
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights on the non-innocent role of electron donors: reversible photocapture of CO2 by RuII-polypyridyl complexes†
Nicolas
Queyriaux‡
 *,
Wesley B.
Swords
,
Hemlata
Agarwala
,
Ben A.
Johnson
,
Sascha
Ott
and
Leif
Hammarström
*,
Wesley B.
Swords
,
Hemlata
Agarwala
,
Ben A.
Johnson
,
Sascha
Ott
and
Leif
Hammarström
Department of Chemistry – Ångström Laboratory, Uppsala University, Box 523, SE 751 20, Uppsala, Sweden. E-mail: nicolas.queyriaux@lcc-toulouse.fr
First published on 18th October 2019
Abstract
The ability of [RuII(tButpy)(dmbpy)(MeCN)]2+ (1-MeCN) to capture CO2, with the assistance of triethanolamine (TEOA), has been assessed under photocatalytically-relevant conditions. The photolability of 1-MeCN has proven essential to generate a series of intermediates which only differ by the nature of their monodentate ligand. In DMF, ligand photoexchange of 1-MeCN to give [RuII(tButpy)(dmbpy)(DMF)]2+ (1-DMF) proceeds smoothly with a quantum yield of 0.011. However, in the presence of TEOA, this process was disrupted, leading to the formation of a mixture of 1-DMF and [RuII(tButpy)(dmbpy)(TEOA)]+ (1-TEOA). An equilibrium constant of 3 was determined. Interestingly, 1-TEOA demonstrated an ability to reversibly catch and release CO2 making it a potentially crucial intermediate towards CO2 reduction.
Introduction
The increase in atmospheric carbon dioxide (CO2) concentration has led many research groups to take an active interest in remediation solutions. Whether to convert CO2 into renewable fuels or to use it as an alternative C1-feedstock in the synthesis of high added-value molecules, different catalytic approaches have been proposed.1–4 Among them, artificial photosynthesis has allowed a wide number of transition metal-based electro- and/or photocatalysts to emerge. In particular, Re(I)-diimine5–7 and Ru(II)-polypyridyl8–10 complexes have been shown to display remarkable efficiency and selectivity. Whereas the reduction of CO2 assisted by Re complexes usually leads to the formation of carbon monoxide (CO) under a wide range of conditions, Ru-based catalysts are more sensitive to the reaction conditions, generating either CO, formic acid (HCOOH) or mixtures of these two-electron reduction products of CO2. Significant efforts have been made to better understand the mechanisms involved in these families of catalysts and have recently been compiled into a comprehensive review.11Most of the studies that have been interested in understanding the catalytic mechanisms involved in CO2 reduction by polypyridyl ruthenium(II) complexes have been focusing on the electrocatalytic approach.10,12–20 While the exact nature of the reaction intermediates responsible for the formation of CO and HCOOH has not yet reached consensus, two key species have been invoked: a η1-CO2 adduct and a hydride complex.11,12,21–23 However, the characteristic times involved in photo-induced electronic transfer processes are singularly different from those involved in heterogeneous electron transfers at the electrode interface. In addition, the nature of the photocatalytic reaction mixture is sufficiently different from the electrocatalytic mixture (absence of a supporting electrolyte or use of large concentration of sacrificial electron donors) to potentially induce significant differences in the mechanistic pathway followed by the catalyst. In that respect, the involvement of adducts generated by the interaction between unreduced Re(I)- or Ru(II)-complexes and sacrificial electron donors based on aliphatic amines, such as triethylamine (TEA) or triethanolamine (TEOA), has been previously evidenced.24–26
In this study, we describe the synthesis and full spectroscopic characterization of the complex [RuII(tButpy)(dmbpy)(MeCN)]2+ (1-MeCN, tButpy = 4,4,4-tri-tButyl-2,2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6,2-terpyridine, dmbpy = 4,4-dimethyl-2,2-dipyridyl). Its ability to undergo ligand photoexchange reactions has been carefully scrutinized under suitable experimental conditions. Under visible light, the remarkable lability of the acetonitrile ligand allows the formation of three intermediates of crucial relevance in the early steps of the photocatalytic process. Amongst these, a peculiar intermediate involving the commonly used sacrificial electron-donor TEOA display the striking feature of reversibly catch and release CO2, making it a potentially crucial intermediate towards CO2 reduction.
