
Insight into the mechanism of CO-release from trypto-CORM using ultra-fast spectroscopy and computational chemistry†‡
Benjamin J.
Aucott§
 a,
Jonathan B.
Eastwood§
a,
L.
Anders Hammarback§
a,
Ian P.
Clark
b,
Igor V.
Sazanovich
b,
Michael
Towrie
b,
Ian J. S.
Fairlamb
*a and
Jason M.
Lynam
*a
a,
Jonathan B.
Eastwood§
a,
L.
Anders Hammarback§
a,
Ian P.
Clark
b,
Igor V.
Sazanovich
b,
Michael
Towrie
b,
Ian J. S.
Fairlamb
*a and
Jason M.
Lynam
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: ian.fairlamb@york.ac.uk; jason.lynam@york.ac.uk
bCentral Laser Facility, STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK
First published on 25th October 2019
Abstract
Photolysis of trypto-CORM, fac-[Mn(tryp)(CO)3(NCMe)] (tryp = tryptophanate) at 400 nm results in controlled CO-release which may be utilised to inhibit the growth of Escherichia coli (E. coli). An investigation into the fundamental processes which underpin the CO-release event is described. Time-dependent density functional theory (TD-DFT) indicates that irradiation at 400 nm results in LMCT from the indole group of the amino acid to orbitals based on the metal as well as the carbonyl and NCMe ligands. Ultra-fast time-resolved infra-red spectroscopy (TRIR) demonstrates that in NCMe solution, photolysis (400 nm) results in loss of CO in under 3 ps with the sequential generation of three new states with two carbonyl ligands and a coordinated tryptophanate. The first species is assigned to vibrationally hot 3[Mn(tryp)(CO)2(NCMe)] which undergoes cooling to give the complex in its v = 0 state. This triplet state then undergoes solvation (τ ≈ 20 ps) with a concomitant change in spin to give [Mn(tryp)(CO)2(NCMe)2] which persists for the remainder of the experiment (800 μs). These data indicate that following the initial photochemically induced loss of CO, any thermal CO loss is much slower. Related experiments with trypto-CORM in a mixture of DMSO and D2O gave analogous data, indicating that this process also occurs in the medium used for the evaluation of biological properties.
Introduction
Despite its deserved reputation as a poison, carbon monoxide has been shown to have well-defined beneficial therapeutic effects. CO is produced naturally in mammals, primarily through the physiological degradation of heme, it acts as a signalling molecule and has been shown to protect against ischemic damage and organ graft rejection, it is antibacterial, antimalarial and may act as a vasodilator.1–3 Although controlled treatment with CO gas may be envisaged as a route to harness these beneficial effects, its lack of selectivity means that alternative delivery methods are needed. This has resulted in the advent of carbon monoxide-releasing molecules (CO-RMs) which are species which liberate CO when subjected to a suitable stimulus.4–7 The vast majority of CO-RMs are transition metal carbonyl complexes which presumably represents the ability of these elements to readily coordinate CO.8 Although there are a vast number of metal carbonyl complexes, control over the spatial and temporal delivery of CO remains a challenge. CO-RMs are needed in which the rate, extent and location of CO-release are controlled. This may be achieved by molecules which selectively localise in certain organs9 or are activated by specific triggers.10 A number of stimuli have therefore been explored to control the CO-release event, such as activation by enzymes,11–13 as well as thermal-,14–18 electrochemical-19 and magnetic heating-induced release.20 However, light offers the benefit of temporal and spatial control over the CO-loss event and therefore the photochemical activation of CO-RMs (photoCO-RMs) has been extensively explored.21–24Complexes based on manganese(I) represent the most common class of photoCO-RMs.25–37 These complexes have antibacterial activity,25,26,34,35 may be woven into materials and activated with a photosensitiser,32 or activated by two-photon excitation.33 We have recently reported the synthesis and anti-bacterial activity of trypto-CORM (Fig. 1) a manganese-based carbonyl complex with a coordinated tryptophanate ligand.38 Trypto-CORM is able to inhibit the growth of E. coli only when exposed to visible (400 nm) light. The behaviour is organism-dependant, for example, trypto-CORM shows some activity against Neisseria gonorrhoeae in the dark.39 Control experiments with leg-haemoglobin (which has an extremely high binding affinity for CO) indicate that this activity is due to the released CO.
