
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–property-reactivity studies on dithiaphospholes†‡
Darren M. C.
Ould
a,
Thao T. P.
Tran
b,
Jeremy M.
Rawson
 *b and
Rebecca L.
Melen
*a
*b and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Cardiff, CF10 3AT, Cymru/Wales, UK. E-mail: melenr@cardiff.ac.uk
bDepartment of Chemistry and Biochemistry, University of Windsor, 401 Sunset Avenue, Windsor, Canada. E-mail: jmrawson@uwindsor.ca
First published on 4th November 2019
Abstract
The reaction of either toluene-3,4-dithiol or benzene dithiol with phosphorus(III) trihalides generates the corresponding benzo-fused 1,3,2-dithiaphospholes, RC6H3S2PX (R = Me (1), R = H (2); X = Cl, Br, I). The P-chloro-dithiaphospholes undergo: (a) halogen abstraction reactions with Lewis acids forming phosphenium cations; (b) substitution with LiHMDS base and; (c) reduction chemistry with sodium metal to generate the P–P σ-bonded dimer, (RC6H3S2P)2. Reduction catalysis of aldehydes with pinacolborane using dithiaphospholes is compared with their dioxaphosphole and diazaphosphole counterparts as pre-catalysts, revealing interesting differences in the reactivity of this series of compounds.
Introduction
In the last few decades there has been a renaissance in main group chemistry stemming from the emerging materials applications of main group compounds1 alongside increasing applications as reagents and catalysts in organic synthesis.2 In all these areas, performance is being enhanced by the inclusion of main group elements or main-group functional groups to tailor electron-donating or withdrawing properties, enhancing donor or acceptor ability, redox behaviour or Lewis acidity inter alia. For example, in materials chemistry responsive organo-boron based luminescent materials3 and main group perovskites for solar cell applications4 have attracted considerable attention. For the later p-block elements the redox active nature of many electron-rich S/N and Se/N-based heterocycles have led to the discovery of some of the highest magnetic ordering temperatures for organic magnets,5 as well as molecule-based conductors6 and as redox active paramagnetic ligands inter alia.7 Alongside this, the ability to tune the Lewis acidity of early p-block elements has attracted attention in main group promoted or catalysed organic transformations8,9 while frustrated Lewis pair (FLP) combinations continue to activate a diverse range of small gas molecules including CO2 and dihydrogen.10Among the many aspects of main group chemistry, there has been increasing use of heterocyclic phosphorus compounds as catalysts for organic transformations, exemplified by work by Zhang et al. on the nucleophilicity of diazaphospholene hydrides.11 Gudat first showed the hydridic nature of the P–H bond in 1,3,2-diazaphospholenes (Scheme 1 top, X = H),12 which have been used in stoichiometric reduction reactions13 and more recently by Kinjo in the catalytic hydrogenation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond with ammonia borane14 as well as the catalytic hydroboration of carbonyl compounds with pinacolborane.15 Speed has also used 1,2,4,3-triazaphospholenes for the catalytic hydroboration of various carbonyl compounds,16 and reported the first use of diazaphospholenes as chiral catalysts by introducing chirality at the R group on the nitrogen atoms.17 The Cramer group has also used 1,3,2-diazaphospholenes for chiral reductive chemistry.18 The related phosphenium cations have also been used as pre-catalysts in the reduction of pyridines and imines with pinacolborane or silanes.19,20
N double bond with ammonia borane14 as well as the catalytic hydroboration of carbonyl compounds with pinacolborane.15 Speed has also used 1,2,4,3-triazaphospholenes for the catalytic hydroboration of various carbonyl compounds,16 and reported the first use of diazaphospholenes as chiral catalysts by introducing chirality at the R group on the nitrogen atoms.17 The Cramer group has also used 1,3,2-diazaphospholenes for chiral reductive chemistry.18 The related phosphenium cations have also been used as pre-catalysts in the reduction of pyridines and imines with pinacolborane or silanes.19,20
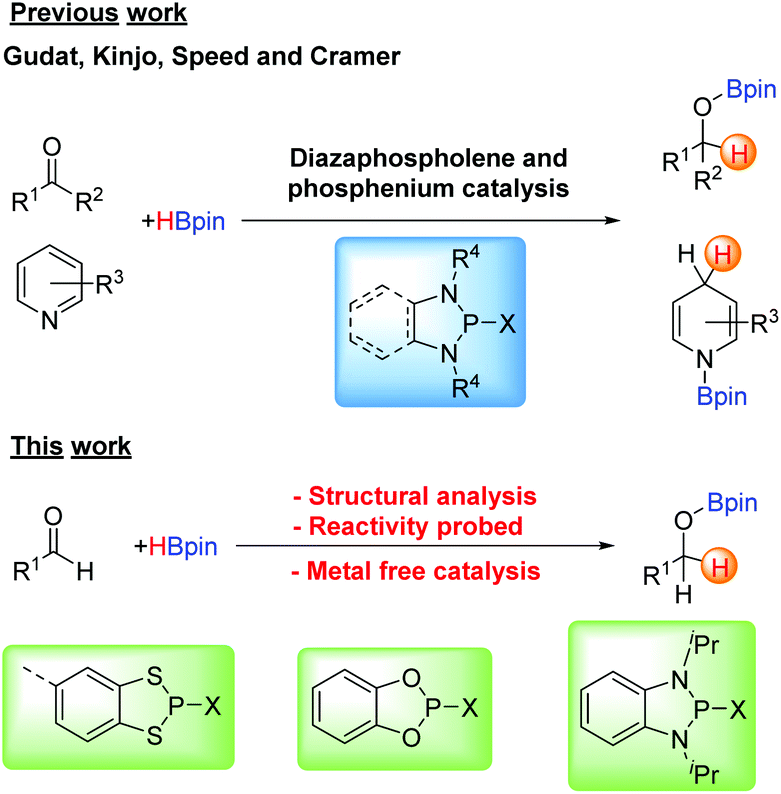 | ||
| Scheme 1 Previous use of diazaphospholenes for catalysis (Top) and this work (Bottom). | ||
The ability to tailor the reaction chemistry through application of the isolobal analogy gives rise to a large number of heterocycles closely related to the diazaphosphole unit. This includes replacement of P by heavier pnictogens such as As21 and one of our groups have recently identified that benzo-fused diaza-chloro-arsole is highly active for the reduction of aldehydes with pinacolborane.22 Conversely replacement of R–N by isolobal chalcogens leads to heterocycles such as the dioxaphospholes and dithiaphospholes (Scheme 1, bottom).
In the current study we report the syntheses, structures and reactivity patterns of benzo-fused 1,3,2-dithiaphospholes, which are closely related to N-heterocyclic phospholenes (NHPs) by replacement of NR by S. Finally we provide a comparative study of these dithiaphospholes with their isolobal dioxaphosphole and diazaphosphole counterparts as pre-catalysts for the hydroboration of aldehydes with pinacolborane.