:6,2-terpyridine, dmbpy = 4,4-dimethyl-2,2-dipyridyl). Its ability to undergo ligand photoexchange reactions has been carefully scrutinized under suitable experimental conditions. Under visible light, the remarkable lability of the acetonitrile ligand allows the formation of three intermediates of crucial relevance in the early steps of the photocatalytic process. Amongst these, a peculiar intermediate involving the commonly used sacrificial electron-donor TEOA display the striking feature of reversibly catch and release CO2, making it a potentially crucial intermediate towards CO2 reduction.
Results and discussion
Synthesis
The complex [RuII(tButpy)(dmbpy)(MeCN)]2+ (1-MeCN, Fig. 1) was synthesized through a typical stepwise procedure (detailed synthetic procedures are presented in the ESI†).19 In a first step, [RuIII(tButpy)Cl3] precursor was prepared by reacting the derivatized-terpyridine ligand with RuCl3·3H2O in EtOH. Further introduction of the bidentate ligand was then realized by heating both reactants in the presence of LiCl and NEt3 in an EtOH/H2O mixture, to yield the chloride derivative, 1-Cl (Fig. 1). Substitution of the chloride ligand was finally achieved by refluxing the complex in a CH3CN/H2O mixture. 1D and 2D 1H-NMR spectra were recorded in poorly-coordinating deuterated acetone, which allowed an optimal dispersion of the well-resolved signals, thus facilitating complete assignment (Fig. S1 and S2 in the ESI†). Chloride ligand exchange by an acetonitrile molecule is easily monitored thanks to dramatic shielding of the signal associated with monodentate ligand-directed proton on the bipyridyl derivative (from 10.14 ppm in 1-Cl to 9.70 ppm in 1-MeCN). ESI-MS spectrum confirmed the calculated mass-to-charge ratios of the expected molecular fragments. The UV-Vis absorption spectra of 1-MeCN and 1-Cl were recorded in acetonitrile, displaying typical features of ruthenium(II) polypyridyl complexes. In particular, intense absorption bands around 300 nm, attributed to ligand-centred π–π* transitions and a broad metal-to-ligand charge transfer (MLCT) transition in the visible region were observed. | ||
| Fig. 1 Molecular structures of 1-Cl and 1-MeCN. | ||
Electrochemical properties
The electrochemical behaviour of 1-MeCN and its chloride counterpart 1-Cl were assessed by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in acetonitrile under inert atmosphere (Fig. 2, Fig. S3 and S4 in the ESI†). The use of acetonitrile solutions as electrolytes allowed direct comparison with literature data. In the anodic domain, CVs feature a reversible, one-electron electrochemical process assigned to the RuII/III