 | ||
| Fig. 1 Structure of trypto-CORM. | ||
An understanding of the pathways that lead to CO loss and the nature of the resulting species has provided important fundamental insight into the structure and bonding of transition metal complexes.40 For example, Perutz and Turner demonstrated that photolysis of [Cr(CO)6] in a xenon matrix results in the formation of square-based pyramidal [Cr(CO)5] with a Cr–Xe interaction.41 This illustrated the high Lewis acidity of such fragments and provided support for the development of bonding models such as the isolobal analogy. The dynamics of the photochemical loss of CO and subsequent solvation has been explored using time-resolved spectroscopy. Light is employed to photo-dissociate a CO ligand and the structure of the resulting transient intermediates probed using electronic or vibrational spectroscopy.42–44 Such studies have assisted with the identification of σ-complexes, and informed the understanding of unusual bonding modes in transition metal species.45–49
Time-resolved vibrational spectroscopy has been used extensively to probe behaviour in organic solvents, however, there are far fewer reports of this method being applied to study the light-induced loss of CO from photoCO-RMs in more biologically relevant media.19,36 Such studies, when coupled with a detailed computational understanding of their photochemistry50 (for example the nature of the electronic transitions and excited state structures which lead to the CO-loss event) will inform the development of new classes of photoCO-RMs.
We have recently employed TRIR spectroscopy to investigate the behaviour of metal complexes fac-[Mn(ppy)(CO)3(L)] (ppy = 2-phenylpyridine, L = ligand) generated by the light-induced dissociation of CO from [Mn(ppy)(CO)4].51,52 Complexes based on this scaffold have found uses as CO-RMs53–55 and in catalysis.56–59
These studies have provided insight into the solvation of unsaturated species and the role of these complexes as catalysts for C–H functionalisation. For example, photolysis (355 nm) of [Mn(ppy)(CO)4] in media such as heptane, toluene, CH2Cl2 and ethers results in CO loss in under 1 ps and the generation of solvent complexes fac-[Mn(ppy)(S)(CO)3] (S = solvent).51 The solvation event is kinetically controlled, for example, in tetrahydrofuran, the solvent initially binds to the manganese through two electrons of a C–H bond which is followed by an ultrafast (ps) rearrangement to the more thermodynamically stable oxygen-bound form.51 Performing the experiments in the presence of substrates used in the direct C–H functionalisation reactions, such as PhC2H, allowed for the observation of the resulting π-alkyne complexes and the migratory insertion reaction leading to C–C bond formation.52
It was anticipated that related studies using trypto-CORM would provide insight into the nature of the species formed following the light-induced loss of CO. A combined computational (DFT) and experimental (TRIR) study of the light-induced dissociation of CO from trypto-CORM is described herein.
Results and discussion
Vibrational and electronic spectra of trypto-CORM
The ground state infra-red spectrum of trypto-CORM exhibits three bands in the metal carbonyl region in MeOH solution at 2037, 1933 and 1921 cm−1, corresponding to the symmetric and (two) asymmetric stretching modes expected for an octahedral complex with three fac-coordinated carbonyl ligands. An additional broader band, assigned to the ester group of the coordinated tryptophan, is observed at 1638 cm−1. This band is red-shifted when compared to the mode in the free amino acid.38 These strong metal- and organic-carbonyl modes are ideal reporting groups to probe changes in the coordination environment of the manganese following photolysis.The lowest energy band in the UV-vis spectrum of trypto-CORM in NCMe has a maximum at 360 nm (ε = 1443 mol−1 dm3 cm−1) which tails into the visible region of the spectrum (Fig. 2a).38 Photolysis of trypto-CORM at 400 nm results in light-induced bleaching of this band and instigates the CO release process.38 The nature of the transitions which constitute this band were modelled with TD-DFT at the PBE0/DGDZVP/def2tzv level of theory, with implicit solvation in NCMe. The calculations at this level indicate that the lowest energy transition should occur at 431 nm and has a 100% HOMO98 → LUMO99 composition. A less intense feature is predicted at 408 mn (100% HOMO98 → LUMO+1100). The HOMO is based on the indole of the tryptophanate ligand whereas the LUMO and LUMO+1 are metal-centred (Fig. 2(b)–(d)). In addition some of the higher energy transitions involve electron density on the ester group of the amino acid ligand (see ESI‡ for complete list) in the occupied orbital. Therefore irradiation at 400 nm is best viewed as resulting in a ligand-to-metal-charge transfer (LMCT).