Results and discussion
Synthesis
P-Halo-benzodithiaphospholes were prepared by Baudler23 in 1973 who showed that reduction led to P–P coupling to form the σ-bonded dimers 4 and 5 (Scheme 2). Burford showed that treatment of these P-halo dithiaphospholes with Lewis acids (AlCl3 or GaCl3) forms the corresponding dithiaphosphenium salts (Scheme 2).24–26 In our study, the syntheses of 1a–1c and 2a–2c essentially followed the protocols first described by Baudler (Scheme 2) via the addition of either toluene-3,4-dithiol or benzene dithiol to the appropriate phosphorus trihalide, PX3, under ambient conditions with the derivatives 1a–2c recovered in very good to excellent yields (73–96%).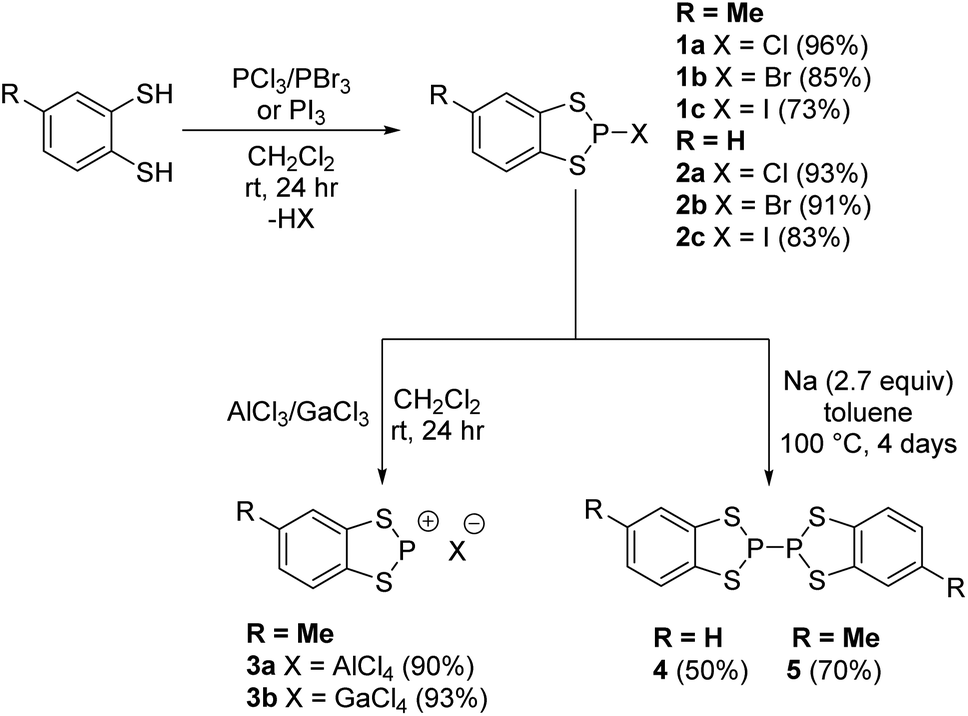 | ||
| Scheme 2 Synthesis of dithiaphosphole derivatives. | ||
31P chemical shift
The dithiaphospholes were initially characterised by in situ31P{1H} NMR spectroscopy, which revealed a singlet resonance centred at δ = 160.4, 163.3, or 155.0 ppm for 1a–1c respectively and complete consumption of the PX3 starting material (ca. δ = 219, 227 and 178 ppm for PCl3, PBr3 and PI3 respectively). The shielding constant σ can be broken down into diamagnetic shielding (σd) and paramagnetic shielding (σp) contributions. For 1H NMR spectroscopy the chemical shift is dominated by the diamagnetic shielding which is related to the electron density and hence sensitive to the electron-withdrawing/releasing nature of substituents attached to the 1H nucleus. Conversely, for other nuclei, paramagnetic shielding is significant. The paramagnetic shielding arises from mixing of the ground state wavefunction with excited state wavefunctions and is therefore closely related to the energy of low-lying states where oxidation state and coordination geometry play an important role. For a series of structurally closely-related complexes such as phosphines empirical correlations can be made. For tertiary phosphines, PR3, the chemical shift follows δP = −62 + ∑σR where σR is a constant for a specific R group.27 The additive nature of the chemical shielding effects for different groups can therefore be used in a chemically meaningful way. For example, the 31P NMR data for 1a–1c (Table 1) do not follow a simple trend based on electronegativity arguments but do correlate with a similar trend observed for PX3. From the reported 31P NMR data for PX3 (X = Cl, Br, I, NMe2, OMe, SMe and Ph), σR values can be derived (Table 1) and chemical shifts estimated based on the chemical environment of the phosphine (Table 1). While imperfect the correlation coefficient (0.85) is good, especially given the approximation that the RC6H3S2 unit can be approximated by two SMe groups.| X | F | Cl | Br | I |
|---|---|---|---|---|
| σ R | 53.3 | 94 | 96.3 | 80 |
| 31P δcalc for (MeS)2PX | 116.3 | 157 | 159 | 143 |
| 31P δobs for [1]X | — | 161.4 | 163.6 | 155.4 |
Crystal structures of 1a–1c and 2a–2c
Crystals of these compounds were obtained by reducing the volume of the crude reaction mixture to form a (super)saturated CH2Cl2 solution and then cooling to −40 °C. Crystals typically formed over 18–36 h. Compounds 1a–1c were found to crystallise in the monoclinic space group P21/c with one molecule in the asymmetric unit. Compound 2a crystallises in the monoclinic space group P21/n while 2b and 2c crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules in the asymmetric unit. Structure determination of 1a–1c revealed the expected three-coordinate phosphorus centre with an exocyclic P–X (X = Cl, Br, or I) bond (Fig. 1). The dithiaphosphole ring is non-planar with a distinct envelope geometry associated with a folding about the S⋯S vector. On descending group 17, there is a decrease in the fold angle θ (defined as the angle formed between S2P and C2S2 planes) and a move towards molecular planarity. The fold angle (θ) varies from of 26.06° (X = Cl) to 24.27° (X = Br) and 19.62° (X = I). Within the series 1a–1c an elongation of the P–X bond is observed in relation to a conventional P–X bond. The bond lengths are 2.1134(7) Å (1a), 2.3153(9) Å (1b), and 2.5730(12) Å (1c), cf. standard P–X bond lengths of 2.008 Å (P–Cl), 2.206 Å (P–Br) and 2.490–2.493 Å (P–I).28 Similar features were found for 2a–2c. The symmetry breaking generated by the presence of the methyl group leads to chirality at the P centre. However, the presence of an inversion centre in all these structures leads to structures containing 50
with two molecules in the asymmetric unit. Structure determination of 1a–1c revealed the expected three-coordinate phosphorus centre with an exocyclic P–X (X = Cl, Br, or I) bond (Fig. 1). The dithiaphosphole ring is non-planar with a distinct envelope geometry associated with a folding about the S⋯S vector. On descending group 17, there is a decrease in the fold angle θ (defined as the angle formed between S2P and C2S2 planes) and a move towards molecular planarity. The fold angle (θ) varies from of 26.06° (X = Cl) to 24.27° (X = Br) and 19.62° (X = I). Within the series 1a–1c an elongation of the P–X bond is observed in relation to a conventional P–X bond. The bond lengths are 2.1134(7) Å (1a), 2.3153(9) Å (1b), and 2.5730(12) Å (1c), cf. standard P–X bond lengths of 2.008 Å (P–Cl), 2.206 Å (P–Br) and 2.490–2.493 Å (P–I).28 Similar features were found for 2a–2c. The symmetry breaking generated by the presence of the methyl group leads to chirality at the P centre. However, the presence of an inversion centre in all these structures leads to structures containing 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixtures of both enantiomers.
:50 mixtures of both enantiomers.
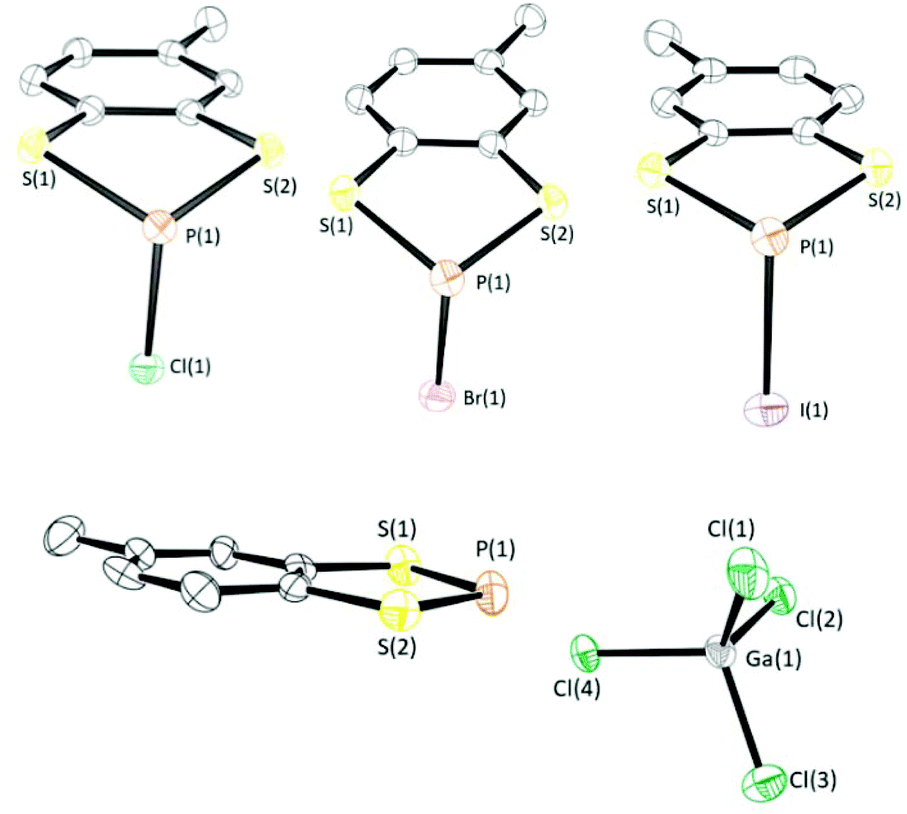 | ||
| Fig. 1 (Top) Solid-state structures of 1a–1c (top left to right respectively), (Bottom) crystal structure of 3b. Thermal ellipsoids drawn at 50% probability and H-atoms removed for clarity. | ||
Computational studies of 1a–1c