couple. The potential of this couple is strongly affected by the nature of the monodentate ligand. The replacement of the chloride ligand by acetonitrile leads to a 500 mV shift towards more positive potentials. On the other hand, cathodic scans of 1-MeCN feature a quasi-reversible, one-electron reduction at −1.79 V vs. Fc+/Fc – characterized by a peak-to-peak difference Δ(Epa − Epc) of 83 mV – and an irreversible reductive event at a more negative potential of −2.08 V vs. Fc+/Fc. Increasing scan rate from 50 mV s−1 to 300 mV s−1 allows partial reversibility recovery (Fig. S5, ESI†). The lack of reversibility of this second process has previously been ascribed to the loss of the acetonitrile ligand, generating a five-coordinate species.19,27 According to earlier reports, both redox processes have been assigned to ligand-centred reductions.16,17,27,28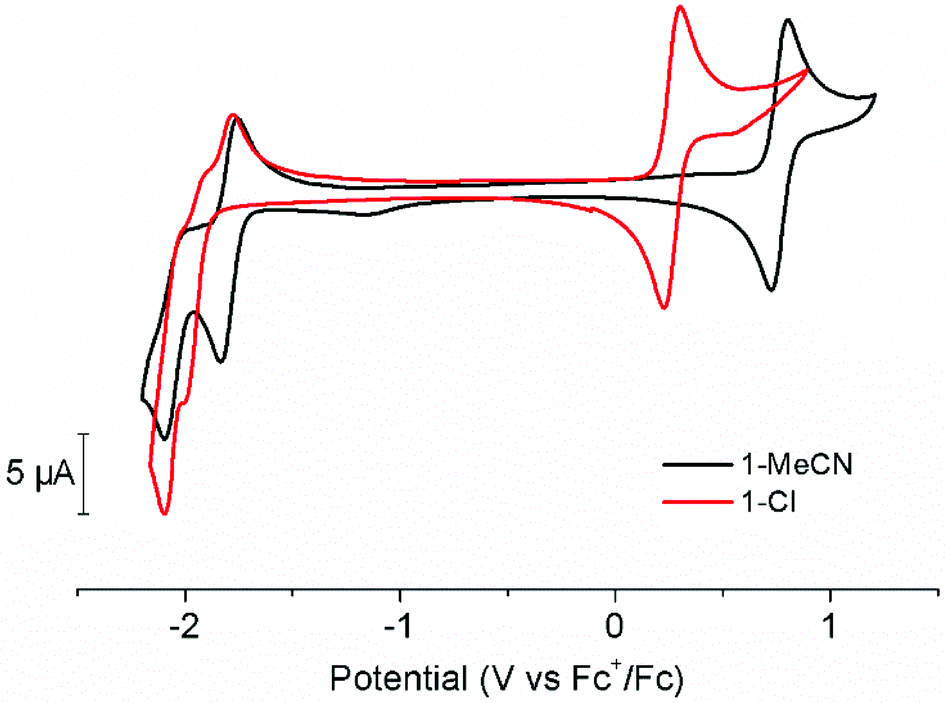 | ||
| Fig. 2 Cyclic voltammograms at 50 mV s−1 of 0.75 mM solution of 1-Cl (red trace) and 1-MeCN (black trace) recorded in MeCN (containing 0.1 M n-Bu4NPF6 as the supporting electrolyte) under an inert atmosphere. Setup: glassy carbon working electrode, Pt counter-electrode and Ag/AgNO3 reference electrode. | ||
Investigating the cathodic behaviour of 1-Cl revealed slightly more complicated electrochemical features. Indeed, neither of the two reductive events displayed a significant reversibility. Moreover, the electrochemical signature from a new species appeared when the potential was switched back towards anodic domain. By comparison, this new oxidation wave could be assigned to the reoxidation of [RuII(tButpy)(dmbpy)(MeCN)]+, generated by Cl−/MeCN exchange induced by the reduction processes. Inverting scan direction directly after the first reduction process provides more detailed information concerning the Cl− lability (Fig. S6 in the ESI†). Under these conditions, partial reversibility of the [Ru(tButpy)(dmbpy)(Cl)]+/0 couple is reached, leading to the simultaneous occurrence of reoxidation processes associated with the MeCN- and Cl−-bearing compounds. Increasing scan rates from 50 mV s−1 to 300 mV s−1 allows for reversibility improvement and reduced contribution from the acetonitrile derivative. Taken together, these results suggest Ru–Cl bond labilization induced by one-electron reduction. The similarity of the potentials associated with the second reduction process in 1-Cl and 1-MeCN is a strong evidence in favour of MeCN coordination upon further applied negative bias, the only two-electron reduction process observed in both cases being associated with the [Ru(tButpy)(dmbpy)(MeCN)]+/[Ru(tButpy)(dmbpy)(MeCN)]0 couple.