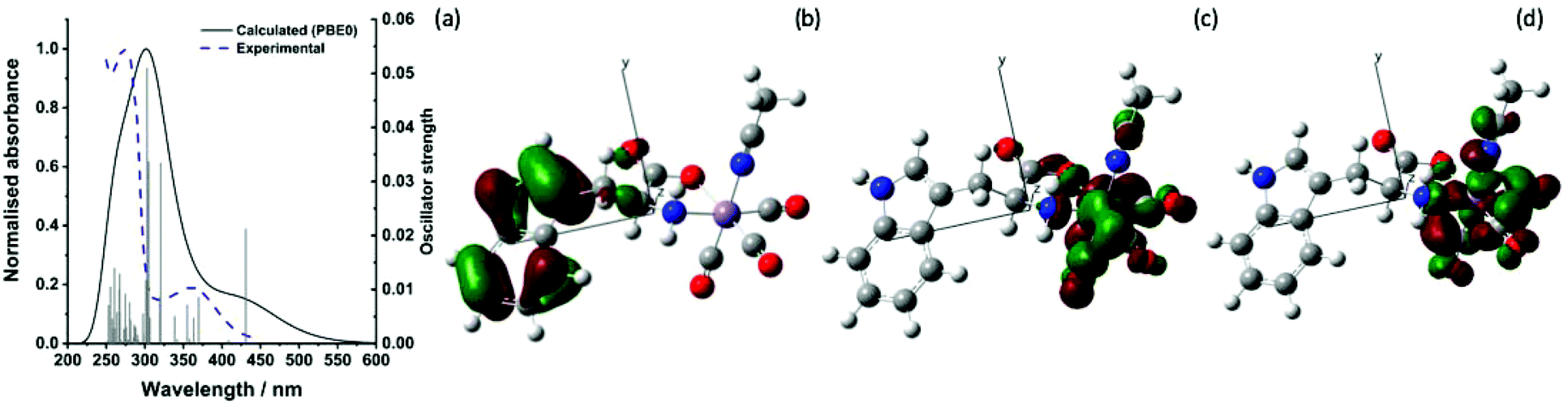 | ||
| Fig. 2 (a) Experimental and predicted electronic spectra of trypto-CORM (b) HOMO98 (c) LUMO99 and (d) LUMO+1100 calculated at the PBE0/DGDZVP/def2tzv level. Isosurface shown at the 0.04 level. | ||
TRIR spectroscopy on trypto-CORM
The TRIR studies were performed using the LIFEtime system60 in the ULTRA facility at the Rutherford Appleton Laboratory using the time-resolved multiple-probe spectroscopy (TRMPS) method.61 This allows for the acquisition of time-resolved spectra with pump–probe delays between 1 ps and 1 ms. A pump wavelength of 400 nm was employed to simulate the conditions used in the biological experiments.38The initial experiments focussed on understanding the photolysis of trypto-CORM in NCMe solution as it was anticipated that prompt photo-dissociation of CO would result in the formation of a vacant coordination site that would be occupied by the coordinating solvent. The TRIR spectroscopic data are presented as difference spectra with negative bands corresponding to the species which are lost on irradiation and the positive bands due to the photoproducts.
