
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral and cationic phosphine and arsine complexes of tin(IV) halides: synthesis, properties, structures and anion influence†
Victoria K.
Greenacre
,
Rhys P.
King
,
William
Levason
 and
Gillian
Reid
*
and
Gillian
Reid
*
School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 8th November 2019
Abstract
The reaction of trans-[SnCl4(PR3)2] (R = Me or Et) with trimethylsilyltriflate (TMSOTf) in CH2Cl2 solution substitutes one chloride to form [SnCl3(PR3)2(OTf)]; addition of excess TMSOTf does not substitute further chlorides. The complexes have been fully characterised by microanalysis, IR and multinuclear NMR (1H, 13C{1H}, 19F{1H}, 31P{1H}, 119Sn) spectroscopy. The crystal structure of [SnCl3(PMe3)2(OTf)] revealed mer-chlorines and trans-phosphines. In contrast, trans-[SnBr4(PR3)2], [SnCl4{Et2P(CH2)2PEt2}], [SnCl4{o-C6H4(PMe2)2}] and [SnCl4{o-C6H4(AsMe2)2}] did not react with TMSOTf in CH2Cl2 solution even after 3 days. The arsine complexes, [SnX4(AsEt3)2] (X = Cl, Br), were confirmed as trans-isomers by similar spectroscopic and structural studies, while attempts to isolate [SnI4(AsEt3)2] were unsuccessful and reaction of SnX4 with SbR3 (R = Et, iPr) resulted in reduction to SnX2 and formation of R3SbX2. trans-[SnCl4(AsEt3)2] is converted by TMSOTf into [SnCl3(AsEt3)2(OTf)], whose X-ray structure reveals the same geometry found in the phosphine analogues, with the triflate coordinated. The salts, [SnCl3(PEt3)2][AlCl4] and [SnCl2(PEt3)2][AlCl4]2 were made by treatment of [SnCl4(PEt3)2] with one and two mol. equivalents, respectively, of AlCl3 in anhydrous CH2Cl2, whereas reaction of [SnCl4(AsEt3)2] with AlCl3 produced a mixture including Et3AsCl2 and [Et3AsCl][Sn(AsEt3)Cl5] (the latter identified crystallographically). In contrast, using Na[BArF] (BArF = [B{3,5-(CF3)2C6H3}4]−) produced [SnCl3(PEt3)2][BArF] and also allowed clean isolation of the arsine analogue, [SnCl3(AsEt3)2][BArF]. [SnCl4{o-C6H4(PMe2)2}] also reacts with AlCl3 in CH2Cl2 to form [SnCl3{o-C6H4(PMe2)2}][AlCl4] and [SnCl2{o-C6H4(PMe2)2}][AlCl4]2. Multinuclear NMR spectroscopy on the [AlCl4]− salts show that δ31P and δ119Sn move progressively to high frequency on conversion from the neutral complex to the mono- and the di-cations, whilst 1J(119Sn–31P) follow the trend: [SnCl3{o-C6H4(PMe2)2}]+ > [SnCl4{o-C6H4(PMe2)2}] > [SnCl2{o-C6H4(PMe2)2}]2+. DFT studies on selected complexes show only small changes in ligand geometries and bond lengths between the halide and triflate complexes, consistent with the X-ray crystallographic data reported and the HOMO and LUMO energies are relatively unperturbed upon the introduction of (coordinated) triflate, whereas the energies of both are ca. 4 eV lower in the cationic species and reveal significant hybridisation across the pnictine ligands.
Introduction
Much of the coordination chemistry of the transition metal ions with neutral ligands is based around metal halide complexes.1 In addition to neutral complexes [MLaXb] (M = transition metal, X = halide, L = neutral ligand), related cations [MLcXb−1]+ or [MLdXb−2]2+ (c, d ≥ a) are commonly known, formed either spontaneously in the presence of excess L, or upon the addition of large, weakly coordinating anions.1 In some cases complexes of the type [MLxXy][MXz] form spontaneously under appropriate conditions, a process sometimes labelled as ‘self-ionisation’. In contrast, complexes of p-block metal and metalloid halides are overwhelmingly neutral [M′LaXb] (M′ = p-block metal) with much stronger M–X than M–L bonding.1 In general, p-block Lewis acids coordinate the neutral ligands relatively weakly, and these are often partially dissociated in solution or undergoing rapid dissociative exchange. The neutral ligands fill ‘free’ coordination sites, but do not compete with the halide ligands for the metal centre. The tin(IV) halides, SnX4, typify this pattern in that the majority of complexes are octahedral with neutral ligands filling two coordination sites.2 Rare exceptions are provided by 1,4,7-trimethyl-1,4,7-triazacyclonane, 1,3,5-trimethyl-1,3,5-triazacyclohexane and 1,4,7-trithiacyclonane (L3), which generate the ‘self-ionisation’ complexes, [SnX3(L3)]2[SnX6] (X = Cl or Br),3 although more flexible acyclic polydentate ligands, such as MeC(CH2AsMe2)3, behave as bidentate ligands, as in [SnX4{κ2-MeC(CH2AsMe2)3}].4Complexes of the tin(IV) halides, SnX4 (X = Cl or Br), with neutral phosphine and arsine ligands have been explored in considerable detail5 and almost all are of the octahedral trans-[SnX4(ER3)2] or cis-[SnX4(bidentate)] (bidentate = diphosphine or diarsine) types. SnI4 is a weaker Lewis acid and forms similar, but less stable adducts,5 whilst complexes of SnF4 have been much less studied.4,5 Reported examples of stibine complexes of p-block halides are extremely rare.5,6
The formation of cationic complexes would be expected to enhance the Lewis acidity of the tin centre, and examples of halo- or organo-tin cations with polydentate and pincer ligands have been described.7–12 MacDonald et al.11 obtained [SnCl3(PMe3)2][AlCl4] and [SnCl2(PMe3)2][AlCl4]2 by treatment of trans-[SnCl4(PMe3)2] with one or two mol. equivalents of AlCl3, respectively. The former has a distorted trigonal bipyramidal geometry with trans axial PMe3 ligands. The dication has trans PMe3 groups, and two cis interactions from the [AlCl4]− anions completing a very distorted octahedron. The Sn–Cl distances decrease along the series [SnCl4(PMe3)2] > [SnCl3(PMe3)2]+ > [SnCl2(PMe3)2]2+. In the present work we have explored the reaction of tin(IV) halides with a series of mono- and bi-dentate phosphine, arsine and stibine ligands and the abstraction/replacement of some of the halides from the resulting complexes by other weakly coordinating anions, including triflate ([CF3SO3]−) via treatment with trimethylsilyltriflate, [AlX4]−via addition of aluminium trihalides and using the large diffuse [BArF]− anion, via addition of its Na+ salt (BArF = [B{3,5-(CF3)2C6H3}4]−).
Experimental
All complex syntheses were carried out using standard Schlenk and vacuum line techniques. Samples were handled and stored in a glove box under a dry dinitrogen atmosphere to exclude moisture. Tin(IV) halides, TMSOTf, and aluminium halides were obtained from Sigma-Aldrich and used as received. Phosphine and arsine ligands were obtained from Sigma-Aldrich or Strem, except for o-C6H4(PMe2)2 and o-C6H4(AsMe2)2, which were prepared by literature methods.13 SbEt3, SbiPr314 and Na[BArF]15 were made as described. CH2Cl2 was dried by distillation from CaH2 and diethyl ether and n-hexane from Na.Infrared spectra were recorded as Nujol mulls between CsI plates using a PerkinElmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 13C{1H}, 19F{1H}, 31P{1H}, 27Al and 119Sn NMR spectra were recorded from CDCl3 or CH2Cl2/CD2Cl2 solutions using a Bruker AV400 spectrometer and referenced to TMS via the residual solvent resonance, CFCl3, 85% H3PO4, [Al(H2O)6]3+ in H2O at pH = 1 or SnMe4 as appropriate. Typically, [Cr(acac)3] was added as a relaxation agent when recording the 119Sn spectra, except for those containing [AlCl4]−, when it appeared to react with the complexes, turning green. In these cases a 2 seconds pulse delay was used. Microanalyses were undertaken by London Metropolitan University.