Photolability investigations
Ishitani and coworkers have opened new perspectives in the understanding of the mechanism of photocatalytic CO2 reduction by demonstrating that constituents of the typical three-components photocatalytic system could have a role exceeding their original purpose. In particular, the tertiary amine TEOA has been shown to displace the acetonitrile ligand of [ReI(bpy)(CO)3(MeCN)]+ or to react with [RuII(dmbpy)2(CO)2]2+ to form stable adducts, even in the absence of any photo- and/or electro-activation.25,26 Blue-light irradiation of [RuII(tpy)(bpy)(DMF)]2+ in THF in the presence of triethylamine (TEA), on the other hand, induce the formation of [RuII(tpy)(bpy)H]+ through a light-elusive TEA-bearing intermediate, [RuII(tpy)(bpy)(TEA)]2+.24 Building on this knowledge, it therefore appeared important to pay a special attention to the nature of the molecular species that could be generated when 1-MeCN was irradiated in the presence of sacrificial electron donors. Due to its relevance in photo-driven CO2 reduction, DMF/TEOA (5:1) mixture was selected as the medium.25,29
 | ||
| Fig. 3 UV-vis spectral changes under 466 nm LED irradiation of (A) an Ar-saturated DMF solution of 1-MeCN (20 μM) and (B) an Ar-saturated DMF/TEOA (5:1) solution of 1-MeCN (20 μM). Spectra were recorded at intervals of 30 seconds (from black trace to red trace). | ||
A significantly different behaviour is observed when irradiation is performed on an Ar-degassed DMF/TEOA (5:1) solution containing 1-MeCN (20 μM). In addition to the previously characterized spectral features associated with the formation of the DMF-bearing complex, a new broad transition centred at 610 nm built up. The appearance of this new absorption band is clearly related to the concomitant formation of new species under irradiation and prevents the observation of clear isosbestic points (Fig. 3B). In order to clarify whether one or multiple new complexes are generated under these conditions, this process was followed by 1H-NMR spectroscopy (Fig. S11, ESI†). Spectra recorded in deuterated DMF displayed the simultaneous appearance of the previously identified signals of 1-DMF and a new set of signals, assigned to the formation of the complex [RuII(tButpy)(dmbpy)(TEOA)]+ (1-TEOA). After about 20 minutes of irradiation, the system reaches a steady-state.
While the initially introduced 1-MeCN has been fully converted, the proportion of the two resulting photoproducts (1-DMF and 1-TEOA) no longer evolves. Interestingly, this equilibrium between the two complexes 1-DMF and 1-TEOA was also observed in the dark (Fig. S12, ESI†), with the steady-state reached in 2 to 3 h. This dark process thus appears to be a relevant phenomenon on the timescale of typical photocatalytic experiments. By adding increasing amounts of TEOA to freshly prepared solutions of photogenerated 1-DMF in deuterated DMF, the equilibrium constant for the dark equilibrium between 1-DMF and 1-TEOA was determined to be Keq = 3 (Fig. S13, ESI†).
Knowing the proportion of the species involved in the final equilibrium, it was then possible to determine the UV-Vis spectrum of 1-TEOA (Fig. S14, ESI†). This is nicely corroborated by its TD-DFT (Time-Dependent Density Functional Theory) calculated spectrum (Fig. S14, ESI†) and further supported by the resulting UV-Vis spectrum obtained when an Ar-degassed MeCN/TEOA (5:1) solution containing 1-MeCN (20 μM) is irradiated (Fig. S15, ESI†). Under these conditions, where photogeneration of 1-DMF does not occur, the new absorption features are well matching with the calculated Ru-TEOA spectra. On this basis, derivation of the speciation of the mixture under LED irradiation was performed (Fig. 4 and Fig. S16–S18, ESI†).