To understand better the structure and bonding in these dithiaphosphole compounds, we turned to computational methods. Natural Bond Order (NBO) analysis (M06-2X and 6-311+G(2d,p)) was performed on 1a–1c (Table 2 and ESI‡), which for 1a revealed a Wiberg P–Cl bond order of 0.86. This reduced bond order can be explained by hyperconjugation between the π electrons in the C2S2 unit and the σ*(P–X) orbital, in turn weakening the P–X bond, which is consistent with observations we have previously reported on related diazaphosphole compounds.29 In addition, NBO analysis showed significant polarisation in this bond, with a natural charge of +0.53 on the P heteroatom, and −0.33 on the Cl atom; the S atoms exhibit natural charges of +0.06. Comparing the results of 1a to 1b and 1c, a reduction in the polarisation of the P–X bond was found to be accompanied by a small increase in bond order. A similar NBO analysis on the cation in 3b (vide infra) was performed which showed a bond order of 1.35 and 1.37 for the P–S bonds and partial charges of +0.53 and +0.30 at P and S respectively.| 1a | 1b | 1c | 3 | |
|---|---|---|---|---|
| All computations used the M06-2X functional and 6-311G+(2d,p) basis set for all atoms except the I atom in 1c, for which the basis set Def2TZVP was implemented.a Average value of the two S atoms taken. | ||||
| P–X bond order | 0.86 | 0.87 | 0.90 | N/A |
| P partial charge | +0.53 | +0.46 | +0.36 | +0.53 |
| X partial charge | −0.32 | −0.27 | −0.17 | N/A |
| S partial chargea | +0.06 | +0.07 | +0.07 | +0.30 |
Reaction with Lewis acids
Stoichiometric treatment of 1a with the Lewis acids AlCl3 and GaCl3 generated the corresponding phosphenium salts 3a and 3b respectively in excellent yield (90–93%) as highly air-sensitive crystalline solids. The solid-state structure of 3a has previously been reported by Cameron and Linden.30 Compound 3b was found to crystallise in the monoclinic space group P21/c with one molecule in the asymmetric unit. As with the previously determined structure of 3a, the solid-state structure of 3b exhibits a planar C2S2P ring and notably shorter P–S bonds (1.998(6) Å and 2.024(5) Å) in relation to 1a (2.0936(7) Å and 2.0954(7) Å), consistent with 3p–3p π conjugation and some delocalisation within the formally 10 π aromatic benzodithiaphosphenium cation. This π-conjugated perspective is supported through the computational studies described above where the P–S bond orders are 1.33 and 1.35 in the cation. The closest cation–anion contact in 3b is a P⋯Cl contact at 3.317(5) Å, which is well within the sum of the van der Waals radii (3.55 Å).Reduction
Reduction of the chloride salt 1a has previously been achieved using sodium metal to form 4.23 However we found that the rate of reduction of both 1a and 2a was particularly sensitive to the size of the sodium pieces employed. Initial studies required two to four days to ensure complete reduction (monitored by 31P NMR spectroscopy) but initial pre-treatment of a suspension of small pieces of sodium metal in toluene (100 °C, 20 min) afforded a fine sodium dispersion which led to shorter reaction times (ca. two days). The P–P coupled product 5 exhibits a diagnostic 31P chemical shift at +40.6 ppm, significantly upfield in relation to the P(III) derivatives 1a and 2a and comparable with [(MeO)2C6H2S2P]2 (δ = 50 ppm).31 The methyl group can give rise to different diastereoisomers but a careful examination of the 31P NMR data failed to provide unambiguous evidence for the presence of both diastereomers. However the presence of two diastereomers could explain the inability, despite multiple attempts, to grow good quality crystals of 5, which is in contrast to 4.During the reduction process an intermediate was observed at approximately 113 ppm which is tentatively attributed to a dithiolate bridged intermediate (Fig. 2). Evidence for the nature of this intermediate comprises good agreement of the observed chemical shift with the closely related dialkoxybenzo-derivatives (δ = 115–120 ppm),15 as well as comparative reduction chemistry of P-chloro-diazaphospholes using Na(K).32–34
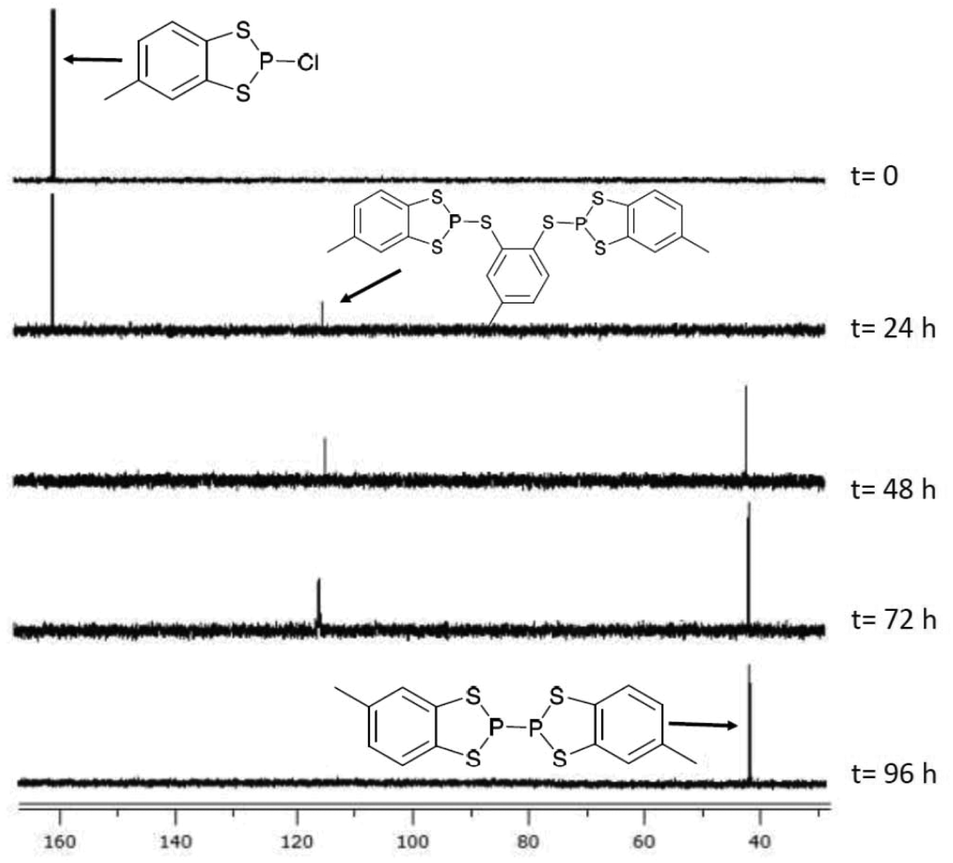 | ||
| Fig. 2 In situ 31P{1H} NMR of the reduction of 1a with Na metal in toluene to form 5. | ||
Compounds 4 and 5 were isolated by filtration to remove insolubles and dried in vacuo to afford yellow solids which, upon slow evaporation of a saturated toluene solution, afforded a polycrystalline mass. In the case of 4 small crystals suitable for X-ray diffraction were cut from the bulk sample. While all samples studied exhibited persistent twinning, a satisfactory solution could be determined in the monoclinic space group P21/n with half a molecule in the asymmetric unit with the structure of 4 located about an inversion centre midway along the P–P bond (Fig. 3). The P–P bond length (2.233(2) Å) is comparable with that observed in the two other crystallographically determined systems containing S2P–PS2 sub-units; the one previously structurally characterised dithiaphosphole,31 [(MeO)2C6H2S2P]2 at 2.2350(16) Å and the binaphtho[1,8-de][1,3,2]dithiaphosphinine reported by Woollins at 2.2306(13) Å.35 It should be noted that there appears to be a strong preference for dimerisation via localised P–P σ-bond formation in these systems whereas the lighter N-atom dithiazolyl analogues dimerise via multi-centre π–π interactions. This is likely a result of the superior strength of the P–P single bond (201 kJ mol−1) in relation to the N–N bond (167 kJ mol−1) coupled with stronger 2p–3p N–S π-bonding in DTAs in relation to 3p–3p P–S π-bonding in 4, i.e. the loss in π-bonding is more than compensated by formation of a localised 2c,2e− bond for 4 and 5.36 The structure of 4 also exhibits a folding of the C2S2P heterocycle about the S⋯S vector with a fold angle of 33.01°, slightly greater than in 1a–1c (26.06–19.62° vide supra) and 2a (28.35°, see ESI‡).
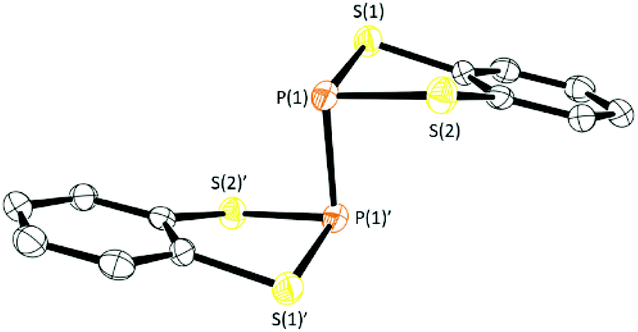 | ||
| Fig. 3 Solid-state structure of 4. Thermal ellipsoids drawn at 50% probability and H-atoms removed for clarity. | ||
Variable temperature solution EPR studies on 4 and 5 showed no tendency for homolytic bond cleavage to form radicals up to 110 °C. This is in contrast to the isolobal diazaphospholes, containing the C2(NR)2P ring system, which generate radicals upon warming through P–P bond cleavage.37–41 The difference is likely due to the steric demands of the N–R group. On the other hand, the P–P bond could be oxidatively cleaved with SO2Cl2, Br2 and I2 reforming 1a–1c based on 31P NMR spectroscopy studies and preparative scale reactions.
