
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeutral, cationic and anionic organonickel and -palladium complexes supported by iminophosphine/phosphinoenaminato ligands†‡
Tomás G.
Santiago
 ,
Carmen
Urbaneja
,
Eleuterio
Álvarez
,
Elena
Ávila
,
Pilar
Palma
and
Juan
Cámpora
*
,
Carmen
Urbaneja
,
Eleuterio
Álvarez
,
Elena
Ávila
,
Pilar
Palma
and
Juan
Cámpora
*
Instituto de Investigaciones Químicas, CSIC-Universidad de Sevilla. CIC-Cartuja, c/Américo Vespucio, 49, 41092, Sevilla, Spain. E-mail: campora@iiq.csic.es
First published on 19th November 2019
Abstract
We report a series of organometallic nickel and palladium complexes containing iminophosphine ligands R2PCH2C(Ph) = N-Dipp (Dipp = 2,6-diisopropylphenyl; R = iPr, La; R = Ph, Lb; and R = o-C6H4OMe, Lc), synthesized by ligand exchange or oxidative addition reactions, and we investigate the capacity of such ligands to undergo reversible deprotonation to the corresponding phosphinoenaminato species. In the attempted ligand exchange reaction of the nickel bis(trimethylsilyl)methyl precursor [Ni(CH2SiMe3)2Py2] with Lb, the iminophosphine acts as a weak acid rather than a neutral ligand, cleaving one of the Ni–C bonds, to afford the phosphinoenaminato complex [Ni(CH2SiMe3)(L′b)(Py)] (L′b = conjugate base of Lb). We disclose a general method for the syntheses of complexes [Ni(CH2SiMe3)(L)(Py)]+ (L = La, Lb or Lc), and demonstrate that iminophosphine deprotonation is a general feature and occurs reversibly in the coordination sphere of the metal. By studying proton exchange reactions of the cation [Ni(CH2SiMe3)(Lb)(Py)]+ with bases of different strength we show that the conjugate phosphinoenaminato ligand in [Ni(CH2SiMe3)(L′b)(Py)] is a base with strength comparable to DBU in THF. The acyl group in the functionalized aryl complex [Ni(p-C6H4COCH3)(Br)(La)] does not interfere in the iminophosphine deprotonation with NaH. The latter reaction affords the unusual anionic hydroxide species [Ni(p-C6H4COCH3)(OH)(L′a)]−Na+, which was isolated and fully characterized.
Introduction
Iminophosphines are very attractive ligands used in the development of new homogeneous catalysts for various processes,1 because their modular design2 allows systematic and independent variation of the steric and electronic properties of the P and N donor centers, and the structural properties of the ligand backbone. Among the most typical examples of this class of hybrid ligands are iminophosphines with a single carbon spacer connecting the phosphine and imino functionalities.3,4 Such iminophosphine ligands often bear bulky o,o′-disubstituted N-aryl substituent groups, like the species A shown in Scheme 1. This substitution pattern is a common feature in many types of catalysts, particularly in those used for late transition metal olefin poylmerization.5 In these catalysts, the o,o′ aryl groups control the molecular weights of the products.6 Not surprisingly, nickel and palladium iminophosphine complexes have attracted attention as ethylene polymerization or oligomerization catalysts but unfortunately, their activities are significantly lower than those of the well-known α-diimine catalysts, and their products tend to exhibit broad or bimodal molecular weight distributions.7,8 One of the potential problems identified in hybrid iminophosphine catalyst systems is the weakly acidic character of the hydrogen atoms in the iminophosphine backbone. As shown in Scheme 1, iminophosphine complexes are deprotonated by strong bases to afford phosphinoenaminates.9 Therefore, it was suggested that polymerization catalysts using this type of ligand could be also deprotonated by aluminum alkyls used as co-catalysts, turning the active species into more electron-rich phosphinoenaminate species (shown as B in Scheme 1).8 However, the weak Brønsted acidity of iminophosphine ligands could be turned into an advantage for other applications. In recent years, there has been growing interest in ligand–metal cooperative behaviour as an alternative strategy to extend or improve the capacities of metals in catalysis.10 Among the various sorts of metal–ligand cooperation, reversible ligand protonation/deprotonation is a central step in some important processes involving hydrogen transfer reactions, such as transfer hydrogenation,11 or aerobic oxidations.10a This context has brought some enolizable ligands such as bis(phosphinomethyl)pyridine (PNP) pincers, closely related to iminophosphines, under the spotlight.10b,e Although enolizable iminophosphine chelates have been less extensively investigated in this regard, Fryzuk has recently demonstrated their ability to promote hydrogen activation through metal–ligand cooperative behaviour in Ir and Ru complexes.12 In addition, first-row transition metal complexes with deprotonated phosphinoamidinates, iminophosphine-type ligands, have found many useful applications in catalysis.13 These reports suggest that iminophosphines could also induce similar cooperative phenomena when coordinated to 16e, square-planar complexes of Ni, enhancing H-transfer reactions that are usually facile in the chemistry of the heavier group 10 metals, particularly Pd.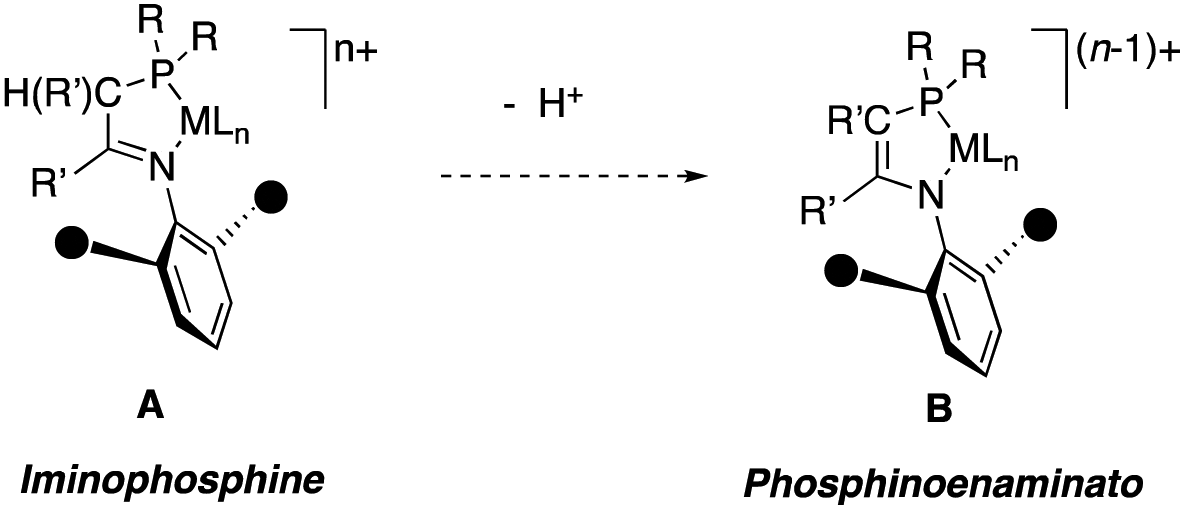 | ||
| Scheme 1 The relationship between iminophosphine and phosphinoenaminato complexes. | ||
One of our current interests focuses on the ability of Ni complexes to emulate Pd in fundamental processes that are relevant to catalysis. Whereas a number of organometallic complexes of the heavier group 10 elements (Pd and Pt) supported by deprotonable iminophosphine ligands have been reported, the chemistry of their analogues was examined in less detail.9 Some years ago, we reported a series of Ni and Pd complexes containing non-enolizable iminophosphinite hybrid ligands,14 similar to iminophosphines, except that the linker between the PR2 unit and the imino group is an oxygen atom. These are excellent ligands for both Ni and Pd organometallic complexes. This prompted us to develop pathways to well-defined organometallic derivatives of Ni ligated by weakly acidic iminophosphines, and to compare these Ni derivatives to their Pd counterparts. Herein we report our initial results on this topic, including the synthesis of organometallic species supported by three bulky iminophosphine ligands, and for the first time we illustrate reversible iminophosphine deprotonation chemistry on the coordination sphere of Ni(II).