[SnCl4(AsEt3)2]
A solution of AsEt3 (0.058 g, 0.36 mmol) in CH2Cl2 (5 mL) was added to a solution of SnCl4 (0.047 g, 0.18 mmol) in CH2Cl2 (5 mL). The colourless solution was left to stir for 1 h before the product was isolated by evaporation of the solvent under vacuum to afford a white solid. Crystals suitable for single crystal X-ray diffraction were grown by slow evaporation of a concentrated CH2Cl2 solution of the compound. Yield 0.093 g, 88%. Required for C12H30As2Cl4Sn (584.7): C, 24.6; H, 5.2. Found: C, 24.5; H, 5.0%. IR (Nujol/cm−1): ν = 288s (Sn–Cl). 1H NMR (CDCl3, 295 K): δ = 2.34 (q, [12H], 3JHH = 7.6 Hz, CH2), 1.42 (t, [18H], 3JHH = 7.6 Hz, CH3). 13C{1H} NMR (CDCl3, 295 K): δ = 8.62 (CH2), 15.04 (CH3). 119Sn NMR (CD2Cl2, 298 K): n.o.; (CD2Cl2, 193 K): δ = −657.4.[SnBr4(AsEt3)2]
A solution of AsEt3 (0.144 g, 0.89 mmol) in CH2Cl2 (5 mL) was added to a solution of SnBr4 (0.195 g, 0.45 mmol) in CH2Cl2 (5 mL). The pale yellow solution was left to stir for 1 h before the product was isolated by removal of the solvent to afford a yellow solid. Crystals suitable for single crystal X-ray diffraction were grown by slow evaporation of a concentrated CH2Cl2 solution of the compound. Yield 0.175 g, 50%. Required for C12H30As2Br4Sn (762.5): C, 18.9; H, 4.0. Found: C, 19.0; H, 4.0%. IR (Nujol/cm−1): ν = 210s (Sn–Br). 1H NMR (CD2Cl2, 295 K): δ = 2.35 (q, [12H], 3JHH = 7.6 Hz, CH2), 1.43 (t, [18H], 3JHH = 7.6 Hz, CH3), 13C{1H} NMR (CD2Cl2, 295 K): δ = 8.5 (CH2), 14.8 (CH3). 119Sn NMR (CD2Cl2, 295 K): δ = −1173.4; (CD2Cl2, 193 K): δ = −1125.3.[SnCl3(AsEt3)2(OTf)]
[SnCl4(AsEt3)2] (0.150 g, 0.26 mmol) was dissolved in CH2Cl2 (10 mL) to form a clear solution. To this TMSOTf (0.057 g, 0.26 mmol) was added in CH2Cl2 (5 mL) and the clear solution was stirred for 2 h. The solvent was removed and the white powder was dried in vacuo. Crystals suitable for single crystal X-ray diffraction were grown from slow evaporation of a solution of the compound in CH2Cl2. Yield 0.138 g, 77%. Required for C13H30As2Cl3F3O3SSn (698.1): C, 22.4, H, 4.3. Found C, 21.9; H, 4.2%. IR (Nujol/cm−1): ν = 294s, 378w (Sn–Cl), 1156m (–OSO2), 1231m, 1200m (CF3). 1H NMR (CD2Cl2, 295 K): δ = 1.39 (t, [18H], 3JHH = 7.8 Hz, CH3), 2.38 (q, [12H], 3JHH = 7.8 Hz, CH2). 13C{1H} NMR (CD2Cl2, 295 K): δ = 9.0 (CH2), 16.0 (CH3). 19F{1H} NMR (CD2Cl2, 295 K): δ = −78.7 (s, CF3). 119Sn NMR (CD2Cl2, 298 K): n.o.; (CD2Cl2, 183 K): δ = −620 (s).[SnCl3(PMe3)2(OTf)]
[SnCl4(PMe3)2]16 (0.300 g, 0.72 mmol) was suspended in CH2Cl2 (5 mL). To this TMSOTf (0.016 g, 0.72 mmol) was added in CH2Cl2 (5 mL). The solution was stirred for 16 h and the solvent and volatiles were removed and the resulting white powder dried in vacuo. The powder was then washed with diethyl ether (3 × 10 mL) and dried in vacuo. Crystals suitable for single crystal X-ray diffraction were grown by the slow evaporation of a saturated solution of the compound in CH2Cl2. Yield 0.215 g, 57%. Required for C7H18Cl3F3O3P2SSn (526.1): C, 16.0, H 3.5. Found C, 15.7; H, 3.4%. IR (Nujol/cm−1): ν = 296m, 306m, 377w (Sn–Cl), 1173m (–OSO2), 1200m, 1235m (CF3). 1H NMR (CD2Cl2, 295 K): δ = 1.83 (m, CH3). 13C{1H} NMR (CD2Cl2, 295 K): δ = 10.09 (t, 1J + 3J31P–13C = 19.4 Hz, CH3). 31P{1H} NMR (CD2Cl2, 298 K): δ = 8.74 (s, 1J31P–117Sn = 2814, 1J31P–119Sn = 2963 Hz). 19F{1H} NMR (CD2Cl2, 295 K): δ −78.6 (s, CF3). 119Sn (CD2Cl2, 298 K): δ = −516 (t, 1JP–119Sn = 2963 Hz).[SnCl3(PEt3)2(OTf)]
[SnCl4(PEt3)2]16 (0.08 g, 0.16 mmol) is dissolved in CH2Cl2 (10 mL). To this TMSOTf (0.036 g, 0.16 mmol) was added in CH2Cl2 (5 mL), resulting in a clear colourless solution which was stirred for 2 h. Solvent and volatiles were removed in vacuo and the resultant white powder was washed with diethyl ether (3 × 10 mL) and then dried in vacuo. Yield 0.065 g, 66%. Required for C13H30Cl3F3O3P2SSn (610.2): C, 25.6; H, 5.0. Found, C, 25.4; H, 5.1%. IR (Nujol/cm−1): ν = 289m, 301w, 374w (Sn–Cl), 1187m (–OSO2), 1235m (CF3). 1H NMR (CD2Cl2, 295 K): δ = 1.30 (m, [18H], CH3), 2.33 (m, [12H], CH2). 13C{1H} NMR (CD2Cl2, 295 K): δ = 7.59 (t, 1J + 3J31P–13C = 2.9 Hz, CH2), 13.95 (t, 1J + 3J31P–13C = 11 Hz, CH3). 19F{1H} NMR (CD2Cl2, 295 K): δ = −78.6 (s, CF3). 31P{1H} (CD2Cl2, 298 K): δ = 28.45. (CD2Cl2, 183 K): δ = 33.58 (s, 1J31P–117Sn = 2617 Hz, 1J31P–119Sn = 2734 Hz). 119Sn NMR (CD2Cl2, 298 K): δ = −535; (CD2Cl2, 183 K): δ = −535 (t, 1J31P–119Sn = 2737 Hz).[SbEt3Cl(OTf)]
SnCl4 (0.100 g, 0.38 mmol) was dissolved in 5 mL of CH2Cl2. To this, SbEt3 (0.160 g, 0.77 mmol) was added in 5 mL of CH2Cl2, resulting in the precipitation of a large amount of white solid. TMSOTf (0.085 g, 0.38 mmol) was added in CH2Cl2 (5 mL), and the reaction stirred for 16 h. The liquid was filtered away from the unidentified solid and concentrated to dryness in vacuo, leading to a white solid. Crystals suitable for single crystal X-ray diffraction were grown by slow evaporation of a concentrated CH2Cl2 solution of the compound. Yield 0.064 g, 30%. Required for C7H15ClF3O3SSb (393.3): C, 21.4; H, 3.8 Found C, 21.7; H, 4.2%. IR (Nujol/cm−1): ν = 329m (Sb–Cl), 1167w (–OSO2), 1198w, 1235w (CF3). 1H NMR (CD2Cl2, 295 K): δ = 1.62 (t, [9H], 3JHH = 7.7 Hz, CH3), 2.80 (q, [6H], 3JHH = 7.7 Hz, CH2). 13C{1H} (CD2Cl2, 295 K): δ = 9.68 (CH2), 28.55 (CH3).[SnCl3{o-C6H4(PMe2)2}][AlCl4]
[SnCl4{o-C6H4(PMe2)2}]4 (0.150 g, 0.33 mmol) was suspended in CH2Cl2 (5 mL), and AlCl3 (0.044 g, 0.33 mmol) was added. This resulted in a pale yellow clear solution which was stirred for a further 2 h. The solution was then concentrated to dryness in vacuo producing a white powder, which was washed with n-hexane (3 × 10 mL) and the solid separated and dried in vacuo to leave a white powder. Yield: 0.120 g, 61%. Required for C10H16AlCl7P2Sn (591.9): C, 20.3; H, 2.7, Found C, 20.4; H, 2.8%. IR (Nujol/cm−1): ν = 322m, 342m (Sn–Cl), 451m, 497br ([AlCl4]−). 1H NMR (CD2Cl2, 295 K): δ = 2.06 (t, 2J + 5JP–H = 4.5 Hz, [12H], CH3), 7.86–7.92 (m, [4H], Ar–H). 13C{1H} NMR (CD2Cl2, 295 K): δ = 11.52 (t, 1J + 3J31P–13C = 16.4 Hz, CH3), 130.7–131.3, 134.2, 135.3 (Ar). 27Al NMR (CD2Cl2, 295 K): δ = 102.8 (s, [AlCl4]−). 31P{1H} NMR (CD2Cl2, 295 K): δ = −24.7 (s, 1J31p–117Sn = 930 Hz, 1J31P–119Sn = 972 Hz). 119Sn (CD2Cl2, 298 K): δ = −469 (t, 1J31–119Sn = 972 Hz).[SnCl2{o-C6H4(PMe2)2}][AlCl4]2