Time-resolved spectra in the region 1780–2100 cm−1 (Fig. 3) exhibited strong negative bands corresponding to the ground state spectrum of trypto-CORM. The observation of these ground state bleach (GSB) bands demonstrates that trypto-CORM is lost on photolysis at 400 nm. Analysis of the resulting spectra indicated the formation of three different species. Two, A and B, were present in spectra recorded with pump–probe delays longer than 3 ps – data recorded with shorter delays were broad and featureless. A spectrum recorded with a pump–probe delay of 10 ps showed the presence of two peaks between 1950 and 1980 cm−1 (Fig. 4, top). Gaussian deconvolution (Table 1) of these bands gave band positions of (1964.8 ± 1.5) cm−1 and (1976.7 ± 0.6) cm−1 assigned to A and B respectively. A further feature was observed centred at ca. 1800 cm−1. This was also successfully deconvoluted into two bands (Fig. 4, bottom) with peak positions of (1813.7 ± 1.7) and (1799.6 ± 3.5) cm−1, assigned to A and B respectively. These features are at a very low frequency for a metal carbonyl stretching mode.
 | ||
Fig. 3 Bottom IR spectrum of trypto-CORM in NCMe in the region 1780–2100 cm−1. Top time-resolved difference IR spectra for the photolysis (400 nm) of trypto-CORM in NCMe with pump–probe delays of 10 ps, 100 ps, and 1 ns showing bands for A and B (■) and C by ( ). The red arrows show the apparent recovery of the GSB for trypto-CORM. ). The red arrows show the apparent recovery of the GSB for trypto-CORM. | ||
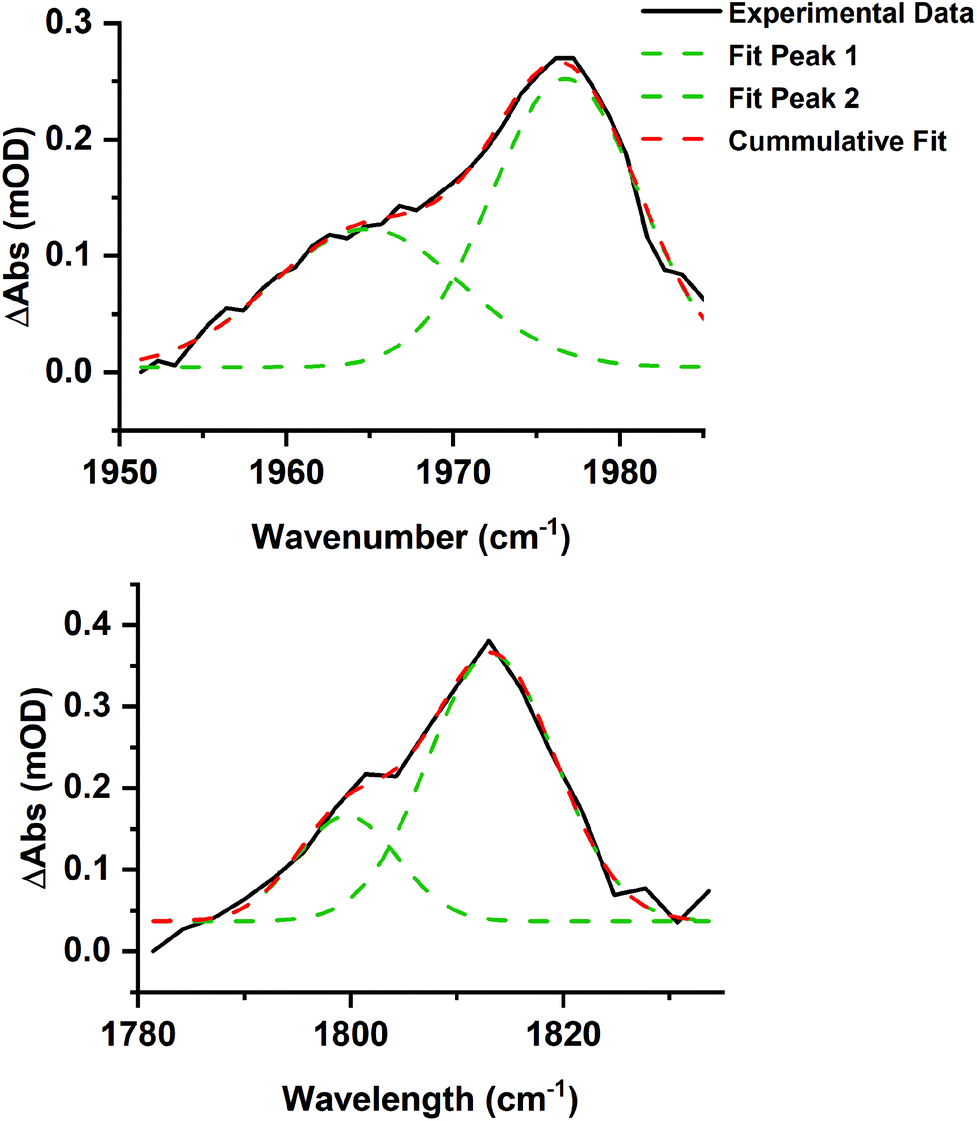 | ||
| Fig. 4 Gaussian deconvolution of the bands at in the region 1950–1990 cm−1 (top, R2 = 0.986) and 1780–1830 cm−1 (bottom, R2 = 0.970) at 10 ps. Parameters are listed in Table 1. | ||
| Band position/cm−1 | Peak width at half height/cm−1 | Integration | Assignment |
|---|---|---|---|
| 1976.7 ± 0.6 | 10.4 ± 0.9 | 2.74 ± 0.42 | B |
| 1964.8 ± 1.5 | 13.4 ± 3.5 | 1.69 ± 0.62 | A |
| 1813.7 ± 1.7 | 14.1 ± 3.3 | 4.93 ± 1.35 | B |