Results and discussion
Iminophosphine ligands
In this study, we chose to focus on the iminophosphine ligands La, Lb and Lc, with an enolizable skeleton based on acetophenone-imine, and differing only in the PR2 fragment (R = i-propyl, phenyl or 2-anisyl, respectively). Such ligands are readily obtained using a one-pot procedure that involves sequential deprotonation of the acetophenone imine, and electrophilic phosphination of the enaminate anion with the corresponding chlorophosphine (Scheme 2). Ligand Lb has been reported in the literature before.9b Deprotonation of acetophenone imines is usually accomplished with lithium diisopropylamide (LDA), but we found that the same can be conveniently achieved using one equivalent of t-BuLi at −80 °C, avoiding the need to generate the lithium amide. The ligands were separated from lithium chloride by extraction in hexane. La and Lb are obtained as pale-yellow oils, but Lc is obtained as a yellow solid that is easier to handle and store. The NMR spectra of all three ligands show that these exist as mixtures of cis/trans isomers, with no detectable amounts of enamine tautomers. | ||
| Scheme 2 Syntheses of iminophosphine ligands. | ||
Ligand exchange reactions
Methods for the synthesis of organometallic derivatives of ligands containing active (acidic) CHn groups usually avoid transmetallation reactions with organolithium or organomagnesium reagents, as these behave as strong bases susceptible to deprotonation of the coordinated ligand. Instead, ligand exchange reactions are usually preferred for this purpose.15 In our previous study, we found that pyridine-stabilized nickel dialkyls, as well as equivalent pyridine or COD-bearing dialkyl and metallacyclic palladium complexes, are very convenient precursors that provide access to a plethora of organometallic derivatives via exchange reactions.16 Since (trialkylsilyl)methyl derivatives often exhibit enhanced stability compared with “normal” alkyl derivatives, we set to synthesize a series of bis(trimethylsilyl)methyl derivatives of Ni and Pd, starting out from the precursor complexes [Ni(CH2SiMe3)2(Py)2]17a (1) and [Pd(CH2SiMe3)2(COD)]17b (2), respectively. Interestingly, the reactions of these nickel and palladium precursors with ligands La, Lb and Lc had divergent results (Scheme 3).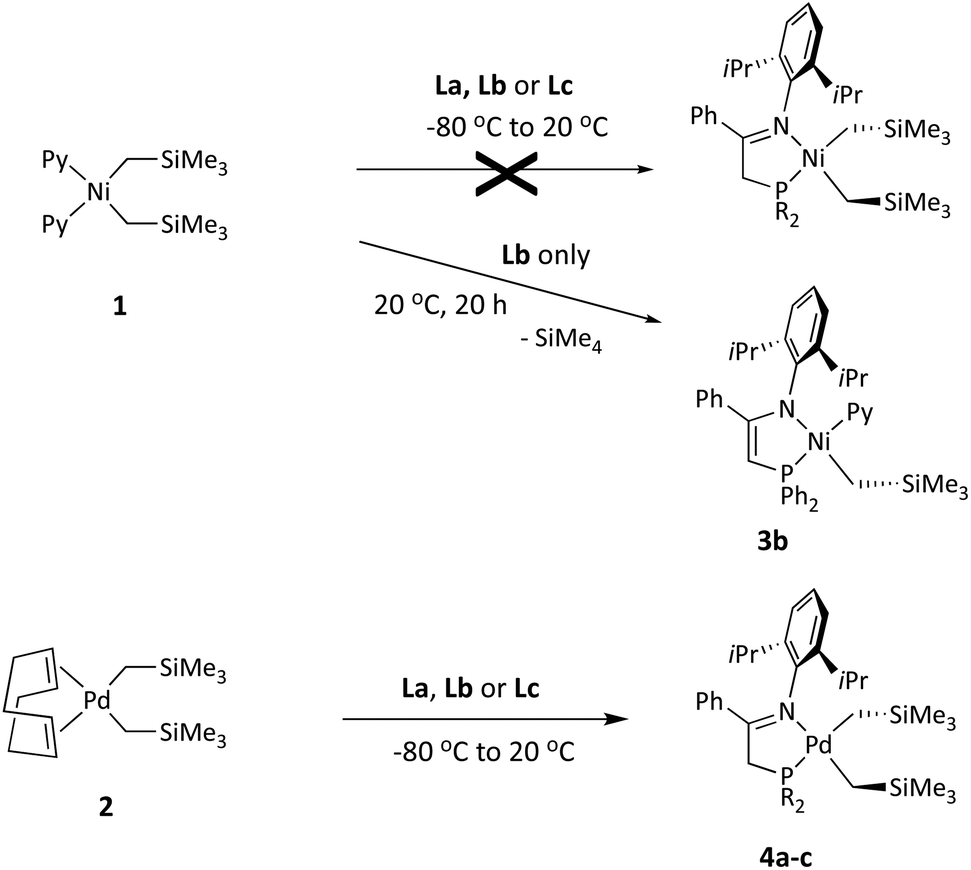 | ||
| Scheme 3 Divergent reactivity of precursors 1 and 2 with iminophosphine ligands. | ||
As discussed below in more detail, the reactions of precursor 1 with La–Lc fail to produce the expected dialkylnickel derivatives. Only in the case of Lb, a different type of product (3b, Scheme 3 and Fig. 1) could be isolated after the reaction mixture was allowed to evolve for 20 h at room temperature. In contrast, the exchange reactions of the iminophosphine ligands with the Pd precursor 2 proceed as expected affording the corresponding dialkyls 4a–c, which were successfully isolated as dark brown solids. Although essentially quantitative conversions were observed in the NMR spectra of the crude reaction mixtures, the high solubility of the products in hydrocarbon solvents (particularly 4a), combined with some thermal instability in solution, hampered their crystallization, thus reducing their final isolated yields. The dialkyl derivative 4c is thermally more stable and quality crystals suitable for X-ray diffraction could be grown; its crystal structure is shown in Fig. 2. The crystal structures of simple Pd(II) dialkyls are not particularly frequent, and the CSD database only contains a couple of examples stabilized with hybrid P,N donors.9a,18 As expected, the stronger trans influence of the P atom is reflected in a significant lengthening of the Pd–C39 bond with regard to the bond in trans to the nitrogen (2.103(6) vs. 2.046(6) Å). The concurrent trans effect could be translated into a significant kinetic labilization of the alkyl sitting in trans to the P donor.