[SnCl4{o-C6H4(PMe2)2}] (0.100 g, 0.22 mmol) was suspended in CH2Cl2 (5 mL), and to this AlCl3 (0.058 g, 0.44 mmol) was added. This resulted in a pale yellow clear solution, which was stirred for a further 2 h. The solution was then concentrated to dryness in vacuo producing a white sticky solid, which was then washed with n-hexane (3 × 10 mL), the solid separated and dried in vacuo to leave a white sticky solid. Yield: 0.81 g 51%. Required for C10H16Al2Cl10P2Sn (725.4): C, 16.7; H, 2.2. Found C, 17.5; H, 2.7%. IR (Nujol/cm−1): ν = 294m, 310m (Sn–Cl), 452sh, 487s ([AlCl4]−). 1H NMR (CD2Cl2, 295 K): δ = 2.18 (t, 2J + 5JPH = 4.5 Hz, [12H], CH3), 7.94–8.30 (m, [4H], Ar–H. 13C{1H} NMR (CD2Cl2, 295 K): δ = 12.28 (t, 1J + 3J31P–13C = 16.1 Hz, CH3), 129.8–130.1, 134.4, 135.9 (Ar–H). 31P{1H} NMR (CD2Cl2, 295 K): δ = −23.7 (1J31P–117Sn = 815 Hz, 1J31P–119Sn = 853 Hz). 27Al NMR (CD2Cl2, 295 K): δ = 102.9 (s, [AlCl4]−). 119Sn NMR (CD2Cl2, 298 K): δ = −429 (t, 1J31P–119Sn = 850 Hz).[SnCl3(PEt3)2][AlCl4]
[SnCl4(PEt3)2] (0.200 g, 0.40 mmol) was suspended in CH2Cl2 (5 mL) and AlCl3 (0.054 g, 0.40 mmol) was added. The suspension was stirred for 2 h leaving a clear and colourless solution; the solution was concentrated to dryness leaving a fine white powder which was washed with Et2O (3 × 10 mL) and dried in in vacuo to yield a white powder. Yield: 0.160 g, 63%. Required for C10H16AlCl7P2Sn (630.04): C, 22.9; H, 4.8. Found C, 23.1; H, 5.3%. IR (Nujol/cm−1): ν = 280m, 289m (Sn–Cl), 484s, 503sh ([AlCl4]−). 1H NMR (CD2Cl2, 295 K): δ = 1.36 (m, [18H], CH3), 2.32 (m, [12H], CH2). 13C{1H} NMR (CD2Cl2, 295 K): δ = 7.34 (t, 2J + 4J31P–13C = 2.93 Hz, –CH3), 14.12 (t, 1J + 3J31P–13C = 22.45 Hz). 31P{1H} NMR (CD2Cl2, 295 K): δ = 35.2 (1J31P–117Sn = 2208 Hz, 1J31P–119Sn = 2317 Hz); (CD2Cl2, 183 K): δ = 27.0 (1J31P–117Sn = 2426 Hz, 1J31P–119Sn = 2520 Hz). 27Al NMR (CD2Cl2, 295 K): δ = 103.2 (s, [AlCl4]−). 119Sn NMR (CD2Cl2, 298 K): δ = −350.2 (br t, 1J31P–119Sn = 2307 Hz).[SnCl2(PEt3)2][AlCl4]2
[SnCl4(PEt3)2] (0.200 g, 0.40 mmol) was suspended in CH2Cl2 (5 ml), to this AlCl3 (0.107 g, 0.80 mmol) was added, the suspension was stirred for 2 h leaving a clear and colourless solution; the solution was concentrated to dryness and washed with Et2O (3 × 10 ml) then dried in vacuo leaving a sticky white solid. Yield 0.201 g (66%) Anal. Required for C12H30Al2Cl10P2Sn (763.39) C, 18.9; H, 4.0. Found C, 19.0; H, 3.8, IR (Nujol/cm−1) = 289m, (Sn–Cl), 484s, ([AlCl4]−). 1H NMR (CD2Cl2, 295 K): δ = 1.39 (m, [18H], CH3), 2.38 (m, [12H], CH2), 13C{1H}NMR (CD2Cl2, 295 K): δ = 7.50 (t, 2J + 4J31P–13C = 2.93 Hz, –CH3), 14.51 (t, 1J + 3J31P–13C = 13.2 Hz), 27Al NMR (CD2Cl2, 295 K): δ = 103.0, 31P{1H} NMR (CD2Cl2, 295 K): δ = 40.8 (1J31P–117Sn = 2172 Hz, 1J31P–119Sn = 2274 Hz). 119Sn NMR (CD2Cl2, 298 K): δ = −301.4 (br, t, 1J31P–119Sn = 2291 Hz).Reaction of [SnCl2(PEt3)2][AlCl4]2 + PEt3
[SnCl2(PEt3)2][AlCl4]2 (0.100 g, 0.13 mmol) was dissolved in CH2Cl2 (3 mL), to this PEt3 (0.015 g, 0.22 mmol) was added in CH2Cl2 (2 mL). To this 10 drops of CD2Cl2 is added. A portion of the solution was used for in situ NMR studies.27Al NMR (CD2Cl2, 295 K): δ = 103.1 (s, [AlCl4]−), 110.8 (d, 1J31P–27Al = 267 Hz, [AlCl3(PEt3)]). 31P{1H} NMR (CD2Cl2, 295 K): δ = −14.8 (sextet, 1J27Al–31P = 264 Hz, [AlCl3(PEt3)]), 32.2 (1J31P–117Sn = 2312 Hz, 1J31P–119Sn = 2394 Hz, new Sn-PEt3 containing cation – see text). 119Sn NMR (CD2Cl2, 298 K): δ = −449 (t, 1J31P–119Sn = 2394 Hz).
[SnCl3(PEt3)2][BArF]
A Schlenk was charged with [SnCl4(PEt3)2] (0.100 g, 0.20 mmol) and Na[BArF] (0.178 g, 0.20 mmol). To this CH2Cl2 (5 mL) was added the resulting cloudy solution was stirred for 2 h, the solution was then filtered and concentrated to dryness in vacuo yielding a white solid. Yield: 0.201 g, 75%. Required for C44H42BCl3F24P2Sn (1324.3): C, 39.9, H, 3.2 Found C, 39.8, H, 3.3%. 1H NMR (CD2Cl2, 295 K): δ = 1.35 (m, [18H], CH3), 2.29 (m, [12H], CH2), 7.57 (s, [4H], [BarF]), 7.72 (m, [8H], [BarF]); 19F{1H} NMR (CD2Cl2, 295 K): δ = −63.0 (s, –CF3) 31P{1H} NMR (CD2Cl2, 295 K): δ = 37.1 (1J31P–117Sn = 2208 Hz, 1J31P–119Sn = 2311 Hz); 119Sn NMR (CD2Cl2, 298 K): δ = −379 (t, 1J31P–119Sn = 2311 Hz).[SnCl3(AsEt3)2][BArF]
A Schlenk was charged with [SnCl4(AsEt3)2] (0.100 g, 0.17 mmol) and Na[BArF] (0.152 g, 0.17 mmol). To this CH2Cl2 (5 mL) was added the resulting cloudy solution was stirred for 2 h and then filtered and concentrated to dryness in vacuo yielding a white solid. Yield: 0.162 g, 67%. Required for C44H42As2BCl3F24Sn (1412.2): C, 37.4, H, 3.0 Found C, 37.3, H, 2.8%. 1H NMR (CD2Cl2, 295 K): δ = 1.44 (t, 3JHH = 7.7 Hz, [18H], CH3), 2.42 (q, 3JHH = 7.7 Hz, [12H], CH2), 7.57 (s, [4H], [BarF]), 7.72 (m, [8H], [BarF]); 19F{1H} NMR (CD2Cl2, 295 K): δ = −63.0 (s, –CF3); 119Sn NMR (CD2Cl2, 183 K): δ = −388 (br s).[SnCl3{o-C6H4(PMe2)2}][BArF]
A Schlenk was charged with [SnCl4{o-C6H4(PMe2)2}] (0.100 g, 0.17 mmol) and Na[BArF] (0.152 g, 0.17 mmol) and to this CH2Cl2 (5 mL) was added. The resulting cloudy solution was stirred for 2 h and then filtered and concentrated to dryness in vacuo yielding a white solid. Yield: 0.162 g, 67%. Required for C42H28BCl3F24P2Sn (1286.2): C, 39.2, H, 2.2 Found C, 39.3, H, 2.0%. 1H NMR (CD2Cl2, 295 K): δ = 2.01 (t, 2J + 5JPH = 4.7 Hz, [12H], CH3), 7.57 (s, [4H], [BArF]), 7.72 (m, [8H], [BArF]), 7.82 (m, [4H], [BArF]);19F{1H} NMR (CD2Cl2, 295 K): δ = −63.0 (s, –CF3); 31P{1H} NMR (CD2Cl2, 295 K): δ = −24.5 (1J31P–Sn = 1007 Hz); 119Sn NMR (CD2Cl2, 183/298 K): δ = n.o.[SnBr3(AsEt3)2][BArF]
A Schlenk was charged with [SnBr4(AsEt3)2] (0.077 g, 0.1 mmol) and Na[BArF] (0.090 g, 0.1 mmol) to this CH2Cl2 (5 mL) was added; the resulting cloudy solution was stirred for 2 h, filtered and concentrated to dryness in vacuo yielding a light yellow gum, which was then dissolved in Et2O (5 mL), then the solution was concentrated to dryness in vacuo yielding a light yellow solid. Yield: 0.091 g, 58%. Required for C44H42As2BBr3F24Sn (1448.2): C, 34.2, H, 2.7. Found C, 34.3, H, 2.9%. 1H NMR (CD2Cl2, 295 K): δ = 1.39 (t, 3JHH = 7.6 Hz, [18H], CH3), 2.42 (br, [12H], CH2), 7.58 (s, [4H], [BarF]), 7.73 (m, [8H], [BarF]); 13C{1H}NMR (CD2Cl2, 295 K): δ = 9.1 (s, –CH3), 15.9 (s, –CH2), 118.1(s), 123.8(s), 126.5(s), 129.3(m), 135.4(s), 162.3(q, 1J11B–13C = 49.9 Hz); 19F{1H} NMR (CD2Cl2, 295 K): δ = −62.8 (s, –CF3). 119Sn NMR (CD2Cl2, 183/298 K): δ = n.o.X-Ray experimental