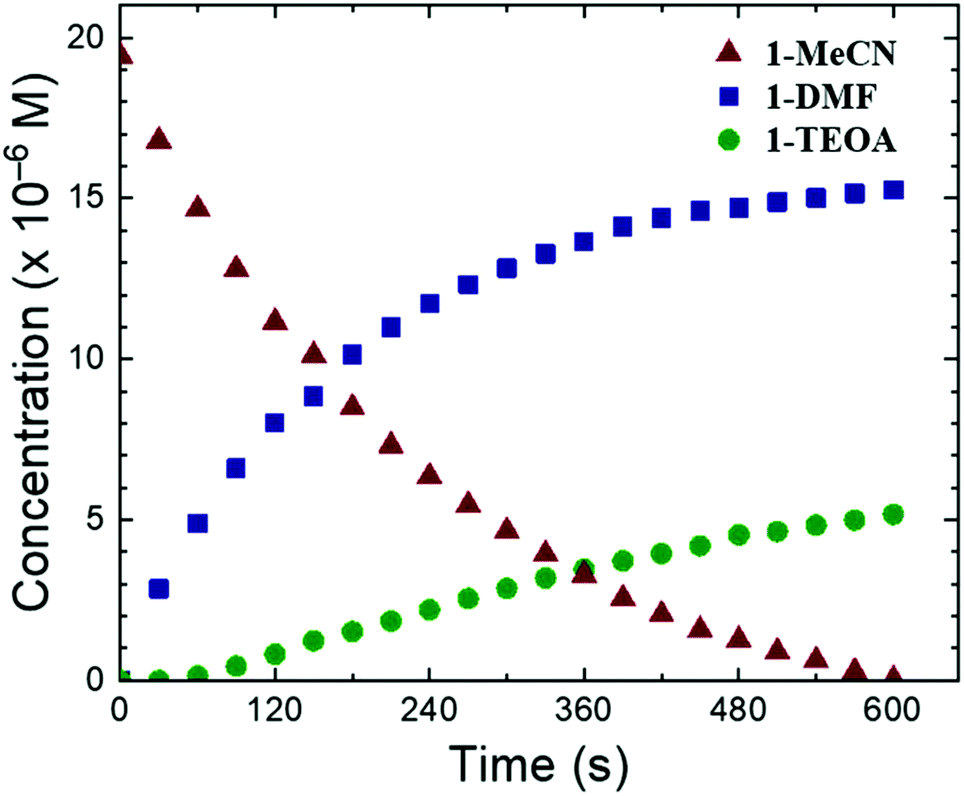 | ||
| Fig. 4 Concentration profile of the three species 1-MeCN, 1-DMF, and 1-TEOA over the course of the photolysis experiment. | ||
Interestingly, when TEOA is replaced by TEA in the reaction mixture, the only detectable process occurring is the MeCN/DMF ligand photoexchange, the kinetics of which is almost identical to the system deprived of any amines (Fig. S19, ESI†). This result suggests that coordination of TEOA to the ruthenium(II) centre in 1-TEOA could preferentially occur through a hydroxyl group. Such coordination mode is expected to facilitate the formation of a CO2 adduct (vide infra). It is however interesting to note that, in closely related complexes, photoinduced generation of triethylamine-bearing adducts was observed but in a significantly less-coordinating solvent (THF).24
:1) solution containing 1-MeCN (20 μM), but this time saturated with CO2, was irradiated at 466 nm. A rapid red shift of the 1MLCT band from 457 nm to 510 nm was observed, although no distinct isosbestic point emerged (Fig. S20, ESI†). No absorption arose around 610 nm, excluding the accumulation of any significant amount of 1-TEOA. Further investigations of this photo-induced process were then conducted using 1H-NMR spectroscopy in deuterated DMF. Spectra of 1-MeCN recorded over the course of irradiation showed quantitative conversion of the signals associated with the starting material to those of a new species within 30 minutes (Fig. S21, ESI†). Using 13C-labelled CO2 as the substrate of the reaction allowed the use of 13C-NMR in order to provide more detailed information on the nature of the newly generated species. In the absence of 1H-decoupling, a triplet centred at 160.4 ppm (JC-H = 3.6 Hz) was observed (Fig. S22, ESI†), strongly suggesting the formation of a coordinated carbonate species. This result mirrors the situation described on Re-based CO2 reduction catalysts where insertion of CO2 to metal complexes bearing a TEOA ligand has been shown.25 It thus appears reasonable to propose the formation of a similar compound, 1-CO2-TEOA (Fig. 5) under the experimental conditions explored in this study. TD-DFT calculated spectrum of 1-CO2-TEOA was obtained (Fig. S23, ESI†): the absence of significant transitions in the 600–650 nm range further supports the CO2 capture by 1-TEOA.
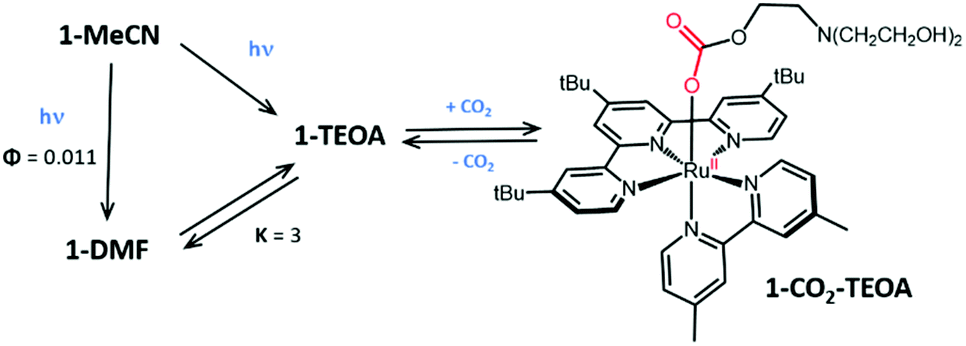 | ||
| Fig. 5 Relevant intermediates involved in dark and photo-induced processes brought to light as early steps of the photocatalytic reduction of CO2 by 1-MeCN. | ||
Taken together, these data support the existence of complex dark and photo-induced processes resulting in the formation of 1-CO2-TEOA as an interesting intermediate under photocatalytically-relevant conditions (Fig. 5). It should also be noted that this capture process is completely reversible and can easily be repeated. By bubbling argon gas for 5 minutes in a solution of 1-CO2-TEOA, the UV-vis spectral features associated to the equilibrated mixture of 1-DMF and 1-TEOA can be recovered, indicating a smooth release of the coordinated CO2 molecule (Fig. S24, ESI†).