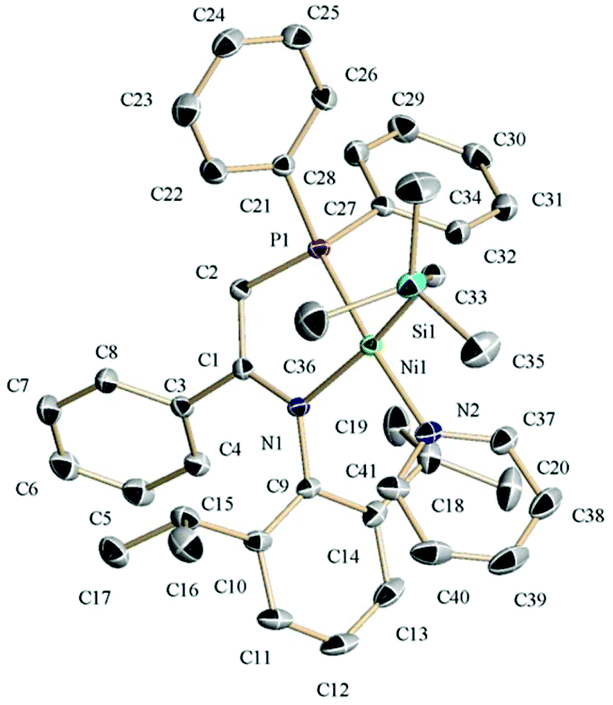 | ||
| Fig. 1 The crystal structure of 3b with 50% probability thermal ellipsoids. The selected bond distances (Å) and angles (°): Ni(1)–C(33), 1.9593(13); Ni(1)–P(1), 2.1153(4); Ni(1)–N(1), 2.0015(11); Ni(1)–N(2), 1.9552(11); C(1)–N(1), 1.3530(16); C(1)–C(2), 1.3842(17); C(33)–Ni(1)–N(2), 89.91(5); and P(1)–Ni(1)–N(1), 85.33(3); torsion N(1)–C(1)–C(2)–P(1), 5.76(17); dihedral between DiPP ring and plane N(1)C(1)Ni(1), 68.4; dihedral between Ph ring and plane C(1)N(1)C(2), 46.2; and dihedral between planes Ni(1)C(33)N(2) and Ni(1)N(1)P(1), 6.4. | ||
 | ||
| Fig. 2 The crystal structure of 4c with 50% probability thermal ellipsoids. The selected bond distances (Å) and angles (°): Pd(1)–C(35), 2.046(6); Pd(1)–C(39), 2.103(6); Pd(1)–N(1), 2.235(5); Pd(1)–P(1), 2.3017(17); C(1)–N(1), 1.301(8); C(1)–C(2), 1.505(8); C(35)–Pd(1)–C(39), 88.0(3); and P(1)–Pd(1)–N(1), 78.50(14); torsion N(1) = C(1)–C(2)–P(1), 41.3(7); dihedral between DiPP ring and plane N(1)C(1)Pd(1), 71.0; dihedral between Ph ring and plane C(1)N(1)C(2), 44.5; and dihedral between planes Pd(1)C(35)C(39) and Pd(1)N(1)P(1), 4.9. | ||
As mentioned above, the reaction of the nickel dialkyl precursor with iminophosphines La–Lc follows an unexpected course. 31P{1H} NMR monitoring showed that the reactions initially afford mixtures of several P-containing species that slowly evolve over a time lapse of ca. one day. Only for Lb the 31P{1H} spectra of the reaction mixture gradually simplified into a single P-containing species that was eventually isolated and identified as the monoalkylnickel phosphinoenaminate complex 3b. The identity of 3b was confirmed by its crystal structure shown in Fig. 1. The molecule exhibits a single alkyl ligand that, as deduced from the solution NMR data, is placed in cis to the P donor, while the pyridine unit occupies the trans position. The anionic phosphinoenaminate ligand is almost planar (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–P, dihedral = 5.6°) and the chelate lacks the characteristic puckering observed in the structure of 4c. The flat geometry of the chelate ring and the C1–C2 and C1–N1 bond lengths within (ca. 1.38 and 1.35 Å, respectively) are typical of a delocalized enamine-type system.
C–P, dihedral = 5.6°) and the chelate lacks the characteristic puckering observed in the structure of 4c. The flat geometry of the chelate ring and the C1–C2 and C1–N1 bond lengths within (ca. 1.38 and 1.35 Å, respectively) are typical of a delocalized enamine-type system.
The complex outcome of the reactions of the dialkyl 1 with iminophosphine ligands was not entirely unexpected. In previous research we found that, whilst exchange reactions of pyridine-stabilized dialkyls [MR2Py2] (M = Ni and Pd; and R = Me, CH2Ph, CH2SiMe3, etc.) with various N,N bidentate donors were in general clean and quantitative, the reaction of 1 with the bulky α-diimine ligand MeC(N–DiPP)–C(N–DiPP)CMe (DiPP = 2,6-diisopropylphenyl) was incomplete, which we attributed to the steric crowding when two CH2SiMe3 groups are simultaneously attached to the relatively small Ni atom.16 Very likely, the reaction of 1 with bulky iminophosphines is also sterically disfavored. This allows that a slow but irreversible process like proteolytic cleavage of a Ni–C bond by the acidic ligand may become competitive, this being selective only in the case of 3b. Nevertheless, the successful isolation of the latter as a stable complex inspired us to achieve the goal of synthesizing its analogues with ligands L′a (3a) and L′c (3c). To this end, we devised a general strategy that would begin with the syntheses of cationic iminophosphine monoalkyl-pyridine derivatives [Ni(CH2SiMe3)(L)(Py)]+ (L = La, Lb or Lc), followed by the removal of one of the weakly acidic H protons from L with a strong base, as reported by Green and Pascu for several Pd and Pt complexes.9 To prepare the desired cationic precursors, we initially considered the reaction of 1 with the conjugate acid of the corresponding ligand, i.e., HL+. Disappointingly, attempts to generate the triflate salt [HLb]+OTf− from Lb and one equivalent of trifluoromethanesulfonic (triflic) acid led to decomposition and a phosphorus-free product was obtained instead, presumably the iminium salt [PhC(NHDiPP)Me]+OTf−. Consequently, we adopted the alternative plan depicted in Scheme 4. Its key feature is a less hindered alkylnickel precursor complex: the tris-pyridine monoalkyl cation 5. It is worth mentioning that no Ni(II) monoalkyl species ligated uniquely by pyridine co-ligands has been reported to date. As shown in Scheme 4, this compound is accessible by reacting 1 with one equivalent of pyridinium triflate. Complex 5 can be isolated in high yield as an analytically pure greenish-yellow solid and, in contrast to the parent dialkyl 1, it is fairly stable in dichloromethane solution. Its 1H NMR and 19F spectra show broad signals at room temperature, which is consistent with rapid exchange with the labile pyridine in trans to the alkyl, while the pair of ligands in the cis position are static, or exchange at a much slower rate. The lability of the pyridine ligand was also noticed in the ESI-MS spectrum of this compound, which only shows a faint molecular ion signal ([Ni(CH2SiMe3)Py3]+, m/z = 382.1), and two much stronger signals corresponding to the loss of one and two pyridine ligands ([Ni(CH2SiMe3)Py2]+ and [Ni(CH2SiMe3)Py]+, m/z = 303.1 and 224.1, respectively), with correct isotope peak distributions.
 | ||
| Scheme 4 General approach to the syntheses of nickel phosphinoenaminate complexes. | ||
As shown in Scheme 4, complex 5 reacts with one equivalent of either La, Lb or Lc, cleanly affording the corresponding cationic complexes 6a–6c, which were isolated as analytically pure products and fully characterized. Their NMR spectra showed that they exist in a single geometric configuration, except 6c that was obtained as a mixture of P,C cis (major) and trans (minor) isomers. The crystal structure of 6b was determined (Fig. 3) for comparison with its neutral analogue 3b. Except for the logical differences within the phosphinoenaminate/iminophosphine systems, the structures of 6b and 3b show a remarkable similarity, both providing nearly identical coordination environments of the Ni(II) center. Thus, albeit the overall positive charge would be expected to strengthen metal–ligand interactions in the cationic derivative, both Ni–N bonds are marginally longer in the cation 6b than in neutral 3b, whilst both Ni–C distances are nearly the same. Only the dative Ni–P bond exhibits a slight shortening in 6b, and both complexes exhibit a similar degree of tetrahedral distortion at the Ni center. The similarity of the coordination sphere of Ni in the cationic and neutral complexes suggests that the influence of the protonated/deprotonated state of the P–N unit is mostly limited to the PN ligand, and has only a minor influence on the electronic state of the metal atom.