Substitution chemistry on 1a
Reaction of 1a with Li[N(SiMe3)2] in a 1:1 mole ratio in toluene afforded 1d (Scheme 3) as a colourless oil which was characterised by multinuclear NMR spectroscopy. The 31P NMR chemical shift for 1d (+94.9 ppm) is a little lower than that predicted based on the additive shielding approach (Table 1) but may reflect the electronic and steric dissimilarity between NMe2 and N(SiMe3)2. Condensation of 1d with two further equivalents of 1a proved sensitive to the solvent employed but reflux in MeCN for 18 h afforded the tri-functionalised product 1e (31P NMR, δ = 86.8 ppm) as a white solid (87%) which was recrystallised by cooling a saturated CH2Cl2 solution to −20 °C. Compound 1e crystallised in the rhombohedral space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (Fig. 4) with the N atom on the three-fold axis and a third of a molecule in the asymmetric unit. The five-membered C2S2P ring in 1e again adopts an envelope geometry with a fold angle of 20.66°. The P–S bond lengths in 1e are 2.105(2) and 2.125(2) Å and the exocyclic P–N bond length (1.7274(2) Å) falls in the normal range for P–N single bonds (1.70–1.77 Å). The PNP bond angle (119.8°) is very close to that expected for an sp2-N suggesting a lone pair of p-character. The structure of 1e is analogous to the previously reported dithia-arsole, (MeC6H3S2As)3N.21 Molecules of 1e aggregate through a series of six symmetry-equivalent S⋯S contacts at 3.512 Å, marginally shorter than the sum of the van der Waals radii (3.60 Å). These contacts generate a supramolecular “S6” chair motif (Fig. 4).
(Fig. 4) with the N atom on the three-fold axis and a third of a molecule in the asymmetric unit. The five-membered C2S2P ring in 1e again adopts an envelope geometry with a fold angle of 20.66°. The P–S bond lengths in 1e are 2.105(2) and 2.125(2) Å and the exocyclic P–N bond length (1.7274(2) Å) falls in the normal range for P–N single bonds (1.70–1.77 Å). The PNP bond angle (119.8°) is very close to that expected for an sp2-N suggesting a lone pair of p-character. The structure of 1e is analogous to the previously reported dithia-arsole, (MeC6H3S2As)3N.21 Molecules of 1e aggregate through a series of six symmetry-equivalent S⋯S contacts at 3.512 Å, marginally shorter than the sum of the van der Waals radii (3.60 Å). These contacts generate a supramolecular “S6” chair motif (Fig. 4).
 | ||
| Scheme 3 Synthesis of 1d and 1e. | ||
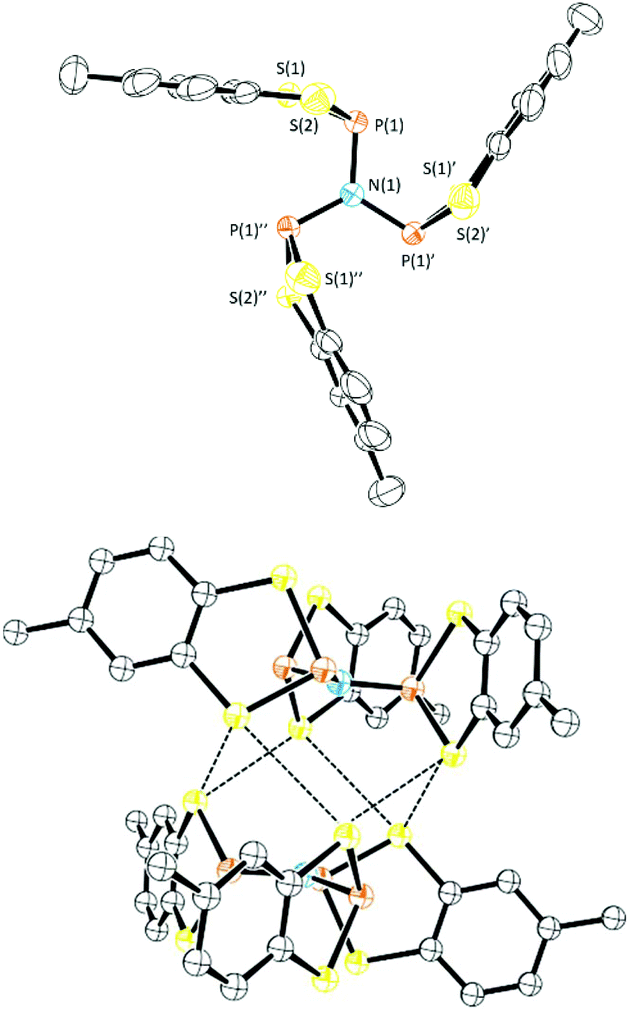 | ||
| Fig. 4 Molecular structure of 1e (Top) and supramolecular dimer linked via S⋯S contacts (Bottom). | ||
Lewis acidity studies on 3b
The phosphenium salts 3a and 3b are extremely air sensitive in solution and particularly prone to hydrolysis reflected in a diagnostic doublet in the 31P NMR spectrum (δ = 57.2 ppm, 1JPH = 699 Hz) which collapses to a singlet in the 31P{1H} NMR spectrum. Prior work by Burford reported26 the 31P chemical shift for these cations around 410 ppm but indicated it was sensitive to both temperature and concentration. They proposed the presence of aggregated species such as [(C6H4S2P)2Cl]+ (formed by disproportionation of 2 MCl4− to form Cl− and M2Cl7−) in equilibrium with [C6H4S2P+]:| 2[C6H4S2P][MCl4] ⇄ [(C6H4S2P)2Cl][M2Cl7]. | (1) |
In this context Russell's work on the related phosphenium ion {(RC)4P}+ led to isolation of the [{(RC)4P}2Cl]+ cation.42 Despite the uncertainty in the exact nature of the active species present in solution, it exhibits some Lewis acid character, based on in situ31P NMR data. For example addition of Ph3P afforded two doublets (δ = 52 ppm, 1JPP = 444 Hz; δ = 8 ppm, 1JPH = 444 Hz) whereas addition of Ph3As afforded a singlet at 47 ppm. When initially working with the cationic phosphenium 3b in THF, a chemical shift in the 31P NMR spectrum at δ = 124 ppm was observed, which more closely resembles a three-coordinate P(III)-centre (cf.1a31P δ = 160 ppm). Indeed, the chemical shift is in agreement with an O-bound S2P–O centre (expected around 130 ppm, Table 1). It was also found that, while visually monitoring the reaction, the THF solution became viscous over one hour and 1H NMR spectra analysis showed the rapid formation of poly(THF) at ambient temperature. Indeed, under more controlled conditions using 0.15 mol% 3b, 80% conversion of THF into poly(THF) occurred in 24 hours, with the reaction rate slowing over time due to the increasing viscosity from the formation of poly(THF). The polymerisation of THF is a facile process and follows the well-known cationic ring opening polymerisation (CROP) mechanism, in which 3b acts as the cationic initiator.43 It should be noted that stirred solutions of (i) GaCl3 with THF and (ii) 1a with THF gave no polymer formation, confirming the role of the phosphenium cation in the polymerisation process. An end group analysis (integral of the terminal CH3 group of the cationic initiator to the CH2 group of the poly(THF) permitted number average molecular weights (Mn) to be determined which fell in the range 1897–3163, corresponding to 23 < n < 41 THF units (see ESI‡). Interestingly, when attempting to use 3a for CROP no formation of poly(THF) occurred. In this case 31P NMR studies revealed a singlet (δ = +161 ppm) which is believed to be due to reformation of 1a and formation of the adduct THF·AlCl3. Similar degradation of the counterion has also been observed for the tetraphenylborate salt which was shown to form MeC6H3S2P–Ph and Ph3B, reflecting the high Lewis acidity of the P+ centre.26 Attempts to polymerise propylene oxide or 1,4-dioxane with 3b under the same conditions afforded a similar downfield shift (31P δ = 127 and 125 ppm respectively) to that observed in THF, consistent with adduct formation but there was no evidence for polymerisation of either of these substrates.