Conclusions
The synthesis and full characterization of the complexes 1-Cl and 1-MeCN have been reported. A comprehensive study of the nature of the intermediates that can be generated in the first stages of photocatalytic experiments have been carried out. Ligand photo-labilization processes, associated with a complex equilibrium between multiple ligand-exchanged species, have been evidenced. In particular, the formation of an intermediate involving the coordination at the metal centre of TEOA, a commonly used sacrificial electron donor, has been demonstrated. Its ability to reversibly capture CO2 is a remarkable feature, that makes it an attractive entry point into photocatalytic processes. This study further enlarges the scope of the previous works from Ishitani and co-workers, highlighting the dramatic role that the choice of solvent or sacrificial electron donor – often-considered as “minor partners” – may play under photocatalytically-relevant conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Foundation Olle Engkvist Byggmästare (grant 2016/3), the Swedish Energy Agency (grant no. 11674-8) and the NordForsk network NordCO2. WBS acknowledges funding from the NSF Graduate Research Opportunities Worldwide (GROW) program and an NSF Graduate Research Fellowship (DGE-1650116).References
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283–9289 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- H. Hori, F. P. A. Johnson, K. Koike, K. Takeuchi, T. Ibusuki and O. Ishitani, J. Chem. Soc., Dalton Trans., 1997, 1019–1024, 10.1039/a607058b.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS.
- Y. Tamaki, K. Koike and O. Ishitani, Chem. Sci., 2015, 6, 7213–7221 RSC.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed, 2016, 55, 1825–1829 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 131–132, 10.1039/C39870000131.
- H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1994, 33, 3415–3420 CrossRef CAS.
- M. Meser Ali, H. Sato, T. Mizukawa, K. Tsuge, M.-a. Haga and K. Tanaka, Chem. Commun., 1998, 249–250, 10.1039/A707363A.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Chem. Commun., 2011, 47, 12607–12609 RSC.
- Z. Chen, J. J. Concepcion, M. K. Brennaman, P. Kang, M. R. Norris, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15606–15611 CrossRef CAS PubMed.
- Z. Chen, P. Kang, M. T. Zhang and T. J. Meyer, Chem. Commun., 2014, 50, 335–337 RSC.
- T. A. White, S. Maji and S. Ott, Dalton Trans., 2014, 43, 15028–15037 RSC.
- B. A. Johnson, H. Agarwala, T. A. White, E. Mijangos, S. Maji and S. Ott, Chemistry, 2016, 22, 14870–14880 CrossRef CAS PubMed.
- H. Tanaka, H. Nagao, S. M. Peng and K. Tanaka, Organometallics, 1992, 11, 1450–1451 CrossRef CAS.
- P. Voyame, K. E. Toghill, M. A. Mendez and H. H. Girault, Inorg. Chem., 2013, 52, 10949–10957 CrossRef CAS PubMed.
- J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86–91 CrossRef CAS.
- Y. Matsubara, H. Konno, A. Kobayashi and O. Ishitani, Inorg. Chem., 2009, 48, 10138–10145 CrossRef CAS PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS PubMed.
- R. N. Garner, L. E. Joyce and C. Turro, Inorg. Chem., 2011, 50, 4384–4391 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed.
- Y. Matsubara, K. Koga, A. Kobayashi, H. Konno, K. Sakamoto, T. Morimoto and O. Ishitani, J. Am. Chem. Soc., 2010, 132, 10547–10552 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt03461g |
| ‡ Current address: CNRS, LCC (Laboratoire de Chimie de Coordination), 31077 Toulouse (France), e-mail: nicolas.queyriaux@lcc-toulouse.fr |
| This journal is © The Royal Society of Chemistry 2019 |