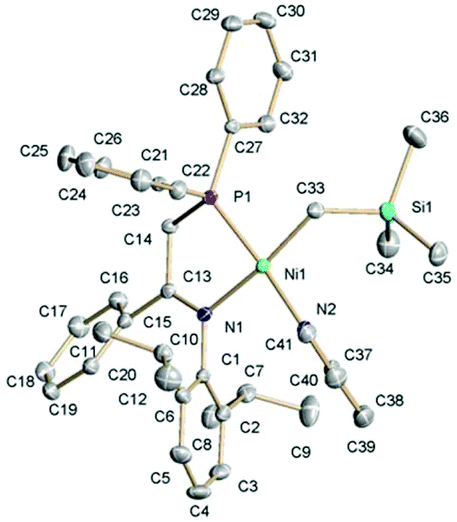 | ||
| Fig. 3 The crystal structure of 6b with 50% probability thermal ellipsoids. The selected bond distances (Å) and angles (°): Ni(1)–C(33), 1.9512(17); Ni(1)–P(1), 2.0943(4); Ni(1)–N(1), 2.0329(13); Ni(1)–N(2), 1.9572(13); C(13)–N(1), 1.298(2); C(13)–C(14), 1.510(2); C(33)–Ni(1)–N(2), 90.48(6); and P(1)–Ni(1)–N(1), 84.26(4); torsion N(1)–C(13)–C(14)–P(1), 16.53(17); dihedral between DiPP ring and plane N(1)C(13)Ni(1), 72.2; dihedral between Ph and plane C(13)N(1)C(14), 41.6; and dihedral between planes Ni(1)C(33)N(2) and Ni(1)N(1)P(1), 12.8. | ||
A suspension of solid NaH in THF deprotonates complexes 6a–c, cleanly affording the corresponding phosphinoenaminates 3a–c. The heterogeneous reaction is slow and takes several hours to complete. However, it is also very selective, cleanly affording a single product in each case, even if the cationic precursor (6c) existed as a cis/trans mixture. In addition, we also investigated the deprotonation of 6b with a set of bases of different strength (see the last point in this section, “reversibility of acid–base reactions”). The spectra of 3b obtained by this method are undistinguishable from those of the sample isolated from the reaction of 1 with Lb.
Oxidative addition of aryl halides to zero-valent Ni and Pd iminophosphine species
Oxidative addition of aryl halides and sulfonates to Ni(0) and Pd(0) complexes is a key step in many catalytic reactions leading to C–C or C-heteroatom bond formation. Although a large number of Ni(II) and Pd(II) aryl complexes containing various ligands (phosphine, N-heterocyclic carbenes, etc.) have been synthesized by oxidative addition reactions, this type of reaction has been rarely used with iminophosphines.19–21 In the case of Ni complexes, the availability of convenient sources of Ni(0) such as Ni(COD)2 considerably facilitates the generation of zero-valent precursors. The oxidative addition of stabilized phosphonium ylides has been used to prepare phenyl-nickel enaminate complexes.19 However, to the best of our knowledge, the oxidative addition of aryl halides or sulfonates to Ni(0)-iminophosphine complexes has not been hitherto demonstrated. Several examples of this type of reaction involving Pd(0) have been reported, using either Pd(DBA)2 (DBA = dibenzylideneacetone) in the presence of the iminophosphine ligand20 or pre-formed Pd(0) species.21 However, these methodologies are usually complicated by the difficulty of removing DBA. In recent years, complex 2 has been used as a versatile source of Pd(0), much as a synthetic equivalent of Ni(COD)2, without the practical drawbacks of DBA complexes.22 Considering these antecedents, we deemed it useful to check whether iminophosphines La–Lc are competent ligands to support oxidative addition reactions leading to stable organometallic complexes of both Ni and Pd.We first investigated the oxidative addition of 4-acetylphenyl halides or triflate, p-AcOC6H4X (X = Br, Cl and OTf), to Ni(0). Preliminary studies showed that iminophosphines La–Lc interact only weakly with Ni(COD)2 in THF at room temperature, forming blue coloured solutions whose 31P{1H} NMR spectra indicate the presence of substantial amounts of free ligands. Therefore, we combined equimolar amounts of Ni(COD)2, ligand (La, Lb or Lc) and the corresponding aryl halide/triflate in THF at low temperatures (−80 °C), and then allowed the mixtures to react at room temperature (Scheme 5).Upon warming, the solutions become dark brown, evolving to somewhat clearer brownish-orange tones within 60–90 min. The 31P{1H} NMR spectra of the reaction mixtures indicate that oxidative additions proceed cleanly for X = Br or Cl, affording in each case a single P-containing species. A workup of these solutions affords the corresponding oxidative addition products as yellow solids in good yields. The spectroscopic properties of these complexes are fully consistent with their expected geometries, with their NMR spectra showing that the functionalized 4-acetylphenyl fragment is incorporated into the Ni(II) complex and is placed in cis to the phosphine donor. These conclusions were confirmed by the crystal structure of 7b (Fig. 4).
 | ||
| Scheme 5 Oxidative addition of aryl halides and triflate to Ni(0)/L combinations. | ||
 | ||
| Fig. 4 The crystal structure of 7b with 50% probability thermal ellipsoids. The selected bond distances (Å) and angles (°): Ni(1)–C(33), 1.8855(19); Ni(1)–P(1), 2.1058(6); Ni(1)–N(1), 2.0255(16); Ni(1)–Br(1), 2.3239(3); C(13)–N(1), 1.289(3); C(13)–C(14), 1.516(3); C(33)–Ni(1)–Br(1), 89.32(6); and P(1)–Ni(1)–N(1), 85.34(5); torsion: N(1)–C(13)–C(14)–P(1), 24.5(2); dihedral between DiPP ring and plane N(1)C(12)Ni(1), 75.7; dihedral between Ph ring and plane C(13)N(1)C(14), 38.4; and dihedral between planes Ni(1)C(33)Br(1) and Ni(1)N(1)P(1), 13.7. | ||
The oxidative addition reactions of the corresponding aryl triflate to Ni(COD)2/L proceed with similar colour changes. However, the triflate complexes 9 proved to be sensitive materials and their isolation is problematic. For each ligand, a broad resonance is observed for the main product in the 31P{1H} spectrum of the mixture but as the reaction advances one or two additional signals gradually gain intensity. We attribute the extra signals to the cationic species arising from the replacement of the triflate anion with solvent (THF), adventitious water, or any other potential donor present in the system. The treatment of such mixtures with lithium chloride causes the quantitative exchange of the labile triflate anion for the halide. The 31P spectrum gets simplified, with all signals being replaced with that of the corresponding chloro-complex (i.e.9 → 8). This confirms that the aryl triflate undergoes oxidative addition similar to its bromo and chloro analogues.
Next, we investigated a similar oxidative addition of 4-acetylphenyl derivatives to prepare Pd aryl complexes. Initial experiments using Pd(DBA)2 and ligands La–Lc showed that the desired products were formed, but purifying them from the DBA released in the reaction proved a tedious and unpractical task. We then turned to complexes 4a–c as seemingly ideal starting materials. NMR tests using pure samples of complexes 4 with 4-bromoacetophenone showed that these reactions proceeded smoothly in THF at 60 °C, but they led to mixtures of two P-containing products (Scheme 6). No other P resonances were generated in the process. The integration of the 31P resonances indicates that the combined yields were close to quantitative. Virtually the same results were obtained when the starting materials 4a–c were generated in situ from 2 and La, Lb or Lc, and, therefore this method was systematically applied in further experiments, as described in the experimental section. Evaporation and washing with hexane afforded solids containing mixtures of two organometallic products. A careful analysis of the NMR spectra of these mixtures showed that the products are as shown in Scheme 6. The identity of the Pd-aryl products 10a–c was confirmed by the presence of the characteristic acetyl signal in the spectra, and by a comparison of the chemical shifts of their 31P resonances with those of the same compounds generated from Pd(DBA)2. The identity of the mono-trimethylsilyl species 13a–c was ascertained from the characteristic high-field signals of the CH2SiMe3 group, and the splitting of the CH2 resonance by coupling with the 31P nucleus (2JHP ≈ 6 Hz). The crude reaction mixtures were analysed by GC-MS, which led to the detection of the homo-coupling product 1,2-bis(trimethylsily)ethane, along with the cross-coupled 4-(trimethylsilyl)methyl-acetophenone. However, neither 4,4′-diacetylbiphenyl nor acetophenone (hydrodehalogenation product) was detected in the mixtures.