Reduction catalysis
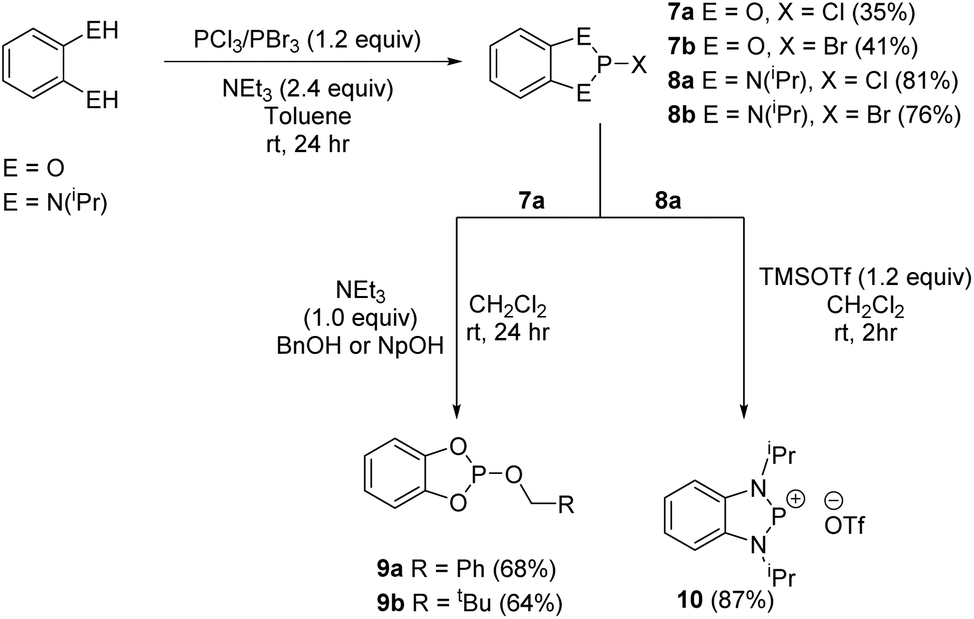
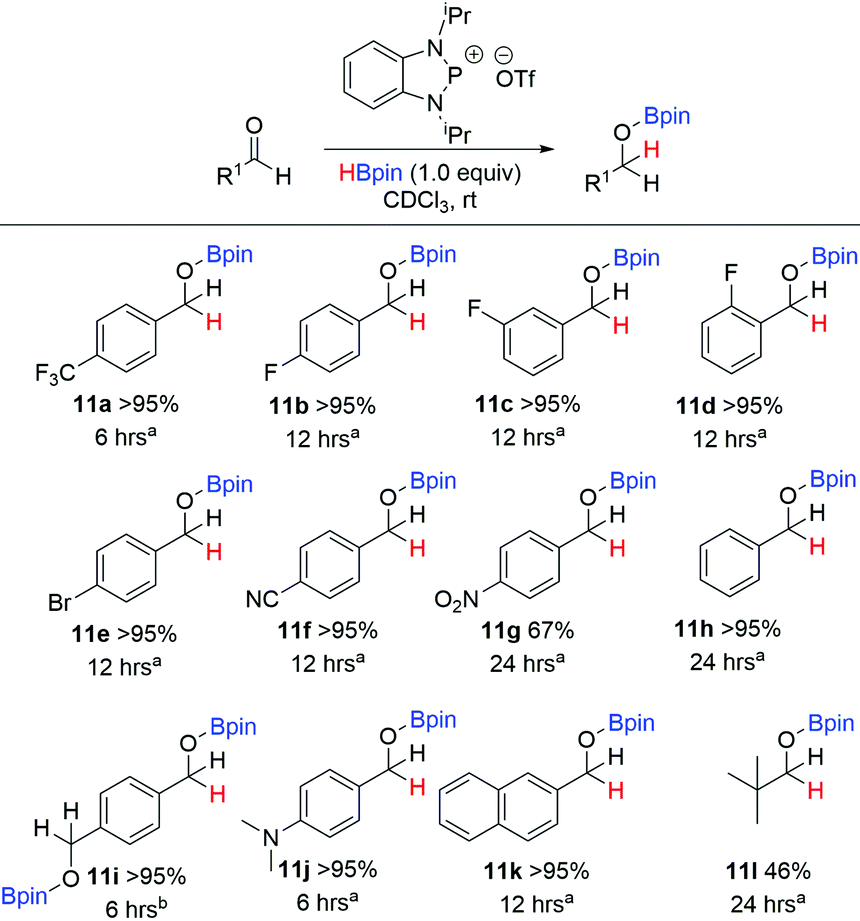
In the last few years phospholes, diazaphospholenes and their derivatives have received attention in catalytic reduction chemistry by a number of different groups, including ourselves.15,22,44,45 We therefore looked to see whether these dithiaphospholes and their derivatives would be active in the reduction of aldehydes with pinacolborane (HBpin). To investigate this, we initially synthesised a library of potential pre-catalysts. Previously the groups of Kinjo and Speed have demonstrated that the use of benzyloxy (Bn) and neopentyloxy (Np) substituted phosphorus systems are effective in reduction catalysis.15,44,45 In this context, the alkoxy derivatives 6a and 6b were prepared by reaction of the dithiaphosphole 1a with either benzyl alcohol or neopentyl alcohol in the presence of base, affording compounds 6a and 6b in moderate yield (63–65%) (Scheme 4). These compounds show upfield signals in the 31P NMR spectrum compared to precursor 1a, with 31P resonances at δ = 124.5 and 123.6 ppm for 6a and 6b correspondingly, in agreement with the computed change in their chemical shift (Table 1). This methodology was also extended to synthesise dioxaphosphole and diazaphosphole pre-catalysts, providing an opportunity to compare the relative catalytic activity of this isolobal series of C2E2P (E = O, NR, S) heterocycles (Scheme 5). To synthesise these compounds, the chloride and bromide pre-cursors were first produced. In the case of the dioxaphospholes, catechol was reacted with phosphorus(III) chloride or phosphorus(III) bromide and triethyl amine to yield 7a and 7b in 36% and 41% yield respectively, whose corresponding 31P NMR chemical shifts are δ = 173.6 and 195.3 ppm. In analogous fashion, reaction of 7a with either benzyl alcohol or neopentyl alcohol afforded 9a and 9b (68% and 64% yield) with upfield 31P NMR chemical shifts of δ = 126.9 and 127.4 ppm respectively. Although the same procedure was attempted for the diazaphosphole compound 8a, addition of benzyl alcohol or neopentyl alcohol gave a mixture of three species in the 31P NMR spectrum, only one of which was the desired product (the others are believed to arise from hydrolysis). Nevertheless, abstraction of the chloride from 8a with TMSOTf to produce the cationic phosphenium 10 was performed successfully. | ||
| Scheme 4 Synthesis of dithiaphosphole pre-catalysts bearing a benzyloxy or neopentyloxy group. | ||
 | ||
| Scheme 5 Synthesis of dioxaphosphole and diazaphosphole compounds. | ||
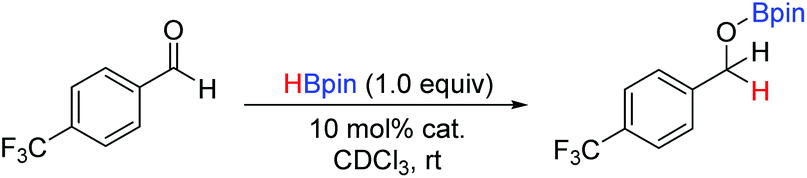
With a range of pre-catalysts in hand, an initial screening test was conducted in order to see which of these compounds was the most active in the reductive catalytic hydroboration of aldehydes. Therefore, 10 mol% of compounds 1, 3, 6, 7, 8, 9 and 10 were added to 4-(trifluoromethyl)benzaldehyde and HBpin in CDCl3. The conversion was then monitored by both 1H and 19F NMR spectroscopy at 2-, 6-, 12- and 24-hour intervals. Table 3 shows the results of the pre-catalyst screening. The dithiaphosphole 1a was found to be catalytically inactive, with <5% conversion observed in the 1H NMR spectrum after 24 hours. A mild improvement was found using 1b, with 14% conversion detected after 24 hours. More interestingly though was the use of 1c, which gave 61% product conversion to 11a after 24 hours. The differences in performance of 1a–1c can be attributed to the varying electronic properties of the structures, as found by NBO analysis (vide supra) and correlate with the P–X bond energy. Noteworthy, when using the benzyloxy and neopentyloxy derived dithiaphospholes 6a and 6b, the catalytic performance fared little better than their chloride precursor, with just 9% and 8% conversion to 11a after 24 hours respectively (see ESI‡ for NBO analysis). In addition to this, the catalytic ability of the phosphenium cations 3a and 3b were examined. Both 3a and 3b afforded quantitative conversion to the hydroborated product 11a in 12 hours. However, knowing that it was possible for dynamic exchange to occur in solution between one of the chlorides on AlCl4− and the phosphorus centre to reform 1a and give free AlCl3 (vide supra), a control reaction was undertaken in which 10 mol% AlCl3 was used as the pre-catalyst for the hydroboration catalysis. Using AlCl3, 1H NMR spectroscopy showed that after 24 hours only 68% conversion to 11a had occurred. This therefore showed that the phosphenium species were indeed responsible for attaining quantitative conversion of 4-(trifluoromethyl)benzaldehyde to 11a.
|
|
|||
|---|---|---|---|
| Entry | Pre-catalyst | Time (h) | Conversiona (%) |
| a Conversion measured by in situ1H NMR spectroscopy. | |||
| 1 | None | 24 | <5 |
| 2 | 1a | 24 | <5 |
| 3 | 1b | 24 | 14 |
| 4 | 1c | 24 | 61 |
| 5 | 3a | 12 | >95 |
| 6 | 3b | 12 | >95 |
| 7 | 6a | 24 | 9 |
| 8 | 6b | 24 | 8 |
| 9 | 7a | 24 | <5 |
| 10 | 7b | 24 | <5 |
| 11 | 8a | 24 | <5 |
| 12 | 8b | 24 | <5 |
| 13 | 9a | 24 | 30 |
| 14 | 9b | 24 | 35 |
| 15 | 10 | 6 | >95 |
| 16 | TMSOTf | 24 | 56 |
| 17 | AlCl 3 | 24 | 68 |
The dioxaphosphole pre-catalysts, 7a and 7b showed little conversion to the hydroborated product, with <5% conversion after 24 hours. However, employing 9a and 9b showed a slight improvement on these results in relation to the sulfur derivatives, giving 30% and 35% conversion to 11a respectively after 24 hours. Given the low conversion results for these pre-catalysts, alternative solvents were explored and the catalysis experiments repeated in both toluene and acetonitrile. Neither afforded appreciable improvements in conversion after 24 hours. In a similar theme, the diazaphosphole compounds 8a and 8b were found to be inactive for the catalysis. Significantly 10 mol% of the cationic phosphenium 10 was found to be highly active, giving quantitative conversion to 11a in 6 hours, as revealed by 1H NMR spectroscopy. As a control reaction, the use of TMSOTf was used as a potential pre-catalyst. Analysis of both the 1H and 19F NMR spectra revealed that 56% conversion was achieved after 24 hours; with multiple products observed.
