly red-shifted compared to [Ru(bpy)2(ppy)]PF6. The heterobimetallic complex pAuRu also shows lower molar extinction coefficients than the monometallic counterparts. This trend can be observed already when going from [Ru(bpy)2(ppy)]PF6 to PCP-Ru. Therefore, we postulate that this decrease in molar exctinction coefficient as well as the red-shift is due to the increase in electron density by the introduction of the electron-rich PCP moiety and the additional electron-rich phosphine respectively.
Excitation of the pAuRu complex at wavelengths defined by the absorption maxima at 485 nm and 548 nm produces emission bands that are substantially shifted to longer wavelengths. At both excitation wavelengths, strong fluorescence at 795 nm is observed. Upon excitation at 485 nm, the compound shows additional emission at 543 nm and 507 nm (Fig. 3).
 | ||
| Fig. 3 Emission spectra at absorption maxima 485 nm (black) and at 548 nm (red) for pAuRu in MeOH/MeCN, 3:1 at 298 K. | ||
No single crystals of pAuRu suitable for X-ray diffraction were obtained. However, the direct precursor 5 did provide us with suitable single crystals and confirmed the molecular structure as shown in Fig. 4. The phenylpyridine moiety (plane through C17–C22) is twisted out of the PCP deck plane (C12–C16) by 41.8°. This angle is significantly larger than comparable PCP pyridyl scaffolds.36 The P–Au and Au–Cl distances are in the expected range with bond lengths of 2.236 Å and 2.292 Å respectively.37 The gold atom is almost linearly coordinated, displaying a gold(I) typical twofold coordination mode [P–Au–Cl: 176.2°]. The P–Au–Cl “tail” is turned towards the PCP scaffold rather than the phenyl substituents to form a 98.6° torsion angle (C5–C4, P–Au).
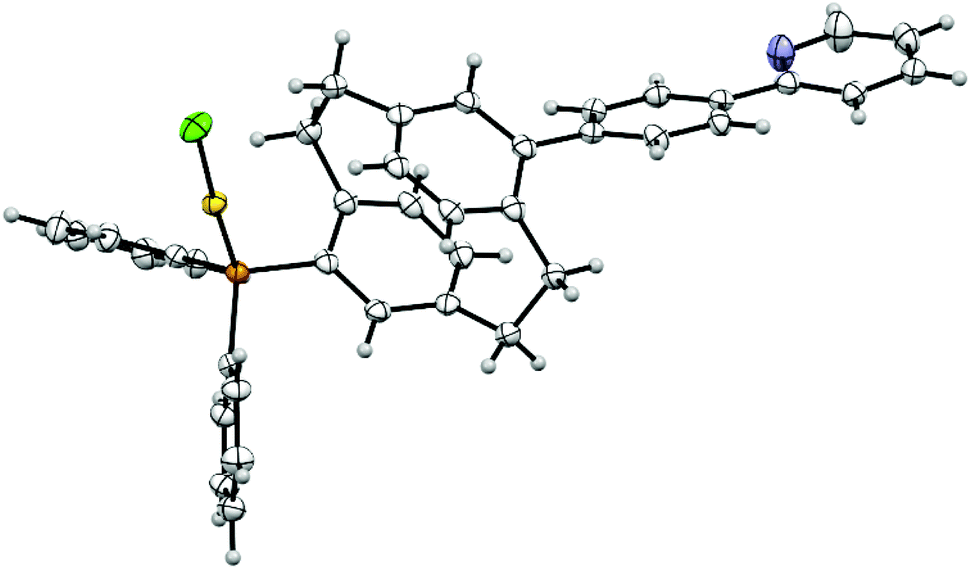 | ||
| Fig. 4 Molecular structure of complex 5 (orange – P, yellow – Au, green – Cl, blue – N, displacement parameters are drawn at 30% probability level). | ||
Catalysis application
The model reaction to test pAuRu was chosen to be the visible-light mediated arylative Meyer–Schuster rearrangement (MSR) which was recently published by three groups: Glorius, Luna and Shin.30–32 The Shin group reported a Au(I) to Ru(II) ratio of 1:1 and was therefore most fit for comparison with our heterobimetallic complex (Scheme 3).