 | ||
| Scheme 6 Oxidative addition reactions using complexes 4 as Pd(0) sources. | ||
As mentioned above, the use of complex 2 as a practical Pd(0) precursor is becoming a rather common option in homogeneous catalysis, with preference over conventional sources of Pd(0). The actual outcome of such “catalyst activation” reactions is seldom investigated, but it has been recently shown that in some cases it can be more complex than foreseen, and affords a number of unexpected side products.23 Therefore, we deemed it worthwhile to perform some additional experiments in our system in order to gain some insight into the mechanism of this reaction. First, we examined the reactions of complexes 4 with p-acetylphenyltriflate and 4-iodoaceto-phenone (the latter only with 4c). 4-Acetylphenyl triflate reacts with 4a–c under the same conditions tested with the bromide and, as observed for Ni, the 31P{1H} spectrum of the final reaction mixture contains several broad signals (up to four), presumably due to the partial replacement of the labile triflate anion of the initial products (11 and 14) with potential ligands present in the mixture (solvent, H2O traces, COD, etc.). As observed with the Ni triflates, the treatment of the final reaction mixtures with solid LiBr in excess simplified the 31P{1H} spectra, with the original broad signals being replaced by the signals of products 10 and 13 previously observed in the reactions with p-bromoacetophenone. Interestingly, the oxidative addition of the aryl triflate yields a larger fraction of the aryl products than observed in the reactions with aryl bromide (14/11 ratios shown in Scheme 6 have been determined after anion exchange with LiBr), but the alkyl/aryl product ratio exhibits the same general trend for X = Br and OTf, decreasing in the order La ≈ Lb < Lc. The oxidative addition of aryl triflate becomes essentially selective for Lc, as after treatment with LiBr, the 31P{1H} spectrum showed a strongly prevalent resonance for 10c that was subsequently isolated as an analytically pure product and fully characterized. Significantly, GC-MS analysis of the corresponding reaction mixture showed the presence of 1,2-bistrimethylsilylethane, but the cross-coupled organic product was not detected. In contrast, the reaction of 4c with p-iodoacetophenone afforded even higher content of the alkyl product than 4-bromoacetophenone. This observation implies that the alkyl ratio increases with the halide or pseudohalide group in the order I > Br > OTf.
We proposed the mechanism shown in Scheme 7 to explain our experimental observations. The process begins with the irreversible reductive elimination of 1,2-bistrimethylsilylethane from complexes 4 to generate a Pd(0) species. Note that the [(P ∼ N)Pd] notation does not mean any explicit guess on the actual structure of such a species. The essentially quantitative spectroscopic yields show that the Pd(0) intermediate is efficiently trapped by the aryl halide. The fact that these intermediates were not detected in the 31P{1H} monitoring indicates that reductive elimination is slower than any subsequent step or, in other words, the rate determining step. The formation of monalkyl complexes (13, 14 and 15) could arise by two different pathways. One of them, represented in the middle line of Scheme 7, is Pd–Pd alkyl-halide exchange (or transmetallation) between the oxidative addition products (10–12) and starting dialkyl 4 to yield an unstable alkyl–aryl intermediate. This rapidly undergoes reductive elimination, affording the cross-coupling organic product and regenerating the Pd(0) species that is recycled through oxidative addition. A similar Pd–Pd aryl-halide transmetallation step was identified by A. Yamamoto in the reaction of palladium diaryls with MeI or aryl halides,24 and Pd(II)–Pd(II) aryl exchanges have been subsequently proposed by other authors to occur in Pd-catalyzed reactions.25 The bimolecular alkyl-halide exchange is probably driven by the special activation of the Pd–C bond placed in trans to the P donor atom in 4. This path is consistent with the observed influence of X (halide or triflate) on the alkyl/aryl ratio, as the better the ability of X to bridge metal centres, the more efficient the exchange (hence the I > Br > OTf trend). The lower alkyl/aryl ratios observed with Lc can be attributed to the lower rate of bimolecular exchange, hampered by the steric hindrance of this bulky ligand. In agreement with the Pd–Pd transmetallation proposal, we confirmed that reacting 1 equiv. of complex 4b with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 10b/13b mixture at 60 °C, in the absence of additional ArBr, caused a 30% enhancement of the signal 13b at the expense of 10b, as expected if alkyl-halide exchange occurs between 10b and 4b concurrently with reductive elimination processes.
:1 10b/13b mixture at 60 °C, in the absence of additional ArBr, caused a 30% enhancement of the signal 13b at the expense of 10b, as expected if alkyl-halide exchange occurs between 10b and 4b concurrently with reductive elimination processes.
 | ||
| Scheme 7 The possible mechanisms leading to the competitive formation of aryl and alkyl products. Top, generation of a Pd(0) precursor, followed by oxidative addition. Center, bimolecular alkyl/halide exchange. Bottom, direct reaction of the dialkyl 4 with the electrophile (not operative). | ||
An alternative route to the alkyl complexes 13–15, shown in the lower part of Scheme 7, would entail a competitive reaction of the starting dialkyl 4 with the aryl halide to afford the corresponding monoalkyl (13–15) and the cross-coupled organic product in a single step. To gather some additional mechanistic information on this regard, we investigated the overall rate of disappearance of 4b in the presence of 0, 2 and 4 equiv. of 4-bromoacetophenone. At 40 °C (the lower temperature was selected in order to facilitate data collection) 4b decays with estimated half-lives of 61, 19 and 17 min, respectively.§ The blank reaction (no electrophile added) leads to a very dark solution probably containing colloidal Pd(0), and a number of low intensity resonances in the 31P{1H} spectrum. Thus, albeit the decomposition of 4b alone is somewhat slower, the half-life of 4b does not change appreciably when the concentration of the electrophile is increased twofold (2 to 4 equiv.), nor does the product ratio (13b/10b = 2.1:1 in the latter two experiments). This is incompatible with the direct reaction of 4b with bromoacetophenone, as in this case the reaction rate should exhibit a direct dependency on the concentration of the aryl bromide. The bimolecular exchange mechanism also accounts for the slower decomposition rate in the blank reaction, since the overall rate of decay of 4b is the sum of the reductive elimination and the bimolecular exchange reactions, and the latter does not occur in the absence of the electrophile. To confirm that the electron-poor aryl halide is not accelerating reductive elimination on 4b, via transient coordination of the electrophile, we carried out two further control experiments. In these experiments complex 4b was allowed to decompose at 40 °C in the presence of 2 and 4 equivalents of methyl acrylate, an electron-poor olefin that could also act as a reductive elimination promotor.26 The half-lives of 4b in these experiments were 60 and 49 min, fairly similar to those measured in the blank experiment, which confirmed that the role of reductive elimination enhancement by the electrophile is marginal, at best.
In closing this section, it is worth highlighting the marked effect of temperature on the 13b/10b ratio: the selectivity of the oxidative addition product actually decreased by a factor of 2 when the reaction temperature was lowered from 60 to 40 °C, giving a cautionary warning on the practical applications of bis(alkyl)Pd(II) complexes as a source of Pd(0). According to our mechanistic proposal, the product ratio is dictated by the relative rates of the alkyl/halide exchange step (a bimolecular reaction) and reductive elimination (a unimolecular process). Bimolecular processes have strongly negative activation entropies, and therefore their rates scale more slowly with temperature compared with those of the unimolecular reactions. Thus, the formation of the alkyl side product is expected to decrease or even to be suppressed as the temperature is increased. A similar argument leads to the conclusion that the alkyl/aryl product ratio will increase with the overall Pd concentration (i.e., the initial concentration of the starting dialkyl 4), as the rate of the comproportionation step has a quadratic dependency on concentration, vs. simple dependency for the reductive elimination rate. Low temperatures and high concentration of the starting dialkyl are conditions favorable for organometallic synthesis, whilst in catalysis very diluted metal complexes and relatively high temperatures are commonly used. The conclusion is that using complex 2 as a Pd(0) equivalent may be appropriate for catalytic applications, but can be inconvenient for synthetic purposes, due to the formation of the mono(trimethylsilyl)methyl product as an awkward byproduct.