Crystals of the complexes were grown from CH2Cl2 solutions allowed to evaporate slowly in the glovebox. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724 + detector mounted at the window of an FR-E + SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 μm focus) with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(S/L)97, SHELX-2013, or SHELX-2014/7.41.18 H atoms bonded to C were placed in calculated positions using the default C–H distance and refined using a riding model. Details of the crystallographic parameters are given in Table S1 in the ESI.† CCDC reference numbers for the crystallographic information files in cif format are CCDC 1916557 [SnCl4(AsEt3)2], CCDC 1916558 [SnBr4(AsEt3)2], CCDC 1916559 [SnCl3(PMe3)2(OTf)], CCDC 1916560 [SnCl3(AsEt3)2(OTf)], CCDC 1916561 [SnCl2(PEt3)3][AlCl4]2, CCDC 1916562 [Et3AsCl][SnCl5(AsEt3)].†Results and discussion
Neutral tin(IV) halide complexes
The neutral tin(IV) phosphine complexes used in this study were made by literature methods or minor modifications thereof, viz- [SnCl4(PEt3)2],17 [SnCl4(PMe3)2],16 [SnBr4(PMe3)2],16 [SnCl4{o-C6H4(PMe2)2}],4 [SnCl4(Et2PCH2CH2PEt2)],4 as was [SnCl4{o-C6H4(AsMe2)2}].16 Their identity and purity were confirmed by comparison with literature data; key spectroscopic data are given in Table 1 for comparison purposes. Tin complexes of triethylarsine were reported as long ago as 1949,19 but lacked any spectroscopic or structural data. The crystal structure of [SnCl4(AsEt3)2] (Fig. 1(a)) reveals it to be the trans isomer and that the d(Sn–Cl) are not significantly different to those in trans-[SnCl4(PEt3)2]17 or trans-[SnCl4(PMe3)2],11 but slightly longer than that in trans-[SnCl4(AsPh3)2]20 (2.415(1), 2.424(1) Å). The d(Sn–As) of 2.65964(16) Å is significantly shorter than that in the triphenylarsine complex (2.7623(3) Å), consistent with the stronger donor power of the trialkylarsine ligand. In trans-[SnBr4(AsEt3)2] (Fig. 1(b)) the d(Sn–As) is 2.6773(3) Å, only very slightly longer than in the chloride.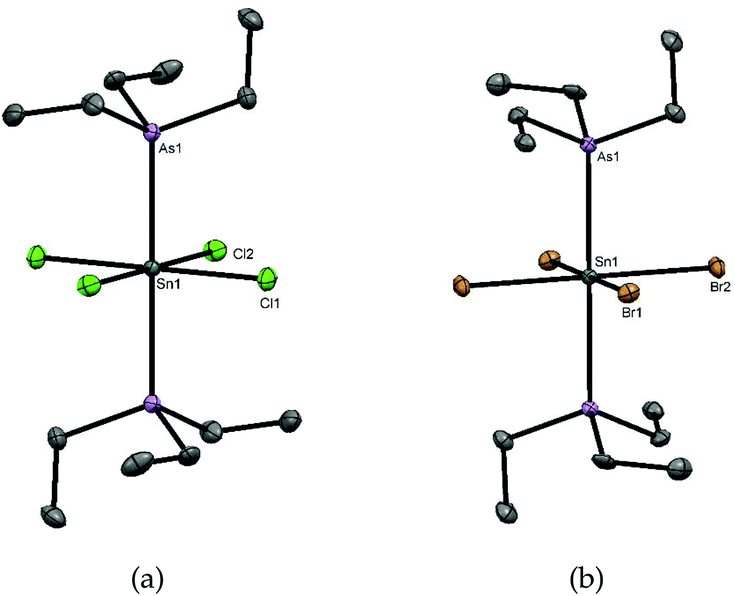 | ||
| Fig. 1 (a) The structure of [SnCl4(AsEt3)2] showing the atom numbering scheme. Ellipsoids are drawn at the 50% level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–Cl1 = 2.4498(4), Sn1–Cl2 = 2.4544(4), Sn1–As1 = 2.65964(16), As1–Sn1–As1 = 180.0, Cl1–Sn1–Cl2 = 90.367(15), Cl1–Sn1–Cl1 = 180.0, Cl2–Sn1–Cl2 = 180.0; (b) the structure of [SnBr4(AsEt3)2] showing the atom numbering scheme. Ellipsoids are drawn at the 50% level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–Br1 = 2.6286(3), Sn1–Br2 = 2.6173(3), Sn1–As1 = 2.6773(3), As1–Sn1–As1 = 180.0, Br1–Sn1–Br2 = 89.298(11), Br1–Sn1–Br1 = 180.0, Br2–Sn1–Br2 = 180.0. | ||
| Compound | δ(31P{1H}) | δ(119Sn) | 1 J(119Sn–31P)/Hz | ν(Sn–X)/cm−1 | Ref. |
|---|---|---|---|---|---|
| a NMR data from CH2Cl2 solution at 293 K, except. b 193 K. | |||||
| [SnCl4(PMe3)2] | +1.6 | −554 | 2635 | 321 | 11 |
| [SnCl4(PEt3)2] | +20.9 | −572 | 2391 | — | 17 |
| [SnCl4{o-C6H4(PMe2)2}] | −28.1 | −616 | 945 | 307, 296 | 4 |
| [SnCl4(Et2PCH2CH2PEt2)] | −4.9 | −615 | 1049 | 318, 307, 282 | 4 |
| [SnCl4{o-C6H4(AsMe2)2}] | — | −675 | — | 323, 315, 304, 300 | 16 |
| [SnCl4(AsEt3)2] | — | −657b | — | 288 | This work |
| [SnBr4(AsEt3)2] | — | −1173b | — | 210 | This work |
| [SnCl3(AsEt3)2(OTf)] | — | −620b | — | 294, 378 | This work |
| [SnCl3(AsEt3)2][BArF] | — | −388 | — | — | This work |
| [SnCl3(PMe3)2(OTf)] | +8.7 | −516 | 2963 | 296, 306 | This work |
| [SnCl3(PEt3)2(OTf)] | +33.4b | −535b | 2737b | 289, 301 | This work |
| [SnCl3(PEt3)2][AlCl4] | +35.5 | −350 | 2307 | 280, 289 | This work |
| [SnCl2(PEt3)2][AlCl4]2 | +40.8 | −301 | 2274 | 289 | This work |
| [SnCl3(PEt3)2][BArF] | +37.1 | −379 | 2311 | 286 | This work |
| [SnCl3(PMe3)2][AlCl4] | +10.8 | −456 | 2725 | — | 11 |
| [SnCl2(PMe3)2][AlCl4]2 | +16.9 | −333 | 2591 | — | 11 |
| [SnCl3{o-C6H4(PMe2)2}][AlCl4] | −24.7 | −469 | 972 | 322, 342 | This work |
| [SnCl2{o-C6H4(PMe2)2}][AlCl4]2 | −23.7 | −429 | 853 | 294, 305 | This work |
Attempts to isolate a complex of AsEt3 with SnI4 were not successful, as noted previously by others.19
Addition of a CH2Cl2 solution of SbR3 (R = Et or iPr) to CH2Cl2 solutions of SnX4 (X = Cl or Br) result in immediate precipitation of grey solids, which contain essentially no C/H (microanalysis), whilst the supernatant solutions contain R3SbX2, based upon comparisons of their 1H NMR spectra with literature data and ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 This suggests that any Sn(IV)–stibine complexes are too unstable to isolate, and redox chemistry occurs [SnX4 + SbR3 → SnX2 + R3SbX2]. Cunningham et al.22 reported that reaction of SbPh3 with SnX4 initially gave yellow solids which became white on washing with pentane. The white solids had 119Sn Mössbauer spectra consistent with SnX2, indicating similar, but slower, redox chemistry occurring with the triarylstibine. The redox chemistry in these tin systems contrasts with the reactions of Group 13 trihalides with trialkylstibines, which gave simple adducts.6
21 This suggests that any Sn(IV)–stibine complexes are too unstable to isolate, and redox chemistry occurs [SnX4 + SbR3 → SnX2 + R3SbX2]. Cunningham et al.22 reported that reaction of SbPh3 with SnX4 initially gave yellow solids which became white on washing with pentane. The white solids had 119Sn Mössbauer spectra consistent with SnX2, indicating similar, but slower, redox chemistry occurring with the triarylstibine. The redox chemistry in these tin systems contrasts with the reactions of Group 13 trihalides with trialkylstibines, which gave simple adducts.6
Reactions of [SnX4(ER3)2] (E = P, As) and [SnCl4(L–L)] (L–L = o-C6H4(PMe2)2, Et2P(CH2)2PEt2 or o-C6H4(AsMe2)2) with TMSOTf
The complexes [SnX4(ER3)2] (X = Cl, ER3 = PMe3, PEt3, AsEt3; X = Br, ER3 = PMe3, AsEt3) were treated with one mol. equivalent of TMSOTf in anhydrous CH2Cl2 solution with a view to removing one chloride ligand (as TMSCl) to create a tin cation. Triflate can behave either as an anion or coordinate to the metal; in practice we found that that it coordinates to the tin in the present study.For the chloride systems the reactions proceeded to completion in ∼2 h to give good yields of moisture-sensitive white powders, identified by microanalysis as [SnCl4(ER3)2(OTf)] (Scheme 1). Curiously, the corresponding reactions with the tin(IV) bromide complexes did not occur, and after 24 h the NMR spectra of these systems showed only unchanged starting materials. Similarly, no reaction occurred between [SnCl4{Et2P(CH2)2PEt2}],4 [SnCl4{o-C6H4(PMe2)2}]4 or [SnCl4{o-C6H4(AsMe2)2}]16 with TMSOTf in CH2Cl2 solution even after 3 d. For the systems which produced mono triflate complexes, use of more TMSOTf failed to give any evidence that a second chloride could be removed.