| 1799.6 ± 3.5 | 11.3 ± 6.1 | 1.55 ± 1.22 | A |
The presence of two bands is consistent with the photochemically induced loss of a CO ligand from trypto-CORM to give species with two mutually cis carbonyl ligands. Analysis of the intensity of the bands with time revealed that A had a greater initial concentration than B. The change in intensity of B with time was fitted to an exponential growth then decay function (τgrowth = (2.5 ± 3.9) ps and τdecay = (15.9 ± 3.9) ps, Fig. 5). This was interpreted as initial formation of A which then transformed into B with a short lifetime. Although it was not possible to obtain a satisfactory fit to first order kinetics for A, qualitatively the loss of A corresponded with the growth of B (see ESI‡).
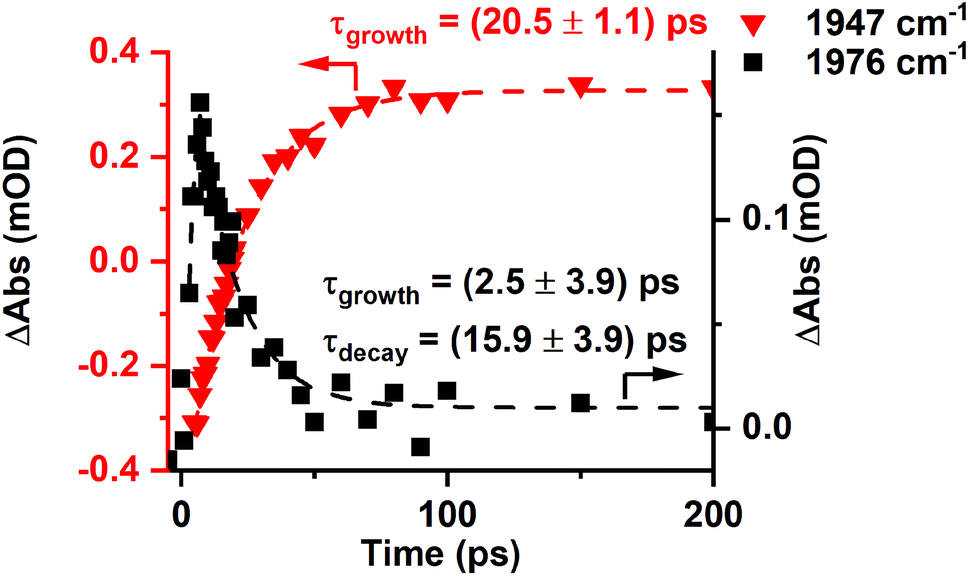 | ||
Fig. 5 Change in intensity of the bands due to B (■) and C ( ) with time. Dashed lines show fits against exponential growth/decay (B) and exponential growth (C) kinetics. ) with time. Dashed lines show fits against exponential growth/decay (B) and exponential growth (C) kinetics. | ||
Over the course of ca. 100 ps, the bands for B decreased in intensity to be replaced by a peak at 1853 cm−1 and a smaller feature at 1947 cm−1: these were assigned to a third photoproduct, C (Fig. 3). Inspection of the bands due to the GSB indicated some apparent recovery in intensity of the band due to trypto-CORM at 1933 cm−1 (highlighted with an arrow in Fig. 3). It was suspected that this was due to the overlap between the GSB bands for trypto-CORM and a peak for C. In order to determine if this was the case, the spectrum of trypto-CORM was subtracted from the difference spectrum recorded with a pump–probe delay of 1 ns. The resulting spectrum (Fig. 6) shows the presence of peak with a band position of 1946 cm−1, leading to the conclusion that C was a cis-dicarbonyl complex. The growth of C proceeded with a statistically identical lifetime to the loss of B, τgrowth = (20.5 ± 1.1) ps, indicating that B → C.