Having discovered that the phosphenium triflate 10 was clearly the most active pre-catalyst, reaction conditions were optimised by first assessing the performance of a range of solvents, while maintaining the catalytic loading of 10 at 10 mol%. The data are summarised in Table 4. Both CH2Cl2 and CDCl3 gave quantitative conversion to the hydroborated product 11a, with the latter occurring at a faster rate than the former. Use of either THF or MeCN failed to give quantitative conversion after 24 hours. In addition, use of toluene as a solvent was attempted but 10 proved sparingly soluble. As CDCl3 proved to be the most effective solvent, the reaction conditions were further optimised. Increasing the equivalence of HBpin to two as opposed to one made little difference to the reaction, with both giving >95% conversion within six hours. Moving from 10 mol% to 5 mol% gave quantitative yields although the rate of product conversion was reduced, with the reaction now requiring 12 hours to reach completion. On the other hand, use of both 2 mol% and 1 mol% failed to generate quantitative conversion within 24 hours, with just 48% and 27% conversion observed respectively. Consequently, it was decided that for the substrate scope, 10 mol% of pre-catalyst 10 would be used in CDCl3 with one equivalent of HBpin. Using these optimised conditions, a substrate scope was performed with a variety of aldehydes in order to evaluate the effectiveness of 10 as a pre-catalyst for hydroboration reduction (Scheme 6). As already discussed, 4-(trifluoromethyl)benzaldehyde was hydroborated within 6 hours using 10 mol% of 10 to give quantitative conversion to the product 11a.
 | ||
| Scheme 6 Substrate scope for the hydroboration of aldehydes using a phosphenium triflate 10. 0.6 ml of CDCl3 solvent, NMR yield calculated from in situ1H NMR spectroscopy. a10 mol% 10, b20 mol% 10, 2.0 equiv. HBpin. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Loading (mol%) | HBpin (equiv.) | Solvent | Time (h) | Conversion (%) |
| a Conversion measured by in situ1H NMR spectroscopy. b Conversion measured by in situ19F NMR spectroscopy. | |||||
| 1 | 10 | 1.0 | CDCl3 | 6 | >95a |
| 2 | 10 | 1.0 | CH2Cl2 | 12 | >95b |
| 3 | 10 | 1.0 | THF | 24 | 76b |
| 4 | 10 | 1.0 | MeCN | 24 | 39b |
| 5 | 10 | 2.0 | CDCl3 | 6 | >95a |
| 6 | 5 | 1.0 | CDCl3 | 12 | >95a |
| 7 | 2 | 1.0 | CDCl3 | 24 | 48a |
| 8 | 1 | 1.0 | CDCl3 | 24 | 27a |
On a similar theme, use of the electron deficient fluorinated aldehydes, 4-, 3- and 2-fluorobenzaldehyde were converted to 11b, 11c and 11d respectively within 12 hours. In addition to fluorinated aldehydes, other electron-withdrawing aldehydes were probed. Use of both 4-bromobenzaldehyde and 4-formylbenzonitrile gave >95% conversion to 11e and 11f respectively within 12 hours. On the other hand, when using 4-nitrobenzaldehyde, only 67% conversion to 11g was detected by 1H NMR spectroscopy. Benzaldehyde was slow to hydroborate, but nevertheless full conversion was observed by 24 hours, while terephthalaldehyde gave full conversion to 11i after 6 hours, albeit 20 mol% of 10 was used given the presence of two aldehyde groups. The electron donating dimethylamino functional group was also tolerated, with 11j found in quantitative yield after 6 hours. 2-Naphthaldehyde was readily reduced to 11k in 12 hours. Lastly, we also tried the aliphatic pivalaldehyde, where 46% conversion to 11l was achieved after 24 hours, the approximate half product conversion we attribute partially to its high steric demand. During the course of performing the substrate scope, 4-methoxybenzaldehyde, 4-methylbenzaldehyde and 2,4,6-trimethylbenzaldehyde substrates were also examined. In these cases 1H NMR spectra gave additional unidentified signals that did not correspond to product formation and consequently although consumption of the starting aldehyde was observed, their results are not included in Scheme 6. The ketone 4′-fluoroacetophenone was also trialled in the substrate scope, giving 57% consumption as detected by 19F NMR spectroscopy. However, additional signals to the reactant/product were identified.
Mechanistic studies on phosphenium based catalysis have been explored by Kinjo and colleagues on related hydroboration of pyridines with HBpin.19 Upon stoichiometric addition of the pre-catalyst 10 to 4-(trifluoromethyl)benzaldehyde, no significant change in the 31P NMR spectrum was observed, indicating that there is no strong interaction between pre-catalyst and aldehyde substrate and consequently 10.
Mechanistically we therefore postulate that this catalysis proceeds in an analogous way to that reported by Kinjo.19 When stoichiometric amounts of 10 were added to HBpin to observe the active catalyst, 31P NMR spectroscopy instead revealed significant decomposition product in the form of PH3, as identified by a quartet signal appearing at δ = −238.5 ppm, with a 1JPH = 189 Hz. Furthermore, the intermediate en route to PH3 was also evident as a primary phosphine in the 31P NMR spectrum (low intensity triplet at δ = −79.5 ppm (1JPH = 199 Hz)), suggesting the formation of a primary phosphine with two P–H bonds via endocyclic cleavage. Similar results have been observed previously by Speed for the catalytic reduction of imines using a diazaphospholene catalyst.44 Lastly, we wished to address why the different phosphole derived pre-catalysts performed so differently, and specifically to address the poor activity of the dithiaphosphole and dioxaphosphole pre-catalysts 6a and 9a. To do this, stoichiometric addition of the pre-catalyst to HBpin in CDCl3 was performed in order to monitor how quickly the respective active hydride species formed.22 In the case of 6a, after 24 hours, both the 31P NMR and 11B NMR spectra showed little appreciable change, with no indication of a hydride species in the 31P NMR spectrum and only the doublet resonance of HBpin at 28.2 ppm was observed in the 11B NMR spectrum. For 9a, again no pre-catalyst formation was observed after 24 hours, with only a small quantity of hydrolysis product detected, as identified by a doublet signal centred at δ = 7.7 ppm (1JPH = 706 Hz) in the 31P NMR spectrum. These studies therefore show that the slow rate of formation of the active catalyst is the cause for the poor activity of the dithiaphosphole and dioxaphosphole pre-catalysts 6a, 6b, 9a and 9b.
Experimental
General experimental
All reactions were carried out under an atmosphere of dinitrogen using standard Schlenk and glove box techniques. With the exception of THF and Et2O, all solvents used were dried by passing through an alumina column incorporated into an MB SPS-800 solvent purification system, degassed and finally stored in an ampoule fitted with a Teflon valve under a dinitrogen atmosphere. THF and Et2O were dried over molten potassium for three days and distilled over argon. Deuterated solvents were distilled and dried over molecular sieves and stored in a glove box before use. Starting materials were purchased from commercial suppliers and used as received. 1H, 13C{1H}, 19F, 11B, 31P, and 27Al NMR spectra were recorded on a Bruker Avance 300, 400, or 500 MHz spectrometer. Chemical shifts are expressed as parts per million (ppm, δ) and are referenced to CDCl3 (7.26/77.16 ppm), C6D6 (7.16/128.06 ppm), or C6D5Br (7.28/122.4 ppm for the most downfield resonance) as internal standards. Multinuclear NMR spectra were referenced to BF3·Et2O/CDCl3 (11B), CFCl3 (19F), H3PO4 (31P), and Al(NO2)3 (27Al). The description of signals includes s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, and m = multiplet. All coupling constants are absolute values and are expressed in Hertz (Hz). IR-Spectra were measured on a Shimadzu IR Affinity-1 photospectrometer. The description of signals includes s = strong, m = medium, w = weak, sh = shoulder, and br = broad. Mass spectra were measured on a Waters LCT Premier/XE or a Waters GCT Premier spectrometer.Synthesis of phosphole compounds
Note: Attempted recrystallisation of 3a from THF afforded 1a and a set of colourless crystals which were identified as AlCl3·2THF by X-ray crystallography (identical to that reported previously).46
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)3). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm: 139.9 (d, 3JPC = 2.9 Hz, 1C, Ar), 136.4 (d, 3JPC = 3.2 Hz, 1C, Ar), 135.8 (1C, Ar), 126.9 (1C, Ar), 124.8 (d, 2JPC = 6.5 Hz, 1C, Ar), 123.9 (d, 2JPC = 6.4 Hz, 1C, Ar), 75.4 (d, 2JPC = 9.9 Hz, 1C, C(CH3)–
3)3). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm: 139.9 (d, 3JPC = 2.9 Hz, 1C, Ar), 136.4 (d, 3JPC = 3.2 Hz, 1C, Ar), 135.8 (1C, Ar), 126.9 (1C, Ar), 124.8 (d, 2JPC = 6.5 Hz, 1C, Ar), 123.9 (d, 2JPC = 6.4 Hz, 1C, Ar), 75.4 (d, 2JPC = 9.9 Hz, 1C, C(CH3)–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 31.6 (d, 3JPC = 2.3 Hz, 1C, (CH3)3–CH2), 26.5 (3C, C(H3)3–CH2), 21.0 (1C, Ar–CH3). 31P NMR (162 MHz, 295 K, CDCl3): δ/ppm 123.6 (t, 3JPH = 6.1 Hz, 1P). IRνmax (cm−1): 1456 (m), 1364 (sh), 1258 (w), 1217 (w), 1117 (w), 972 (s), 789 (m), 727 (m) and 687 (sh). HRMS (EI+) m/z calculated for [M]+ [C12H17OPS2]+: 272.0458, found: 272.0452.