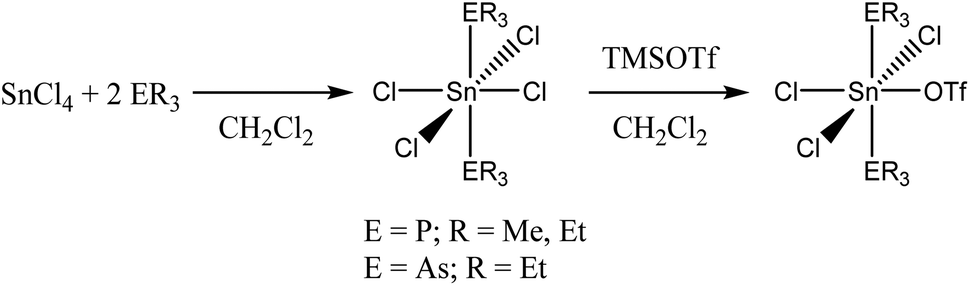 | ||
| Scheme 1 Reactions with TMSOTf. | ||
Colourless crystals of [SnCl3(PMe3)2(OTf)] and [SnCl3(AsEt3)2(OTf)] were grown by slow evaporation of solutions in anhydrous CH2Cl2, and their X-ray structure analyses revealed the geometry shown in Fig. 2. The structures show six-coordinate tin centres with mer-chlorines, and trans pnictines with the triflate trans to chlorine. Comparison of the two structures shows that the d(Sn–CltransCl) are not significantly different and in both cases are longer than d(Sn–CltransOTf). The d(Sn–CltransCl) and d(Sn–As) are also similar to those in [SnCl4(AsEt3)2]. The d(Sn–O) distances are ∼2.26 Å, which fall at the lower end of the range found in octahedral Sn(IV) triflates (∼2.2–2.6 Å)23 and may be compared to the sum of the covalent radii of Sn and O (2.15 Å).24
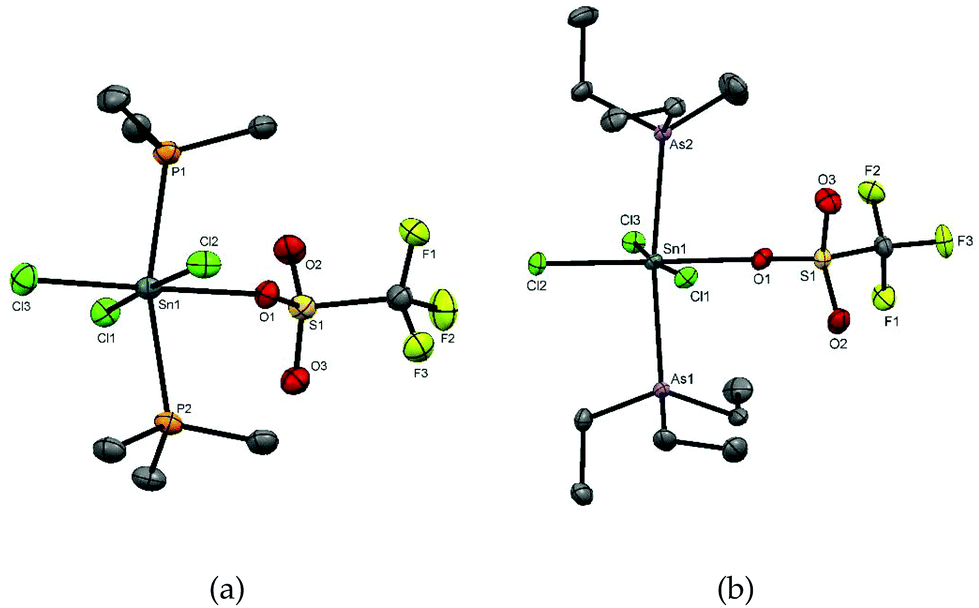 | ||
| Fig. 2 (a) The structure of [SnCl3(PMe3)2(OTf)] showing the atom numbering scheme. Ellipsoids are drawn at the 50% level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–Cl1 = 2.4457(9), Sn1–Cl2 = 2.4558(9), Sn1–Cl3 = 2.4085(10), Sn1–P1 = 2.5496(9), Sn1–P2 = 2.5506(9), Sn1–O1 = 2.266(3), P1–Sn1–P2 = 164.16(3), Cl1–Sn1–Cl2 = 174.66(3), Cl3–Sn1–Cl1 = 92.67(3), Cl3–Sn1–Cl2 = 92.68(4), O1–Sn1–Cl1 = 89.25(7), O1–Sn1–Cl2 = 85.41(7), O1–Sn1–P1 = 82.17(7), O1–Sn1–P2 = 82.51(7); (b) the structure of [SnCl3(AsEt3)2(OTf)]: selected bond lengths (Å) and angles (°) are: Sn1–Cl1 = 2.4480(7), Sn1–Cl2 = 2.4168(8), Sn1–Cl3 = 2.4533(7), Sn1–As1 = 2.6451(4), Sn1–As2 = 2.6530(4), Sn1–O1 = 2.259(2), As1–Sn1–As2 = 172.239(12), Cl1–Sn1–Cl2 = 92.83(3), Cl3–Sn1–Cl1 = 173.76(3), Cl3–Sn1–Cl2 = 93.17(3), O1–Sn1–As2 = 86.28(6), O1–Sn1–As1 = 85.96(6), O1–Sn1–Cl1 = 88.46(6), O1–Sn1–Cl3 = 85.52(6). | ||
Comparison of the NMR spectroscopic data (Table 1) shows that conversion of [SnCl4(PR3)2] into [SnCl3(PR3)2(OTf)] results in modest high frequency shifts of the 119Sn and 31P resonances and a more marked increase in 1JSn–P. The 119Sn chemical shift also moves to high frequency in [SnCl3(AsEt3)2(OTf)] compared to [SnCl4(AsEt3)2].
The reaction of SnCl4, SbEt3 and TMSOTf in CH2Cl2 again resulted in redox chemistry, with [Et3SbCl(OTf)] isolated as one product in ∼30% yield, along with much insoluble grey solid, which had no significant C/H content and is most likely SnCl2 (cf.ref. 22). Crystals of the former were grown from CH2Cl2 solution and although the crystal quality was poor, confirmed them to be [Et3SbCl(OTf)], which has a trigonal bipyramidal geometry with equatorial ethyl groups (see ESI†).
Reactions of [SnX4(ER3)2] and [SnCl4(L–L)] with AlX3
No examples of ‘self-ionisation’, such as [SnX3(ER3)2]2[SnX6] or [SnX2(L–L)2][SnX6], have been reported in reaction of SnX4 with pnictines,5i.e. SnX4 is not a sufficiently strong Lewis acid to abstract a halide from a second tin centre in these systems. This is also consistent with ligands such as MeC(CH2AsMe2)3 failing to displace a halide from a neutral [SnX4{κ2-MeC(CH2AsMe2)3}],4 to form [SnX3{κ3-MeC(CH2AsMe2)3}]+. However, using the stronger Lewis acid, AlCl3, has been successful in removing a chloride in some cases.11The [SnCl3(PEt3)2][AlCl4] and [SnCl2(PEt3)2][AlCl4]2 were made by treatment of [SnCl4(PEt3)2] with one and two mol. equivalents, respectively, of AlCl3 in anhydrous CH2Cl2 (Scheme 2), and behave similarly to the previously reported11 [SnClx(PMe3)2][AlCl4]y (x = 3, y = 1; x = 2, y = 2).