Reversible acid–base reactions of nickel iminophosphine/enaminate complexes
In addition to NaH, other strong bases such as tBuOK, Cs2CO3 and DBU deprotonate 6b in THF (Scheme 8). When these reactions were monitored using 31P{1H} spectra, the resonance of the starting material at 42.5 ppm was replaced in all cases with that of 3b at 33.2 ppm (both in THF). In contrast, triethylamine, diisopropyl(ethyl)amine (Hunig's base) or proton sponge fail to induce any change in the spectrum, either at room temperature or after heating to 60 °C. The addition of one equivalent of HOTf to solution of 3b generated with either NaH or tBuOK caused the disappearance of its 31P resonance and restored that of 6b, demonstrating the full reversibility of these acid–base processes.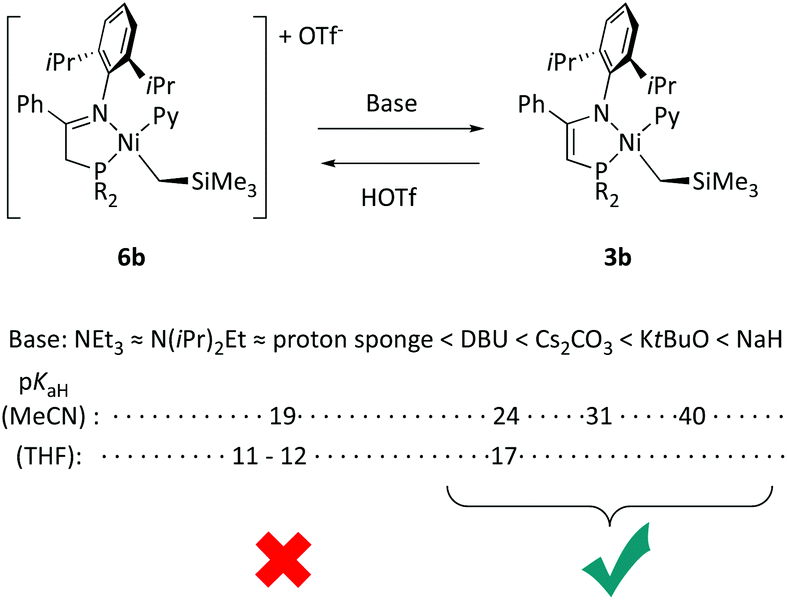 | ||
| Scheme 8 Reversible deprotonation of cationic nickel iminophosphine complexes to neutral phosphinoenaminate with different base strengths. | ||
As an insoluble, hence heterogeneous base, NaH is efficient but slow. In contrast, soluble bases (tBuOK and DBU) or partially soluble ones (Cs2CO3) deprotonate 6b rapidly. Upon treatment with such bases, 6b is consumed within the time required to carry the sample into the NMR probe. Whereas reactions with NaH, tBuOK, and Cs2CO3 are essentially quantitative, the intensity of the signal of 3b in the experiment with 1 equiv. of DBU was ca. 25% of the expected value, though it was the only detected resonance in the final spectrum. An excess of this base (5 equiv.) causes irreversible decomposition, the final spectrum showing a number of low intensity 31P resonances different from those of 3b or 6b, those of the free ligand Lb amongst them.
A basicity scale is displayed at the bottom of Scheme 8 (pKaH is the pKa of the acid conjugates in organic, non-protic solvents¶27). Consistent with our qualitative observations, bases placed below DBU in the scale fail to perform the deprotonation of 6b, at least to a noticeable extent. Thus, it can be assumed that the basicity of 3b is comparable with that of DBU. Very likely, the decomposition problems observed with this base relate to incomplete deprotonation of the iminophosphine ligand, allowing some other undisclosed processes to compete and, eventually, causing decomposition of the material (note that DBU is a non-innocent base, as it is also a potent coordinating ligand towards Ni28). Interestingly, whilst the transformations associated with the stronger bases NaH or KtBuO generate a narrow signal of 3b, the same signal appears broad in the spectra generated with Cs2CO3 or DBU, suggesting that in these cases reversible proton exchange equilibria are established.
In a further test, we explored the capacity of iminophosphine ligands to support reversible deprotonation in a different coordination environment, carrying out the reaction of a nickel aryl complex 7a with NaH. Note that 7a has at least two potentially reactive points for a strong base, the iminophosphine ligand, and the acetyl group. In addition, the deprotonation of a neutral complex should lead to an anionic species. The latter could eliminate insoluble sodium bromide leaving an unstable, coordinatively unsaturated complex. Thus, we were pleased to observe that 7a reacts cleanly in THF solution with a large excess of solid NaH (>10 eq.), affording a single 31P-containing product 16. This was isolated as a highly sensitive brick-red solid, nearly insoluble in C6D6, that decomposed in CD2Cl2. The 1H NMR spectrum, recorded in THF-d8, showed a signal for the enaminate CH at 3.04 ppm with a small coupling to 31P (1.5 Hz), and the usual 3H-intensity of the p-acetyl resonance at 2.33 ppm that confirmed that only the iminophosphine ligand had been deprotonated. These spectroscopic properties, in addition to its insolubility in hydrocarbon solvents, suggested that 16 might be an anionic complex of the type [Ni(L′a)(X)(p-C6H4COCH3)]−Na+, with L′a representing the conjugate base of ligand La, as shown in Scheme 9. Surprisingly, the 1H NMR spectrum also shows a high-field resonance (−2.78 ppm) integrating for 1H, which suggests that X could be a hydroxide ligand. Accordingly, the IR spectrum of 16 shows a weak, but sharp band at 3584 cm−1, overlapping an intense but broader one centered at 3422 cm−1, which is consistent with a partially associated hydroxide ligand. The hydroxide could come from Br exchange with a small amount of NaOH in sodium hydride, used in a large excess (10-fold), or may be generated by the interaction of NaH with moisture traces. As a matter of fact, we confirmed that 7a is also cleanly deprotonated by a suspension of NaOH in anhydrous THF, directly yielding the same product 16 in essentially quantitative yield. Complex 16 was crystallized by layering THF solution with diethyl ether, and its crystal structure was determined (Fig. 5).
 | ||
| Scheme 9 Deprotonation of complex 7a. | ||
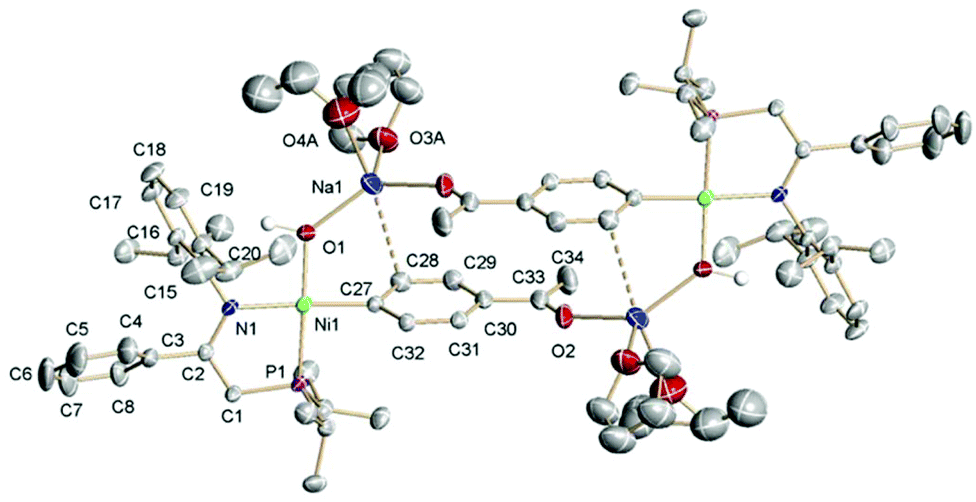 | ||
| Fig. 5 The crystal structure of complex 16·(THF, Et2O) with 50% probability thermal ellipsoids. The selected bond distances (Å): Ni1–O1, 1.8916(18); Ni1–C27, 1.896(3); Ni1–N1, 1.981(2); Ni1–P1, 2.1438(12); N1–C2, 1.363(3); C(33)–O(2), 1.214(4); C(33)–C(30), 1.483(4); C(33)–C(34), 1.510(5); O1–Na1, 2.160(2); O2–Na1′, 2.271(3); O3A–Na1, 2.335(4); O4A–Na1, 2.384(4); and Na1–C(28), 3.080(3). The selected angles (°): O1–N1–C27, 87.41(19) and N1–Ni1–P1, 85.49(6); torsion: N1C2–C1–P1, 4.0(3); and angle between planes O1–Ni1–C27 and N1–Ni1–P1, 9.2. | ||
The structure of 16 confirmed the presence of the anionic enaminato(aryl)nickel moiety, with a hydroxo ligand occupying the fourth coordination position. In the solid state, the sodium cation coordinates to the hydroxo ligand, through a strong (short) Na–O bond (2.160(2) Å), and completes its coordination sphere with two ether molecules (one THF and one Et2O), and a dative interaction with the acetyl oxygen atom belonging to a second coordination unit. This gives rise to a dimeric molecular entity in the solid state. This arrangement presents a contact (3.080(3) Å) between the sodium atom and C28, one of the carbon atoms of the aromatic ring bound to the Ni center. The coordination environment of Na in this molecule is most likely contingent upon the choice of the solvent and crystallization conditions. In solution, it would be reasonable to assume that 16 has the ion pair structure shown in Scheme 9, with an external Na+ cation fully solvated with THF. The Ni–O distance (1.889(2) Å) is similar to that found in other square-planar, monomeric Ni(II) hydroxides reported in the literature, either neutral or anionic.29
It is remarkable that the deprotonation of 7a should afford selectively complex 16, as NaH is one of the bases commonly used to generate enolates from ketones. The low acidity of the acetyl group of this organometallic complex is probably due to the influence of the electron-rich Ni fragment connected to the para position in the aryl ring. Since 7a arises from the oxidative addition of a functional aryl halide to a Ni(0) species, the outcome of this experiment is relevant for potential applications of the nickel-iminophosphine system in cross-coupling catalysis.