(CH3)2), 1.69 (dd, 3JHH = 6.6 Hz, 4JPH = 1.0 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 136.9 (d, 2JPC = 10.5 Hz, 2C, Ar), 121.3 (2C, Ar), 111.6 (d, 3JPC = 1.6 Hz, 2C, Ar), 48.1 (d, 2JPC = 12.7 Hz, 2C, H(CH3)2), 22.3 (4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 147.1 (s, 1P). Values in agreement with literature.29
(CH3)2), 1.76 (dd, 3JHH = 6.6 Hz, 4JPH = 1.0 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 137.2 (d, 2JPC = 10.2 Hz, 2C, Ar), 122.3 (2C, Ar), 112.4 (d, 3JPC = 1.6 Hz, 2C, Ar), 49.0 (d, 2JPC = 11.8 Hz, 2C, H(CH3)2), 21.8 (d, 3JPC = 1.6 Hz, 4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 169.2 (s, 1P). IRνmax (cm−1): 1477 (m), 1369 (m), 1288 (w), 1159 (m), 1007 (m), 932 (m) and 752 (m). HRMS (EI+) m/z calculated for [M]+ [C12H18BrN2P]+: 300.0391, found: 300.0384. Melting point 100–106 °C.
2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 146.0 (d, 2JPC = 7.6 Hz, 2C, Ar), 136.7 (d, 3JPC = 2.9 Hz, 2C, Ar), 128.7 (Ar), 128.4 (Ar), 127.7 (Ar), 123.0 (Ar), 112.2 (Ar), 65.9 (d, 2JPC = 2.0 Hz, 1C, C(CH3)3–H2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 126.9 (s, 1P). IRνmax (cm−1): 1474 (s), 1373 (w), 1229 (s), 980 (m), 916 (w), 824 (s), 729 (s), 692 (s) and 625 (m). HRMS (EI+) m/z calculated for [M]+ [C13H11O3P]+: 246.0446, found: 246.0441.
2), 0.83 (s, 9H, CH2C3). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 146.0 (d, 2JPC = 7.7 Hz, 2C, Ar), 122.7 (2C, Ar), 111.9 (2C, Ar), 73.6 (d, 2JPC = 2.0 Hz, 1C, C(CH3)3–H2), 31.9 (d, 3JPC = 2.7 Hz, 1C, (CH3)3–CH2), 26.2 (3C, (CH3)3–CH2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 127.4 (s, 1P). IRνmax (cm−1): 1476 (s), 1366 (w), 1333 (w), 1231 (s), 1003 (s), 824 (s), 739 (m), 698 (m), 623 (m) and 536 (w). HRMS (EI+) m/z calculated for [M]+ [C11H15O3P]+: 226.0759, found: 226.0756.
(CH3)2), 1.87 (dd, 3JHH = 6.6 Hz, 4JPH = 1.3 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 138.6 (d, 2JPC = 6.0 Hz, 2C, Ar), 127.2 (2C, Ar), 114.3 (2C, Ar), 52.6 (d, 2JPC = 10.8 Hz, 2C, H(CH3)2), 23.9 (4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 216.0 (s, 1P). 19F{1H} NMR (376 MHz, 295 K, CDCl3): δ/ppm −78.4 (s, 3F, O3SCF3−). Values in agreement with literature.29
H2), 31.6 (d, 3JPC = 2.3 Hz, 1C, (CH3)3–CH2), 26.5 (3C, C(H3)3–CH2), 21.0 (1C, Ar–CH3). 31P NMR (162 MHz, 295 K, CDCl3): δ/ppm 123.6 (t, 3JPH = 6.1 Hz, 1P). IRνmax (cm−1): 1456 (m), 1364 (sh), 1258 (w), 1217 (w), 1117 (w), 972 (s), 789 (m), 727 (m) and 687 (sh). HRMS (EI+) m/z calculated for [M]+ [C12H17OPS2]+: 272.0458, found: 272.0452.
(CH3)2), 1.69 (dd, 3JHH = 6.6 Hz, 4JPH = 1.0 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 136.9 (d, 2JPC = 10.5 Hz, 2C, Ar), 121.3 (2C, Ar), 111.6 (d, 3JPC = 1.6 Hz, 2C, Ar), 48.1 (d, 2JPC = 12.7 Hz, 2C, H(CH3)2), 22.3 (4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 147.1 (s, 1P). Values in agreement with literature.29
(CH3)2), 1.76 (dd, 3JHH = 6.6 Hz, 4JPH = 1.0 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 137.2 (d, 2JPC = 10.2 Hz, 2C, Ar), 122.3 (2C, Ar), 112.4 (d, 3JPC = 1.6 Hz, 2C, Ar), 49.0 (d, 2JPC = 11.8 Hz, 2C, H(CH3)2), 21.8 (d, 3JPC = 1.6 Hz, 4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 169.2 (s, 1P). IRνmax (cm−1): 1477 (m), 1369 (m), 1288 (w), 1159 (m), 1007 (m), 932 (m) and 752 (m). HRMS (EI+) m/z calculated for [M]+ [C12H18BrN2P]+: 300.0391, found: 300.0384. Melting point 100–106 °C.
2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 146.0 (d, 2JPC = 7.6 Hz, 2C, Ar), 136.7 (d, 3JPC = 2.9 Hz, 2C, Ar), 128.7 (Ar), 128.4 (Ar), 127.7 (Ar), 123.0 (Ar), 112.2 (Ar), 65.9 (d, 2JPC = 2.0 Hz, 1C, C(CH3)3–H2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 126.9 (s, 1P). IRνmax (cm−1): 1474 (s), 1373 (w), 1229 (s), 980 (m), 916 (w), 824 (s), 729 (s), 692 (s) and 625 (m). HRMS (EI+) m/z calculated for [M]+ [C13H11O3P]+: 246.0446, found: 246.0441.
2), 0.83 (s, 9H, CH2C3). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 146.0 (d, 2JPC = 7.7 Hz, 2C, Ar), 122.7 (2C, Ar), 111.9 (2C, Ar), 73.6 (d, 2JPC = 2.0 Hz, 1C, C(CH3)3–H2), 31.9 (d, 3JPC = 2.7 Hz, 1C, (CH3)3–CH2), 26.2 (3C, (CH3)3–CH2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 127.4 (s, 1P). IRνmax (cm−1): 1476 (s), 1366 (w), 1333 (w), 1231 (s), 1003 (s), 824 (s), 739 (m), 698 (m), 623 (m) and 536 (w). HRMS (EI+) m/z calculated for [M]+ [C11H15O3P]+: 226.0759, found: 226.0756.
(CH3)2), 1.87 (dd, 3JHH = 6.6 Hz, 4JPH = 1.3 Hz, 12H, CH(C3)2). 13C{1H} NMR (101 MHz, 295 K, CDCl3): δ/ppm 138.6 (d, 2JPC = 6.0 Hz, 2C, Ar), 127.2 (2C, Ar), 114.3 (2C, Ar), 52.6 (d, 2JPC = 10.8 Hz, 2C, H(CH3)2), 23.9 (4C, CH(H3)2). 31P{1H} NMR (162 MHz, 295 K, CDCl3): δ/ppm 216.0 (s, 1P). 19F{1H} NMR (376 MHz, 295 K, CDCl3): δ/ppm −78.4 (s, 3F, O3SCF3−). Values in agreement with literature.29
Polymerisation of THF
In a typical reaction THF (50 mL, 616 mmol, 1369 equiv.) was added to a sample of 3b (0.018 g, 0.45 mmol, 1.0 equiv.) (equivalent to 9.06 mmol L−1 or 1:2469 w/w). The solution immediately turned colourless and the progress of the polymerisation was followed by 1H NMR spectroscopy.