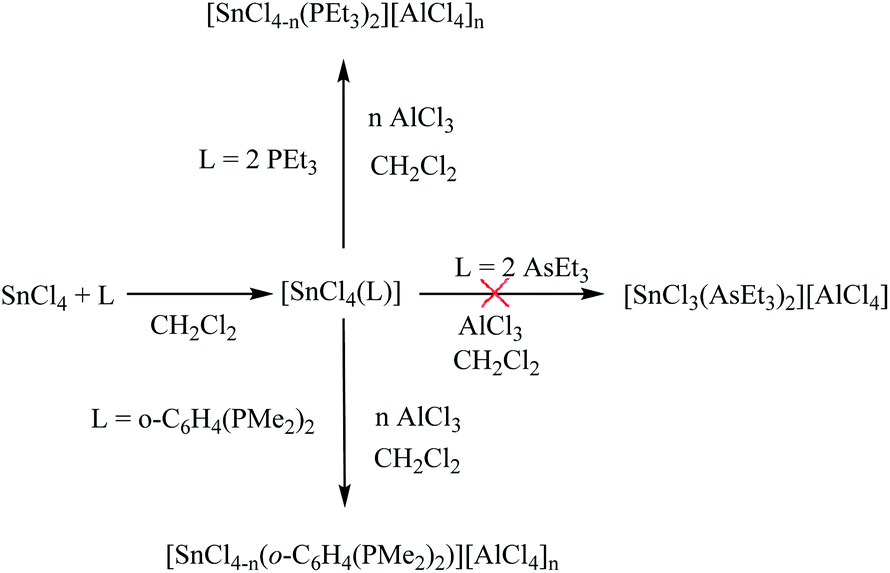 | ||
| Scheme 2 Reactions of the tin complexes with AlCl3. | ||
Attempts to grow crystals from a CH2Cl2 solution of [SnCl2(PEt3)2][AlCl4]2 produced a few small colourless crystals, one of which was found on X-ray structure solution to be [SnCl2(PEt3)3][AlCl4]2 containing a tris(phosphine) dication (Fig. 3), presumably as a result of rearrangement in solution during recrystallisation.
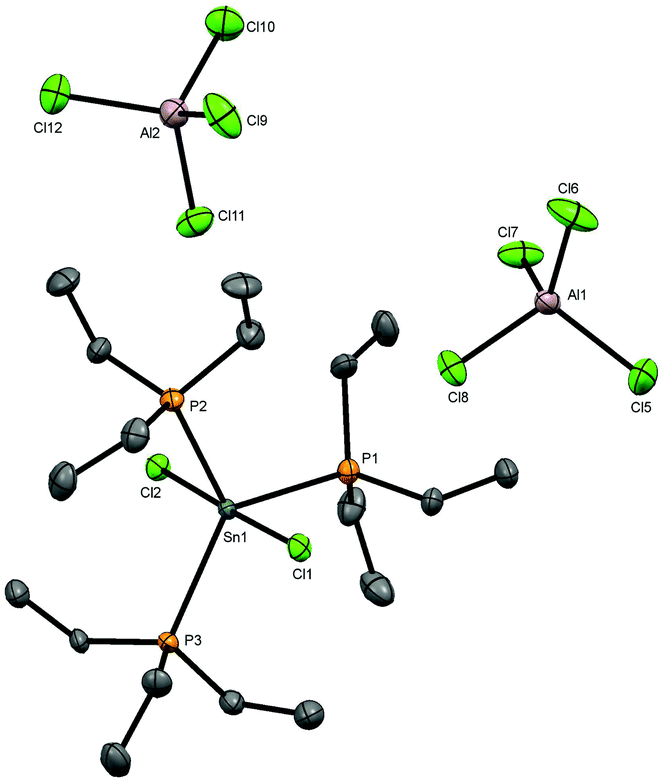 | ||
| Fig. 3 The structure of [SnCl2(PEt3)3][AlCl4]2 showing the atom numbering scheme. Note there are two crystallographically independent [SnCl2(PEt3)3][AlCl4]2 units in the asymmetric unit. Ellipsoids are drawn at the 50% level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–Cl1 = 2.4531(13), Sn1–Cl2 = 2.4589(13), Sn1–P1 = 2.5679(14), Sn1–P2 = 2.5610(13), Sn1–P3 = 2.5525(13), Cl2–Sn1–P3 = 91.10(4), Cl2–Sn1–P2 = 92.07(5), Cl2–Sn1–P1 = 86.01(4), Cl1–Sn1–Cl2 = 178.28(4), Cl1–Sn1–P3 = 90.52(4), Cl1–Sn1–P2 = 87.51(4), Cl1–Sn1–P1 = 92.63(4), P3–Sn1–P2 = 123.83(4), P3–Sn1–P1 = 122.85(4), P2–Sn1–P1 = 113.31(5). | ||
The structure contains a trigonal bipyramidal cation with axial chlorides, the bond angles showing only small deviations from the idealised values, and in contrast to [SnCl2(PMe3)2][AlCl4]2, there is no interaction of the tin centre with the tetrachloroaluminate anions. This is likely due to the steric effect caused by the presence of the third PEt3 ligand. The d(Sn–Cl) are very similar to the d(Sn–CltransCl) in the OTf complex in Fig. 3. There were insufficient crystals produced to obtain any spectroscopic data, so direct synthesis of a bulk sample was explored. However, addition of one mol. equivalent of PEt3 to [SnCl2(PEt3)2][AlCl4]2 in anhydrous CH2Cl2 solution does not form the new cation [SnCl2(PEt3)3]2+. In situ NMR studies reveal a mixture of three main species (Experimental section); [AlCl4]−, [AlCl3(PEt3)] (identified by its characteristic NMR spectra25) and a new Sn-PEt3 containing species. The 119Sn NMR spectrum shows a triplet 1J(119Sn–31P) coupling and hence indicates only two phosphines coordinated to the tin centre, e.g. [SnClx(PEt3)2]n+. The 31P and 119Sn chemical shifts do not correspond to those of [SnCl3(PEt3)2][AlCl4].
The solution NMR data (Table 1) show that along the series [SnCl4(PEt3)2], [SnCl3(PEt3)2][AlCl4], [SnCl2(PEt3)2][AlCl4]2, the chemical shifts move to higher frequency in both the 31P and 119Sn spectra. The 31P{1H} and 119Sn chemical shifts of the cations also vary significantly with temperature and the tin spectra have rather broad resonances, which probably reflects a variation in the interaction of the cations with the [AlCl4]− anions under different conditions.
The reaction of [SnCl4(AsEt3)2] with AlCl3 in CH2Cl2 did not proceed similarly to the reactions involving PEt3. Monitoring the reaction by NMR spectroscopy identified a mixture of species present including starting materials and Et3AsCl2.26 A few crystals grown from this mixture contained the chlorotriethylarsonium(V) cation and a pentachloro(triethylarsine)stannate(IV) anion, [Et3AsCl][Sn(AsEt3)Cl5] (Fig. 4).