Conclusions
In this article we explored the ability of iminophosphine ligands to stabilize a variety of organonickel and organopalladium complexes, supporting reversible deprotonation and oxidative addition reactions. The syntheses of such complexes have been adapted to each of these metals. Thus, ligand exchange reactions from suitable bis(trimethylsilyl)methyl precursor complexes with La–Lc have divergent results for M = Ni (1) or Pd (2) probably due to the different sizes of the metal atoms. The exchange is straightforward for the latter affording dialkylpalladium complexes 4a–c. In contrast, dialkyl precursor 1 reacts with iminophosphine Lb affording the phosphinoenaminate complex 3b. We devised a general synthesis of nickel phosphinoenaminates 3, starting from the new cationic monoalkylnickel precusor 5. Neutral arylnickel complexes were readily obtained via oxidative additions to mixtures Ni(COD)2 + L. The use of complexes 4a–c as Pd(0) sources for the analogous oxidative addition reactions was found unpractical for synthetic purposes, due to the formation of undesired side products arising from competitive Pd(II)–Pd(II) alkyl/halide exchange. The weak acidic character of the phosphino-imine ligand is a source of interesting reactivity with potential applications in catalysis. We have demonstrated the reversibility of the deprotonation of this ligand for Ni, a potential route for cooperative metal–ligand behavior. Tests with bases of different strengths indicate that the iminophosphine/phosphinoenaminate pair in the coordination sphere of Ni is an acid/base pair comparable to H-DBU+/DBU pair. Interestingly, neutral halo(aryl) complexes arising from the oxidative addition of 4-bromoacetophenone are selectively deprotonated at the iminophosphine ligand, even with strong bases, affording an unusual type of anionic arylnickel complex. These results open interesting prospects in the application of nickel complexes as catalysts for coupling reactions such as the Heck or Suzuki reactions, with iminophosphine ligands acting as auxiliaries in proton transfer steps, or base-promoted activation of the electrophile.Experimental section
General considerations
All manipulations were carried out under an inert atmosphere, either using the standard Schlenk technique in a vacuum line operating with purified argon, or a nitrogen-filled glove box equipped with a pump for solvent evaporation, an externally cooled (with liquid N2) cavity, a freezer and an analytical balance. The following paragraphs provide a summary of the general procedures, and a full description of the syntheses and complete spectroscopic and analytical data are given in the ESI‡ for all ligands and complexes.N(DiPP) (0.560 g, 2 mmol) in THF (10 mL), stirred at −80 °C, was added 1.6 mL of a 1.7 M solution of tBuLi in pentane (2.72 mmol). The stirring was continued for 30 minutes, and then the cooling bath was removed allowing the mixture to warm to room temperature. Then, the volume corresponding to 1 equivalent of the corresponding chlorophosphine (2 mmol) was added, and the stirring was continued, monitoring the reaction by 31P NMR spectroscopy. Usually, the reactions were complete within ca. 30 min. The solvent was removed under vacuum and the residue was then extracted with n-hexane (40 mL) and the extract was filtered through Celite. Evaporation under reduced pressure leaves a pale-yellow oil that can be stored at 5 °C under an Ar atmosphere.
Ligand exchange reactions
Isolation of 3b. To a solution of complex 1 (0.115 g, 0.3 mmol) in 10 mL of THF, stirred at −80 °C, was added an equimolar amount of the corresponding ligand (La, Lb or Lc), dissolved in 1.5 mL of THF. The mixture was stirred for 5 min, and then allowed to stir at room temperature. The 31P{1H} spectra of the reaction mixtures were recorded periodically. The initial spectra, recorded after 1 h, showed several signals, including some unreacted free ligands. After 20 h, the spectrum of the mixture generated from Lb showed a single signal at 32.3 ppm (together with a small amount of unreacted Lb), but those generated from La and Lc still showed a number of peaks that revealed the existence of a number of different products. After this time, the mixtures were evaporated, the residues taken up in Et2O (15 mL) and the solutions filtered through Celite. These were concentrated and some hexane was added until becoming slightly turbid. Cooling in the freezer (−25 °C) led to the crystallization of 3b (from Lb), but no solid products crystallized from the mixtures generated from La or Lc. The red-orange crystals of 3b were collected by filtration, and a small sample of crystals was reserved for X-ray analysis. The remaining product was washed with some hexane and dried in vacuo. Yield, 0.084 g, 0.12 mmol, 40%. Complete spectroscopic data for 3b can be found in the ESI.‡
Syntheses of neutral alkylnickel complexes [Ni(CH2SiMe3)(L′)(Py)] (L′ = deprotonated L), 3a–c
Oxidative addition of 4-X-C6H4COMe to Ni(0)/L (L = La, Lb or Lc; and X = Br or Cl)
General procedure. A 100 mL round-bottom Schlenk flask loaded with Ni(COD)2 (ca. 1 mmol), THF (ca. 10 mL) and a stirring bar was cooled to −80 °C. To the stirred suspension was added a solution containing equimolar amounts of the corresponding ligand (La, Lb or Lc) and the aryl halide. After 5 min, the cooling bath was removed. As the reaction mixture approached the room temperature, the colour became dark brown. As the stirring was continued at room temperature, the color turned gradually to dark yellow. After 2 h, the mixture was evaporated under reduced pressure. The oily residue was extracted with CH2Cl2, and the solution was filtered through a Celite plug. The clear solution was evaporated and the oily residue cooled to −80 °C and vigorously washed with 2 × 20 mL of diethyl ether. The ether washings were discarded, and the resulting powdery solid was dried under vacuum. The powders were spectroscopically pure, but they were recrystallized by slow diffusion of Et2O or hexane into a cold, concentrated CH2Cl2 solution, in the glove box freezer.
Oxidative addition of 4-TfO-C6H4COMe to Ni(0)/L (L = La, Lb or Lc)
In order to confirm that the final products were chloro derivatives, the oxidative addition reaction of Ni(COD)2 with Lb and 4-TfOC6H4COCH3 was carried out on a preparative scale (1 mmol), following the same procedure described above for the oxidative addition of 4-haloacetophenones, except that a ten-fold excess of LiCl was added once the oxidative addition reaction was complete. The usual workup yielded the expected complex 8b, which was identical to the sample obtained from 4-chloroacetophenone. The 31P{1H} spectrum of the intermediate triflate complex 9b (THF solution, external PPh3/C6D6 reference) showed two broad signals at δ 37.3 (major) and 40.4 (minor), and the 19F spectrum showed a prominent signal at δ 80.3, probably due to the coordinated or ion-paired triflate anion. All attempts to isolate the intermediate triflate complex 9b were unsuccessful.