Oxidation reactions with dimer 5
General experimental for hydroboration catalysis
In a glovebox under a dinitrogen atmosphere, three separate vials were charged with the phosphorus catalyst (10, 5, 2 or 1 mol%), aldehyde (0.1 mmol, 1 equiv.), and HBpin (12.8 mg, 0.1 mmol, 1 equiv.). By syringe, solvent (0.6 mL) was added to the vial containing HBpin and then mixed between the three vials at least twice. The solution was transferred to a J. Young NMR tube and multinuclear NMR spectra were acquired at 2 h, 6 h, 12 h and 24 hours. Product conversion was calculated from the in situ1H NMR spectrum by integrating the aldehyde signal and new resonance resulting from the hydride from HBpin.Characterisation of hydroborated products
Conclusions
This work has provided a series of crystallographic studies on a range of dithiaphosphole compounds. The solid-state structures of the halogen-containing species has revealed elongation in the P–X bond (where X = halogen), with computational supporting extensive polarisation of the P–X bond. These P-halo dithiaphospholes have then be used in a series of additional reactions. Crystallographic studies were then performed on these products. This was followed by a comparative study of the catalytic activity of these dithiaphospholes in relation to the corresponding dioxaphosphole and diazaphosphole compounds. Among those examined as metal-free pre-catalysts for the hydroboration of a series of aldehydes with HBpin, the phosphenium cation 1,3-diisopropyl-benzodiphosphenium triflate showed excellent conversion rates under mild conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RLM would like to acknowledge the EPSRC for an Early Career Fellowship (EP/R026912/1). JMR would like to thank NSERC for an operating grant (NSERC Discovery Grant RGPIN-2010-05523). We thank Dr A. D. Bond (Cambridge University) for assistance with the structure of compound 3.Notes and references
- Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jakle, Wiley, 2018 Search PubMed.
- Main Group Metals in Organic Synthesis, ed. H. Yamamoto and K. Oshima, Wiley, 2006 Search PubMed.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
- Q. Tai, K. Tang and F. Yan, Energy Environ. Sci., 2019, 12, 2375–2405 RSC.
- J. M. Rawson and C. P. Constantinides, in Chapter 4 in Spin in Organics, ed. J. S. Miller, World Scientific, 2018, vol. IV Search PubMed.
- A. W. Cordes, R. C. Haddon and R. T. Oakley, Adv. Mater., 1994, 6, 798–802 CrossRef CAS.
- K. E. Preuss, Coord. Chem. Rev., 2015, 289–290, 49–61 CrossRef CAS.
- R. L. Melen, Science, 2019, 484, 479–484 CrossRef PubMed.
- S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef.
- J. Zhang, J. Yang and J. Cheng, Angew. Chem., Int. Ed., 2019, 58, 5983–5987 CrossRef CAS PubMed.
- D. Gudat, A. Haghverdi and M. Nieger, Angew. Chem., Int. Ed., 2000, 39, 3084–3086 CrossRef CAS PubMed.
- S. Burck, D. Gudat, M. Nieger and W. Du Mont, J. Am. Chem. Soc., 2006, 128, 3946–3955 CrossRef CAS PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 3342–3346 CrossRef CAS PubMed.
- C. C. Chong, H. Hirao and R. Kinjo, Angew. Chem., Int. Ed., 2015, 54, 190–194 CrossRef CAS PubMed.
- C. H. Tien, M. R. Adams, M. J. Ferguson, E. R. Johnson and A. W. H. Speed, Org. Lett., 2017, 19, 5565–5568 CrossRef CAS PubMed.
- M. R. Adams, C. Tien, R. Mcdonald and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 16660–16663 CrossRef CAS PubMed.
- S. Miaskiewicz, J. H. Reed, P. A. Donets, C. C. Oliveira and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 4039–4042 CrossRef CAS PubMed.
- B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed.
- T. Lundrigan, E. N. Welsh, T. Hynes, C. Tien, M. R. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, J. Am. Chem. Soc., 2019, 141, 14083–14088 CrossRef CAS PubMed.
- T. T. P. Tran, D. M. C. Ould, L. C. Wilkins, D. S. Wright, R. L. Melen and J. M. Rawson, CrystEngComm, 2017, 19, 4696–4699 RSC.
- D. M. C. Ould and R. L. Melen, Chem. – Eur. J., 2018, 24, 15201–15204 CrossRef CAS PubMed.
- M. Baudler, A. Moog, K. Glinka and U. Kelsch, Z. Naturforsch. B, 1973, 28, 363–369 CAS.
- N. Burford, J. Passmore and M. J. Schriver, J. Chem. Soc., Chem. Commun., 1986, 140–142 RSC.
- N. Burford, B. W. Royan, A. Linden and T. S. Cameron, J. Chem. Soc., Chem. Commun., 1988, 842–844 RSC.
- N. Burford, B. W. Royan, A. Linden and T. S. Cameron, Inorg. Chem., 1989, 28, 144–150 CrossRef CAS.
- O. Kuhl, Phosphorus-31 NMR spectroscopy: A concise introduction for the synthetic organic and organometallic chemistry, Springer, Berlin, 2008 Search PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Typical interatomic distances: organic compounds, International Tables for Crystallography, 2006, ch. 9.5, vol. C, pp. 790–811 Search PubMed.
- D. M. C. Ould, A. C. Rigby, L. C. Wilkins, S. J. Adams, J. A. Platts, S. J. A. Pope, E. Richards and R. L. Melen, Organometallics, 2018, 37, 712–719 CrossRef CAS.
- T. S. Cameron and A. Linden, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 41, 75–81 CrossRef CAS.
- S. C. Kosnik, M. C. Nascimento, J. M. Rawson and C. L. B. Macdonald, Dalton Trans., 2017, 46, 9769–9776 RSC.
- I. V. Komlev, A. I. Zavalishina, I. P. Chernikevich, D. A. Predvoditelev and E. E. Nifantiev, Zh. Obshch. Khim., 1972, 42, 802–807 CAS.
- E. E. Nifantiev, S. F. Sorokina, L. A. Vorobieva and A. A. Borisenko, Dokl. Akad. Nauk SSSR, 1982, 263, 900–910 Search PubMed.
- E. E. Nifantiev, N. S. Vyazankin, S. F. Sorokina, L. A. Vorobieva, O. A. Vyazankina, D. A. Bravo-Zhivotovsky and A. R. Bekker, J. Organomet. Chem., 1984, 277, 211–225 CrossRef.
- C. Kirst, B. E. Bode, D. B. Cordes, P. S. Nejman, A. M. Z. Slawin, K. Karaghiosoff and J. D. Woollins, Dalton Trans., 2016, 45, 6348–6351 RSC.
- E. G. Awere, N. Burford, R. C. Haddon, S. Parsons, J. Passmore, J. V. Waszczak and P. S. White, Inorg. Chem., 1990, 29, 4821–4830 CrossRef CAS.
- V. Naseri, R. Edge, R. J. Less, E. J. L. Mcinnes, K. Mu, J. M. Rawson and D. S. Wright, Chem. Commun., 2009, 1691–1693 Search PubMed.
- D. Förster, H. Dilger, F. Ehret, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2012, 3989–3994 CrossRef.
- O. Puntigam, I. Hajdók, M. Nieger, M. Niemeyer, S. Strobel and D. Gudat, Z. Anorg. Allg. Chem., 2011, 637, 988–994 CrossRef CAS.
- O. Puntigam, D. Förster, N. A. Giffin, S. Burck, J. Bender, F. Ehret, A. D. Hendsbee, M. Nieger, J. D. Masuda and D. Gudat, Eur. J. Inorg. Chem., 2013, 2041–2050 CrossRef CAS.
- N. A. Gi, A. D. Hendsbee, T. L. Roemmele, M. D. Lumsden, C. C. Pye and J. D. Masuda, Inorg. Chem., 2012, 51, 11837–11850 CrossRef PubMed.
- J. M. Slattery, C. Fish, M. Green, T. N. Hooper, J. C. Jeffery, R. J. Kilby, J. M. Lynam, J. E. Mcgrady, D. A. Pantazis, C. A. Russell and C. E. Willans, Chem. – Eur. J., 2007, 13, 6967–6974 CrossRef CAS PubMed.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- M. R. Adams, C. H. Tien, B. S. N. Huchenski, M. J. Ferguson and A. W. H. Speed, Angew. Chem., Int. Ed., 2017, 56, 6268–6271 CrossRef CAS PubMed.
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841–844 CrossRef CAS.
- A. H. Cowley, R. E. Davis and P. E. Riley, Inorg. Chem., 1981, 20, 1179–1181 CrossRef CAS.
- W. Wang, X. Shen, F. Zhao, H. Jiang, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, J. Org. Chem., 2018, 83, 69–74 CrossRef CAS PubMed.
- M. K. Barman, K. Das and B. Maji, J. Org. Chem., 2019, 84, 1570–1579 CrossRef CAS PubMed.
Footnotes |
| † Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2019.0084454664 |
| ‡ Electronic supplementary information (ESI) available: X-ray data, NMR spectra. CCDC 1951113–1951115, 1951125–1951127, 1951132, 1430534 and 824860. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt03577j |
| This journal is © The Royal Society of Chemistry 2019 |