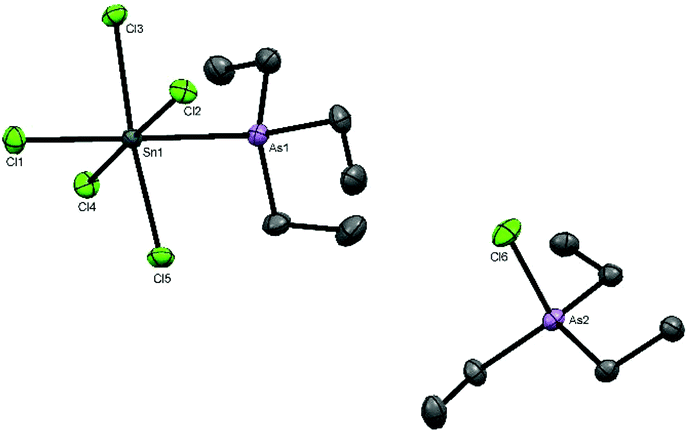 | ||
| Fig. 4 The structure of [Et3AsCl][SnCl5(AsEt3)] showing the atom numbering scheme. Ellipsoids are drawn at the 50% level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) are: Sn1–As1 = 2.6696(5), Sn1–Cl3 = 2.4453(8), Sn1–Cl4 = 2.4304(9), Sn1–Cl2 = 2.4365(8), Sn1–Cl1 = 2.3930(9), Sn1–Cl5 = 2.4254(8), Cl3–Sn1–As1 = 87.09(2), Cl4–Sn1–As1 = 85.21(2), Cl4–Sn1–Cl3 = 91.59(3), Cl2–Sn1–As1 = 92.00(2, Cl2–Sn1–Cl3 = 88.20(3), Cl1–Sn1–Cl3 = 90.72(3, Cl1–Sn1–Cl4 = 90.90(3), Cl1–Sn1–Cl2 = 91.88(3), Cl1–Sn1–Cl5 = 93.86(3), Cl5–Sn1–As1 = 88.54(2), Cl5–Sn1–Cl4 = 91.32(3), Cl5–Sn1–Cl2 = 88.66(3). | ||
Arsine substituted halostannate anions do not seem to have been reported previously,5 and the nearest literature example is the zwitterionic phosphine complex [SnCl5(Ph2PCH2PPh2H)].27 Comparison of the key bond lengths with those in trans-[SnCl4(AsEt3)2] shows that the d(Sn–As) and d(Sn–CltransCl) are little different, although d(Sn–CltransAs) is ∼0.05 Å shorter. The reaction of trans-[SnBr4(AsEt3)2] with AlBr3 also produced a mixture of products, including Et3AsBr2.26
The reactions of [SnCl4{o-C6H4(PMe2)2}] with one and two mol. equivalents of AlCl3 in anhydrous CH2Cl2 were also investigated. The products were identified by microanalysis as [SnCl3{o-C6H4(PMe2)2}][AlCl4] and [SnCl2{o-C6H4(PMe2)2}][AlCl4]2. These very moisture-sensitive complexes were poorly soluble in CH2Cl2 and attempts to grow crystals have been unsuccessful. Obtaining multinuclear NMR data was also hindered by their poor solubility in weakly coordinating organic solvents. However, the trends in chemical shifts and coupling constants within the series (Table 1) replicate those observed for [SnClx(PEt3)2][AlCl4]y (x = 4, y = 0; x = 3, y = 1; x = 2, y = 2), with δ119Sn and δ31P NMR moving to high frequency on going from the neutral complex to the mono- and the di-cations. The 1J(119Sn–31P) also follow the same trend as the PMe3 complexes, with 1J31P–119Sn [SnCl3{o-C6H4(PMe2)2}]+ > [SnCl4{o-C6H4(PMe2)2}] > [SnCl2{o-C6H4(PMe2)2}]2+.
[SnCl4{o-C6H4(AsMe2)2}] also reacted with AlCl3 in anhydrous CH2Cl2 to form a poorly soluble white product, but the NMR spectra indicated a mixture of species present, which we were unable to identify.
Reactions of [SnX4(EEt3)2] and [SnCl4(L–L)] with Na[BArF]
Addition of Na[BArF] to a CH2Cl2 solution [SnX4(EEt3)2] (E = P; X = Cl or Br; E = As; X = Cl) or [SnCl4{o-C6H4(PMe2)2}] (Scheme 3) readily formed the [BArF]− salts, [SnX3(EEt3)2][BArF] and [SnCl3{o-C6H4(PMe2)2}][BArF], with precipitation of NaX. The NMR spectroscopic data for the cations match those of the [AlCl4]− salts discussed above, and using Na[BArF] as the halide abstractor allowed generation of the [SnBr3(AsEt3)2]+ cation that proved elusive with the other halide abstractors. Using excess Na[BArF] did not lead to further halide abstraction. | ||
| Scheme 3 Reactions of the tin complexes with Na[BArF]. | ||
DFT studies
Computational studies of the tin complexes were undertaken to supplement the experimental data. Ground state geometries of [SnCl3(PMe3)2][AlCl4], [SnCl4(AsEt3)2], [SnCl3(AsEt3)2(OTf)], [SnCl4(PEt3)2],17 [SnCl4(PMe3)2],11 [SnCl3(PMe3)2(OTf)], [SnCl3(PMe3)2]+ were optimised alongside the uncoordinated ligands AsEt3 and PEt3 using DFT with the B3LYP functional and a basis set with a Stuttgart–Dresden effective core potential on Sn and As atoms and with an all-electron double-ζ basis set on all electrons in H, C, O, F, P, Al, S, and Cl atoms. Comparison of DFT results for the [SnCl3(PMe3)2][AlCl4] and [SnCl4(PMe3)2] against those reported,11 was used to test the suitability of the basis set – see ESI.†The bond lengths from the calculated ligands are similar to those from available experimental data (Table S2†) and show that the average C–E–C angles from AsEt3 and PEt3 (98.5 and 100.4°, respectively) are slightly wider than those found in AsMe3 (97.0°) and PMe3 (98.8°). The average As–C bond distances from the DFT analysis (∼1.98 Å) are similar to those from experimental data (∼1.94 Å) for the tin complexes, showing a shortening in comparison to ‘free’ AsEt3 (As–C = 2.01 Å). The P–C bond distances also follow this trend, with a shortening from the phosphine complexes (DFT: 1.87–1.89 Å; X-ray: ∼1.79 Å), compared to the free PMe3 and PEt3 (1.90–1.92 Å; X-ray: 1.84 Å). This, when combined with the widening of the C–As–C (DFT: 105.33–106.2°; X-ray: 105.9–106.0°) and C–P–C angles (DFT: 106.3–109.1°; X-ray: 107.1–109.1°) of the complexes, against the values for the ‘free’ ligands (AsEt3: 98.5°; PMe3: 98.8° and PEt3: 100.4°), matches the trends seen for trialkylstibine ligands upon coordination to Group 13 metal halides,6 although the effect here is much smaller. The widening of the C–E–C angle corresponds to an increased s-character in the E–C bond and increased p-character in the lone pair. The comparable data from the halo tin-arsine and phosphine complexes shows the free ligands have s-character of AsEt3: 12.20; PMe3: 15.08; PEt3: 15.57%, which doubles to 25.3–25.9% upon coordination.
Calculating the ground state structure of the [SnCl4(AsEt3)2] and [SnCl3(AsEt3)2(OTf)] complexes, alongside DFT calculations of the ionic [SnCl3(AsEt3)2]+ complex, show that there is very little change in the bond lengths/angles and this is consistent with reported data for [SnCl3(PMe3)2]+. Burford's complexes are also noted for a significant decrease in the P–Sn–P angle with increasing charge,11 the neutral [SnCl4(AsEt3)2] complex (X-ray: 180; DFT: 179.9°) and the cationic [SnCl3(AsEt3)2]+ species show a similar trend (DFT: 174°).
Notably, while the HOMO and LUMO energies in the neutral tetrahalide and triflate complexes are not significantly different, those of the cationic species are some 4 eV lower in energy, consistent with the latter displaying higher Lewis acidity (Table S7†). Furthermore, using the AsEt3 complexes as examples, the HOMO is concentrated on the SnCl4 unit in the [SnCl4(AsEt3)2], with some delocalisation onto the AsEt3 ligands in [SnCl3(AsEt3)2(OTf)], and this is even more obvious in [SnCl3(AsEt3)2]+ (Fig. 5).
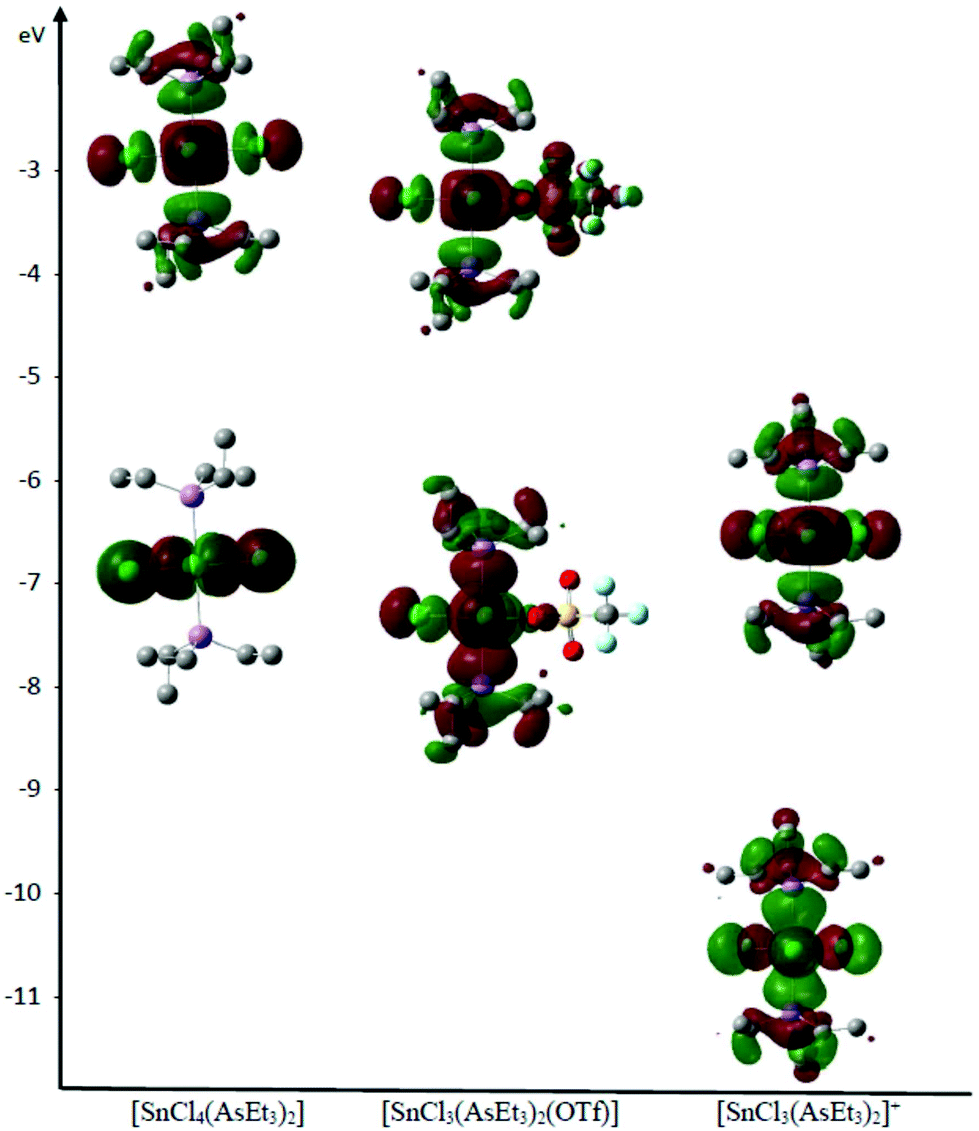 | ||
| Fig. 5 Representation of the LUMO and HOMO of [SnCl4(AsEt3)2], [SnCl3(AsEt3)2(OTf)] and [SnCl3(AsEt3)2]+ determined by DFT analysis, showing their relative energies. | ||
Conclusions
This study has confirmed the high stability of tin tetrahalide complexes with neutral phosphine and arsine ligands and has demonstrated the formation of some complexes with SnIV:X ratio of 1:3 or 1:2.