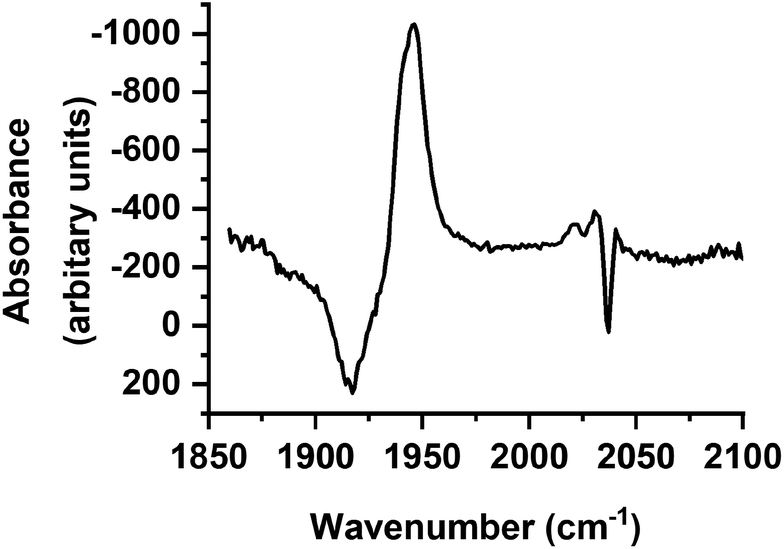 | ||
| Fig. 6 Difference spectrum in the region 1850–2100 cm−1 recorded with a pump–probe delay of 1 ns with ground state bleach signal subtracted. | ||
Difference spectra recorded in the region 1500–1700 cm−1 exhibited a strong GSB at ca. 1639 cm−1 (Fig. 7), consistent again with the loss of trypto-CORM on photolysis. Bands that could be assigned to the ester group in A, B and C were observed at short (<20 ps) and long (>100 ps) pump–probe delays respectively. The small changes in the band position of the ester mode was taken to indicate that the environment of the tryptophanate ligand had not changed significantly from either the parent trypto-CORM, or between A, B and C.