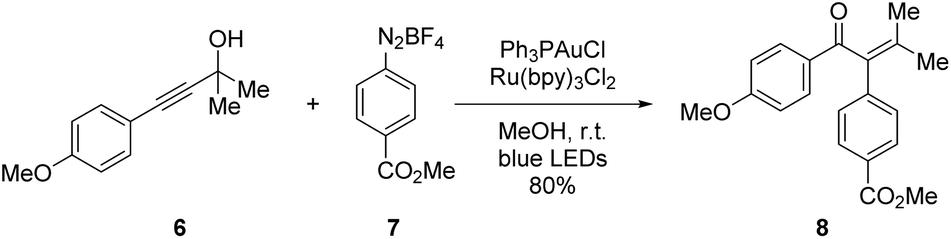 | ||
| Scheme 3 Literature reaction conditions reported by Shin et al. | ||
With our setup, the conditions reported by Shin et al., afforded the arylated enone 11 in 76% yield (Table 2, entry 1). During our investigations, we found that a mixture of methanol and acetonitrile provided overall more consistent results (entry 2). We attribute this to the complete solution of the diazonium salt 10 which is added in four-fold excess and is not well-soluble in methanol.
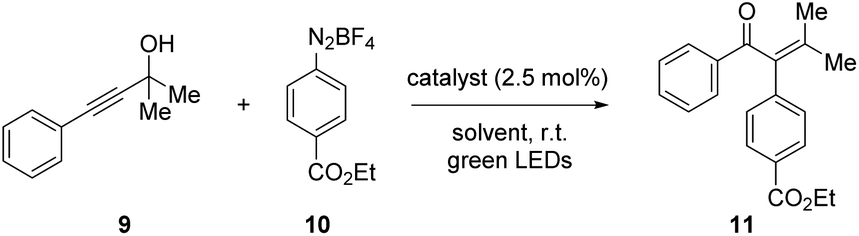
In order to exclude possible electronic effects induced by the different coordination sphere that arises from the 2-phenylpyridine unit within the pAuRu, the literature procedure by Shin et al. was additionally conducted with the monocationic Ru(bpy)2(ppy)PF6 instead of the dicationic Ru(bpy)3Cl2 (entry 3). Furthermore, we investigated the impact of the sterically demanding PCP backbone on the overall reaction. The corresponding monometallic PCP derivatives PCP-Au and PCP-Ru were applied in a test reaction (entry 4). All of those modifications on the catalytic system consistently provided the same results which leads to the conclusion that neither the different coordination sphere that results from exchanging a bpy ligand with a ppy ligand, nor the PCP backbone influence the overall reactivity.
The single-molecule, heterobimetallic pAuRu complex could successfully catalyze the Meyer–Schuster rearrangement and afforded the arylated enone 11 in 63% yield with a reaction time of 25 minutes (see ESI† for kinetics) (entry 5). A comparison of reaction kinetics for the monometallic PCP-Au and PCP-Ru with those measured for pAuRu show no significant difference in induction period or conversion rate. While no mechanistic investigations were performed, this suggests that the reaction follows the mechanistic proposals in literature.30–32
Since both steric and electronic influences that may result from the slightly different coordination environment through the PCP backbone can be excluded, the observed drop in yield when using pAuRu could suggest a through-space metal–metal communication and deactivation. This communication would be enabled by the π-conjugated PCP backbone. However, other factors (e.g. reduced extinction coefficient) could not be excluded and might influence the outcome of this reaction as well.
The lack of understanding about how heterobimetallic complexes are to be designed for applications in cooperative catalysis demands new frameworks on which to study these complexes systematically. In this work we demonstrated the synthesis and catalytic viability of the heterobimetallic complex pAuRu featuring [2.2]paracyclophane as a rigid backbone. The performance of pAuRu complex in photoredox catalysis was evaluated and compared to the monometallic approach.
The remarkable stability, high yielding and facile synthetic access of pAuRu make it an invaluable first-of-its-kind PCP heterobimetallic complex. The investigated arylative Meyer–Schuster rearrangement reaction is successfully catalyzed in 63% yield with pAuRu. Compared with the respective monometallic complexes, a drop in yield was observed, suggesting metal-to-metal interaction by through-space transannular communication.
We believe that pAuRu is the first in a line of distance-modulated bimetallic complexes exploiting the isomeric geometry features of PCP. We hope this will aid in the design and the understanding of metal–metal interactions in bimetallic complexes in catalysis applications. Synthetic routes towards further isomers and PCP-based other heterobimetallic complexes are underway in our laboratories.
Conflicts of interest
No conflicts of interest to declare.Notes and references
- M. Schlangen, D. Schröder and H. Schwarz, Angew. Chem., Int. Ed., 2007, 46, 1641–1644 CrossRef CAS PubMed.
- G. B. Shul'pin, Dalton Trans., 2013, 42, 12794–12818 RSC.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895–904 CrossRef CAS PubMed.