In the case of SbEt3, we found that SnCl4 causes chlorination of the stibine, consistent with the greater stability of Sb(V) over As(V) (although P(V) is also more stable than As(V), the stronger basicity of the PR3 ligands means that coordination is favoured over chlorination in this system).
Replacement of chloride by triflate to form [SnCl3(ER3)2(OTf)] is possible for ER3 = PMe3, PEt3, AsEt3, but not with complexes of chelating bidentate ligands. This may indicate that reversible dissociation of the neutral ligand plays a role in the substitution. The failure of corresponding [SnBr4(ER3)2] to react with TMSOTf was unexpected, although formation of TMSBr is less favoured energetically.
Aluminium trichloride is able to abstract chloride ligands from [SnCl4(PEt3)2] and [SnCl4{o-C6H4(PMe2)2}] to form mono- and di-cationic Sn(IV) complexes, but these reactions do not occur with arsine ligands, possibly due to the weaker donor arsines having less affinity for the hard tin(IV) cations. On the other hand, using the large, weakly coordinating [BArF]− anion does lead smoothly to the formation of [SnCl3(EEt3)2][BArF] for E = P and As, [SnBr3(AsEt3)2][BArF], as well as affording [SnCl3{o-C6H4(PMe2)2}][BArF].
In practice, the Na[BArF] works better as a halide abstractor to form the monocations (but does not form dications), whereas AlCl3 can compete for coordination to the weakly bound arsine ligand, whilst it is clear that the triflate anion can coordinate readily to Sn(IV).
DFT studies have shown that bonding and orbital components in the triflate complexes are very similar to those for the tetrachlorides, while the HOMO and LUMO energies are somewhat lower (by ca. 4 eV) for the cations compared to the neutral species, indicating that the cationic species are stronger Lewis acids.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank EPSRC for support via EP/P025137/1 and EP/N035437/1 and for a studentship to R. P. K. (EP/N509747/1).References
- Comprehensive Coordination Chemistry, ed. G. Wilkinson, J. A. McCleverty and R. D. Gillard, Pergamon, Oxford, 1988 Search PubMed; Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004 Search PubMed.
- P. G. Harrison, Comprehensive Coordination Chemistry, 1988, 3, 183 CrossRef CAS PubMed; J. Parr, Comprehensive Coordination Chemistry II, 2004, 3, 545 CrossRef CAS PubMed; The Chemistry of Tin, ed. P. J. Smith, Chapman & Hall, London, 1998 CrossRef CAS PubMed; M. Gawron, C. Dietz, M. Lutter, A. Duthie, V. Jouikov and K. Jurkschat, Chem. – Eur. J., 2015, 21, 16609 CrossRef CAS PubMed.
- G. R. Willey, T. J. Woodman, U. Somasundaram, D. R. Aris and W. Errington, J. Chem. Soc., Dalton Trans., 1998, 2575 Search PubMed; G. R. Willey, A. Jarvis, J. Palin and W. Errington, J. Chem. Soc., Dalton Trans., 1994, 255 RSC.
- M. F. Davis, M. Clarke, W. Levason, G. Reid and M. Webster, Eur. J. Inorg. Chem., 2006, 2773 CrossRef CAS.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65 CrossRef CAS.
- V. K. Greenacre, W. Levason and G. Reid, Organometallics, 2018, 37, 2123 CrossRef CAS.
- A. C. T. Kuate, M. Schürmann, D. Schollmeyer, W. Hiller and K. Jurkschat, Chem. – Eur. J., 2010, 16, 8140 CrossRef PubMed.
- E. MacDonald, L. Doyle, N. Burford, U. Werner-Zwanziger and A. Decken, Angew. Chem., Int. Ed., 2011, 50, 11474 CrossRef CAS PubMed.
- J. Hanning, H. Schubert, K. Eichele, F. Winter, R. Pöttgen, H. A. Meyer and L. Wesemann, Inorg. Chem., 2012, 51, 5787 CrossRef PubMed.
- M. Driess, C. Monse, K. Merz and C. van Wüllen, Angew. Chem., Int. Ed., 2000, 39, 3684 CrossRef CAS PubMed.
- E. MacDonald, L. Doyle, S. S. Chitnis, U. Werner-Zwanziger, N. Burford and A. Decken, Chem. Commun., 2012, 48, 7922 RSC.
- R. Suter, A. Swidan, H. S. Zijlstra, C. L. B. Macdonald, J. S. McIndoe and N. Burford, Dalton Trans., 2018, 47, 16729 RSC.
- E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877 CrossRef CAS; R. D. Feltham, A. Kasenally and R. S. Nyholm, J. Organomet. Chem., 1967, 7, 285 CrossRef.
- W. Taylor, U. H. Soto, V. M. Lynch and M. J. Rose, Inorg. Chem., 2016, 55, 3206 CrossRef CAS PubMed.
- M. Brookhart, B. Grant and A. F. Volpe Jr., Organometallics, 1992, 11, 3920 CrossRef CAS.
- A. R. J. Genge, W. Levason and G. Reid, Inorg. Chim. Acta, 1999, 288, 142 CrossRef CAS.
- G. G. Mather, G. M. McLaughlin and A. Pidcock, J. Chem. Soc., Dalton Trans., 1973, 1823 RSC; W. McFarlane and N. H. Rees, Polyhedron, 1989, 8, 2047 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 11 CrossRef PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- J. A. C. Allison and F. G. Mann, J. Chem. Soc., 1949, 2915 RSC.
- M. F. Mahon, N. I. Moldovan, K. C. Molloy, A. Muresan, I. Silaghi-Dumitrescu and L. Silaghi-Dumitrescu, Dalton Trans., 2004, 4017 RSC.
- H. J. Breunig and W. Kanig, Phosphorus Sulfur Relat. Elem., 1982, 12, 149 CrossRef CAS.
- D. Cunningham, M. J. Fraser and J. D. Donaldson, J. Chem. Soc. A, 1971, 2049 RSC.
- A. P. M. Robertson, J. N. Friedmann, H. A. Jenkins and N. Burford, Chem. Commun., 2014, 50, 7979 RSC; P. Svec, R. Olejnik, Z. Padelkova, A. Ruzicka and L. Plasseraud, J. Organomet. Chem., 2012, 708–709, 82 CrossRef CAS; B. Kasna, L. Dostal, R. Jirasko, I. Cisarova and R. Jambor, Organometallics, 2008, 27, 3743 CrossRef.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- J. Burt, W. Levason, M. E. Light and G. Reid, Dalton Trans., 2014, 43, 14600 RSC.
- L. Verdonck and G. P. Van Der Kelen, Spectrochim. Acta, Part A, 1977, 33, 601 CrossRef CAS; M. F. Davis, W. Levason, G. Reid and M. Webster, Dalton Trans., 2008, 2261 RSC.
- A. S. Batsanov, R. M. K. Deng, K. B. Dillon, A. E. Goeta, J. A. K. Howard, J. Meldrum, P. K. Monks, H. Puschmann and H. J. Shepherd, Heteroat. Chem., 2009, 20, 136 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Multinuclear NMR and IR spectra associated with each of the new compounds described, together with the X-ray crystallographic parameters and full details of the DFT calculations. CCDC 1916557–1916562. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03683k |
| This journal is © The Royal Society of Chemistry 2019 |