Studies on the oxidative addition of p-XC6H4COCH3 to Pd(0) using complex 2 as a precursor
In a separate experiment, a solution of complex 4b in THF was generated as indicated above, and then split in two equal parts. To one of them was added the stoichiometric amount of p-bromoacetophenone, and heated at 60 °C until completion. A 31P{1H} NMR spectrum showed that the corresponding mixture of complexes 10b and 13b had been formed. To this mixture was added the second half of the 4b solution, and the combination was heated again to 60 °C, until the 31P resonance of 4b disappeared. The final 31P spectrum showed an enhancement of the signal of 13b (ca. 30%) and a decrease of that of 10b by a similar amount.
The available NMR data for the mixtures 10/13 are provided in the ESI.‡
NMR studies on the reversible deprotonation of [Ni(CH2SiMe3)(py)(Lb)]+[OTf]− (6b) with different bases
Deprotonation of complex 7a with NaH or NaOH
C–N), 182.4 (d, 2JCP = 43.0 Hz, ipso-Cq, Ni-Ar), 197.2 (–COMe). IR (nujol mull, cm−1): 3584 (sh, w, (O–H)), 3422 (br, st, (O–H)), 3053 (sh, w, ν(C–H arom.), 1669, 1657 (ν(CO, νCC). Anal. calcd for C34H45NNaNiO2P: C, 66.68; H, 7.41; N, 2.29. Found: C, 66.93; H, 7.68; N, 1.96.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Spanish Ministries of Economy and Innovation (MINECO) and Ciencia, Innovación y Universidades (MCIU/AEI) and the EU FEDER funds of the European Union supported this work through grants CTQ2015-68978-P and PGC2018-095768-B-I00.Notes and references
-
(a)
R. Tannert and A. Pfaltz, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, ed. P. C. Kamer and P. W. N. M. van Leeuwen, Wiley, 2012, ch. 7, pp. 233–286 Search PubMed
; (b) W.-H. Zhang, S. W. Chen and T. S. A. Hor, Coord. Chem. Rev., 2011, 255, 1991–2024 CrossRef CAS
- V. Vasilenko, T. Roth, C. K. Blasius, S. N. Intorp, H. Wadepohl and L. Gade, Beilstein J. Org. Chem., 2016, 12, 856–953 Search PubMed
-
(a) F. Speiser, P. Braunstein and L. Saussine, Acc. Chem. Res., 2005, 36, 784–793 CrossRef PubMed
- S. Wang, W.-H. Sun and C. Redshaw, J. Organomet. Chem., 2014, 751, 717–741 CrossRef CAS
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS
-
(a) L. Guo, S. Dai, X. Sui and C. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS
-
(a) W. Keim, S. Killat, C. F. Nobile, G. P. Suranna, U. Englert, R. Wang, S. Mecking and D. L. Schröder, J. Organomet. Chem., 2002, 662, 150–171 CrossRef CAS
- O. Daugulis and M. Brookhart, Organometallics, 2002, 21, 5926–5934 CrossRef CAS
-
(a) K. S. Coleman, M. L. H. Green, S. I. Pascu, N. H. Rees, A. R. Cowley and L. H. Rees, J. Chem. Soc., Dalton Trans., 2001, 3384–3395 RSC
- For some relevant reviews on this topic, see:
(a) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed
- For example, see: C. Tian, L. Gong and E. Meggers, Chem. Commun., 2016, 52, 4207–4210 RSC
-
(a) T. C. Wambach, J. M. Ahn, B. O. Patrick and M. D. Fryzuk, Organometallics, 2013, 32, 4431–4432 CrossRef CAS
-
(a) A. J. Ruddy, C. M. Kelly, S. M. Crawford, C. A. Wheaton, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Organometallics, 2013, 32, 5185–5188 CrossRef
-
(a) L. Ortiz de la Tabla, I. Matas, P. Palma, E. Álvarez and J. Cámpora, Organometallics, 2012, 31, 1006–1016 CrossRef CAS
-
(a) S. Mecking, Coord. Chem. Rev., 2000, 203, 325–351 CrossRef CAS
- J. Cámpora, M. M. Conejo, K. Mereiter, P. Palma, C. Pérez, M. L. Reyes and C. Ruiz, J. Organomet. Chem., 2003, 683, 220–239 CrossRef
-
(a) E. Carmona, F. González, M. L. Poveda, J. L. Atwood and R. D. Rogers, J. Chem. Soc., Dalton Trans., 1981, 777–782 RSC
- D. A. Smith, A. S. Batsanov, K. Costuas, R. Edge, D. C. Apperley, D. Collison, J. F. Halet, J. A. K. Howard and P. W. Dyer, Angew. Chem., Int. Ed., 2010, 49, 7040–7044 CrossRef CAS PubMed
-
(a) P.-L. Yi and L.-C. Liang, Inorg. Chem., 2008, 47, 749–758 CrossRef PubMed
- K. R. Reddy, K. Surekha, G. H. Lee, S. M. Peng and S. T. Liu, Organometallics, 2000, 19, 2637–2639 CrossRef CAS
-
(a) B. Crociani, S. Antonaroli, V. Beghetto, U. Matteoli and A. Scrivanti, Dalton Trans., 2003, 2194–2202 RSC
-
(a) B. P. Fors, D. A. Watson, M. R. Briscoe and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 13552–13554 CrossRef CAS PubMed
- T. L. Andersen, S. Kramer, J. Overgaard and T. Skrydstrup, Organometallics, 2017, 36, 2058–2066 CrossRef CAS
-
(a) F. Ozawa, M. Fujimori, T. Yamamoto and A. Yamamoto, Organometallics, 1986, 5, 2144–2149 CrossRef CAS
-
(a) L. A. Casado, J. A. Casares and P. Espinet, Organometallics, 1997, 16, 5730–5736 CrossRef
-
(a)
J. F. Hartwig, Organotransition Metal Chemistry. From Bonding to Catalysis, University Science Books, Sausalito, California, 2010 Search PubMed
-
(a) A. Kutt, S. Selberg, I. Kaljurand, S. Tshepelvitsh, A. Heering, A. Darnell, K. Kaupmees, M. Piirsalu and I. Leito, Tetrahedron Lett., 2018, 59, 3738–3748 CrossRef CAS
-
(a) H. Hoberg, Y. Peres, C. Kruger and Y. H. Tsay, Angew. Chem., Int. Ed., 1987, 26, 771–773 CrossRef
-
(a) J. Cámpora, P. Palma, D. del Río and E. Álvarez, Organometallics, 2004, 23, 1652–1655 CrossRef
Footnotes |
| † Dedicated to the memory of Prof. Richard A. Andersen. |
| ‡ Electronic supplementary information (ESI) available: Complete experimental details, spectroscopic and crystallographic data; CIF files for compounds CCDC 1944621 (3b), 1944622 (4c), 1944623 (6b), 1944624 (7b) and 1944625 (16). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/C9DT04062E |
| § The half-lives were estimated from approximate first-order kinetic plots, but this reaction is not expected to obey a simple kinetic law. At this stage, a comparison of overall reaction rates suffices to draw some valuable conclusions regarding the influence of the aryl bromide concentration on the reaction rate and product distribution. |
| ¶ The approximate basicity scales shown in Scheme 8 are based on the pKa values measured for the corresponding conjugate acids in different nonaqueous acids. The largest series of pKa values are available in MeCN (DBU-H+, 24.3; H-proton sponge+, 18.6; and H-NEt3+, 18.6) (see ref. 28a). Other values can be estimated from other solvents using empirical correlations for X-OH acids (ref. 28b): pKa in MeCN = pKa in DMSO +10.5 (used for tBuOH, pKa = 29.4 in DMSO, ref. 28c) and pKa in MeCN = 1.6·pKa in H2O + 14.9 for HCO3− (pKa = 10.3 in H2O) as an estimation for Cs2CO3. Similar correlations are not available for THF but the pKa values in this solvent are also given in ref. 28a for HNEt3+ (12.5), H-proton sponge+ (11.1) and DBU (16.6). |
| This journal is © The Royal Society of Chemistry 2020 |