- A. Padwa and D. J. Austin, Angew. Chem., Int. Ed. Engl., 1994, 33, 1797–1815 CrossRef.
- D. M. Knoll, H. Šimek, Z. Hassan and S. Bräse, Eur. J. Org. Chem., 2019, 6198–6202 CrossRef CAS.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- J. M. Lee, Y. Na, H. Han and S. Chang, Chem. Soc. Rev., 2004, 33, 302–312 RSC.
- R. M. Haak, S. J. Wezenberg and A. W. Kleij, Chem. Commun., 2010, 46, 2713–2723 RSC.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- M. Comotti, C. D. Pina and M. Rossi, 9th Int. Symp. Act. Dioxygen Homog. Catal. Oxid., 2006, vol. 251, pp. 89–92.
- K. Worm, F. Chu, K. Matsumoto, M. D. Best, V. Lynch and E. V. Anslyn, Chem. – Eur. J., 2003, 9, 741–747 CrossRef CAS PubMed.
- K. Lai, K. I. Dave and J. R. Wild, J. Biol. Chem., 1994, 269, 16579–16584 CAS.
- W. Wang, L. Zhao, H. Lv, G. Zhang, C. Xia, F. E. Hahn and F. Li, Angew. Chem., Int. Ed., 2016, 55, 7665–7670 CrossRef CAS PubMed.
- M. Böhmer, F. Kampert, T. T. Y. Tan, G. Guisado-Barrios, E. Peris and F. E. Hahn, Organometallics, 2018, 37, 4092–4099 CrossRef.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2014, 115, 28–126 CrossRef PubMed.
- J. Busch, D. M. Knoll, C. Zippel, S. Bräse and C. Bizzarri, Dalton Trans., 2019, 48, 15338–15357 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- C. R. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, John Wiley & Sons, 2018 Search PubMed.
- J. Twilton, C. (Chip) Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448–2462 RSC.
- S. Taschinski, R. Döpp, M. Ackermann, F. Rominger, F. de Vries, M. F. S. J. Menger, M. Rudolph, A. S. K. Hashmi and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2019, 58, 16988–16993 CrossRef CAS PubMed.
- M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed.
- A. Molina-Ontoria, M. Wielopolski, J. Gebhardt, A. Gouloumis, T. Clark, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2011, 133, 2370–2373 CrossRef CAS PubMed.
- I. Majerz and T. Dziembowska, J. Phys. Chem. A, 2016, 120, 8138–8147 CrossRef CAS PubMed.
- Y. Morisaki and Y. Chujo, Macromolecules, 2003, 36, 9319–9324 CrossRef CAS.
- A. Tlahuext-Aca, M. N. Hopkinson, R. A. Garza-Sanchez and F. Glorius, Chem. – Eur. J., 2016, 22, 5909–5913 CrossRef CAS PubMed.
- J. Um, H. Yun and S. Shin, Org. Lett., 2016, 18, 484–487 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, E. Busto and A. Luna, Adv. Synth. Catal., 2016, 358, 1526–1533 CrossRef CAS.
- P. G. Bomben, K. C. D. Robson, P. A. Sedach and C. P. Berlinguette, Inorg. Chem., 2009, 48, 9631–9643 CrossRef CAS PubMed.
- P. J. Pye, K. Rossen, R. A. Reamer, R. P. Volante and P. J. Reider, Tetrahedron Lett., 1998, 39, 4441–4444 CrossRef CAS.
- A. D. Ryabov, V. S. Sukharev, L. Alexandrova, R. Le Lagadec and M. Pfeffer, Inorg. Chem., 2001, 40, 6529–6532 CrossRef CAS PubMed.
- C. Braun, E. Spuling, N. B. Heine, M. Cakici, M. Nieger and S. Bräse, Adv. Synth. Catal., 2016, 358, 1664–1670 CrossRef CAS.
- C. Sarcher, A. Lühl, F. C. Falk, S. Lebedkin, M. Kühn, C. Wang, J. Paradies, M. M. Kappes, W. Klopper and P. W. Roesky, Eur. J. Inorg. Chem., 2012, 2012, 5033–5042 CrossRef CAS.
- (a) Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947–6963 RSC; (b) Z. Hassan, E. Spuling, D. M. Knoll and S. Bräse, Angew. Chem., Int. Ed., 2019, 58, 2–17 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, crystallographic data and kinetic data. CCDC 1958713 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt04366g |
| This journal is © The Royal Society of Chemistry 2019 |