
Coordination-induced O–H bond weakening in Sm(ii)-water complexes†

Abstract
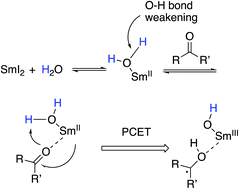
The combination of Sm(II)-based reductants with water provides reagent systems that promote a range of reductions and bond-forming reactions that are endergonic based on known redox potentials. Despite the utility of these reagent systems, little work has been done to elucidate the basis for this unusual reactivity. Herein we present recent work designed to explore the underlying mechanistic basis for the unexpected reactivity of the Sm(II)-water reducing system and demonstrate that O–H bond weakening induced by coordination of water to low valent samarium enables proton coupled electron transfer to substrates. We also demonstrate that coordination-induced bond weakening can be exploited and used to create extremely powerful reductants capable of reducing functional groups typically recalcitrant to reduction through single electron transfer alone.
- This article is part of the themed collection: 2019 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

