
Designed asymmetric coordination helicates with bis-β-diketonate ligands†
Mohanad
Darawsheh,  a
Leoní A.
Barrios,
ab
Anna
Sadurní,
c
Jordi
García,
c
Paul
Lloyd-Williams,
c
Simon J.
Teat,
d
Olivier
Roubeau,
e
David
Aguilà
*ab
and
Guillem
Aromí
*ab
a
Leoní A.
Barrios,
ab
Anna
Sadurní,
c
Jordi
García,
c
Paul
Lloyd-Williams,
c
Simon J.
Teat,
d
Olivier
Roubeau,
e
David
Aguilà
*ab
and
Guillem
Aromí
*ab
a
Leoní A.
Barrios,
ab
Anna
Sadurní,
c
Jordi
García,
c
Paul
Lloyd-Williams,
c
Simon J.
Teat,
d
Olivier
Roubeau,
e
David
Aguilà
*ab
and
Guillem
Aromí
*ab
Abstract
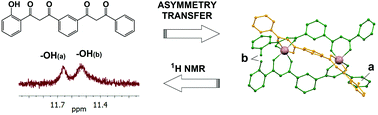
A new bis-(β-diketone) ligand featuring built-up structural asymmetry yields non-symmetric Fe(III) and Ga(III) dinuclear, triple-stranded helicates by design. Their structural properties have been studied, both in solid state and in solution, and compared with their corresponding symmetric analogues. The robustness observed shows the potential of this synthetic strategy to develop non-symmetric helicoidal motifs with specific functional groups.
 Please wait while we load your content...
Please wait while we load your content...