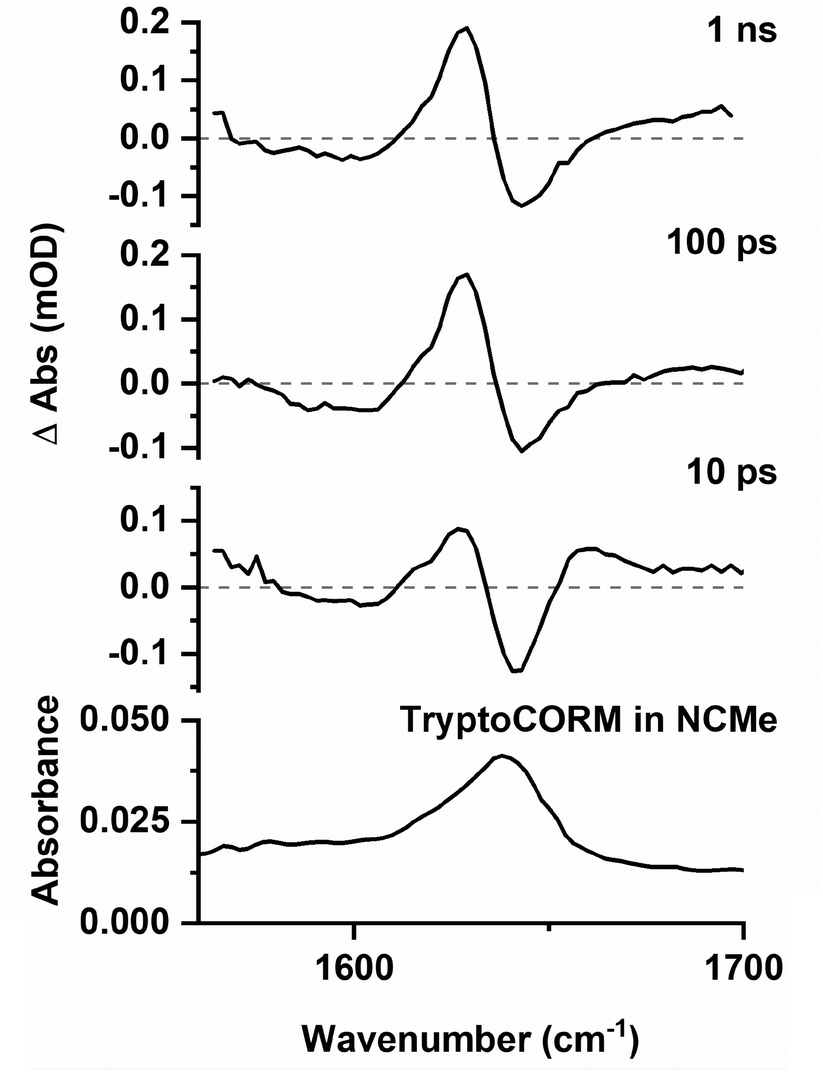 | ||
| Fig. 7 Bottom IR spectrum of trypto-CORM in NCMe in the region 1560–1700 cm−1. Top time-resolved difference IR spectra for the photolysis (400 nm) of trypto-CORM in NCMe with pump–probe delays of 10 ps, 100 ps, and 1 ns. | ||
In order to evaluate the behaviour of trypto-CORM in an aqueous medium, the experiment was repeated in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (1:9 v:v) solution. The lower solubility of the complex in this medium compared to NCMe entailed that the resulting spectra were less intense. However, it was evident from the resulting data that related processes were occurring in both media. In the region between 1800 and 2100 cm−1 bands were observed at 1985 and 1812 cm−1 (Fig. 8) which were assigned to the DMSO analogues of A and B. After 100 ps these bands had been replaced by a new peak at 1826 cm−1 and, as was the case for NCMe, the GSB for the trypto-CORM at ca. 1935 cm−1 had recovered in intensity (Fig. 7, red arrows), indicating a band from a species analogous to C overlapping with that from trypto-CORM.
:D2O (1:9 v:v) solution. The lower solubility of the complex in this medium compared to NCMe entailed that the resulting spectra were less intense. However, it was evident from the resulting data that related processes were occurring in both media. In the region between 1800 and 2100 cm−1 bands were observed at 1985 and 1812 cm−1 (Fig. 8) which were assigned to the DMSO analogues of A and B. After 100 ps these bands had been replaced by a new peak at 1826 cm−1 and, as was the case for NCMe, the GSB for the trypto-CORM at ca. 1935 cm−1 had recovered in intensity (Fig. 7, red arrows), indicating a band from a species analogous to C overlapping with that from trypto-CORM.
 | ||
Fig. 8 Bottom IR spectrum of trypto-CORM in DMSO:D2O (1:9 v:v) in the region 1780–2100 cm−1. Top time-resolved difference IR spectra for the photolysis (400 nm) of trypto-CORM in NCMe with pump–probe delays of 10 ps, 100 ps, and 1 ns showing bands for A (■) and C by ( ). The red arrows show the apparent recover of the GSB for trypto-CORM. ). The red arrows show the apparent recover of the GSB for trypto-CORM. | ||
Spectra recorded in the region 1560–1700 cm−1 (Fig. 9) were weak, however, it was possible to observe a GSB and that the peaks for transient species present at 10 ps. A band assigned to C was present at 1 ns.
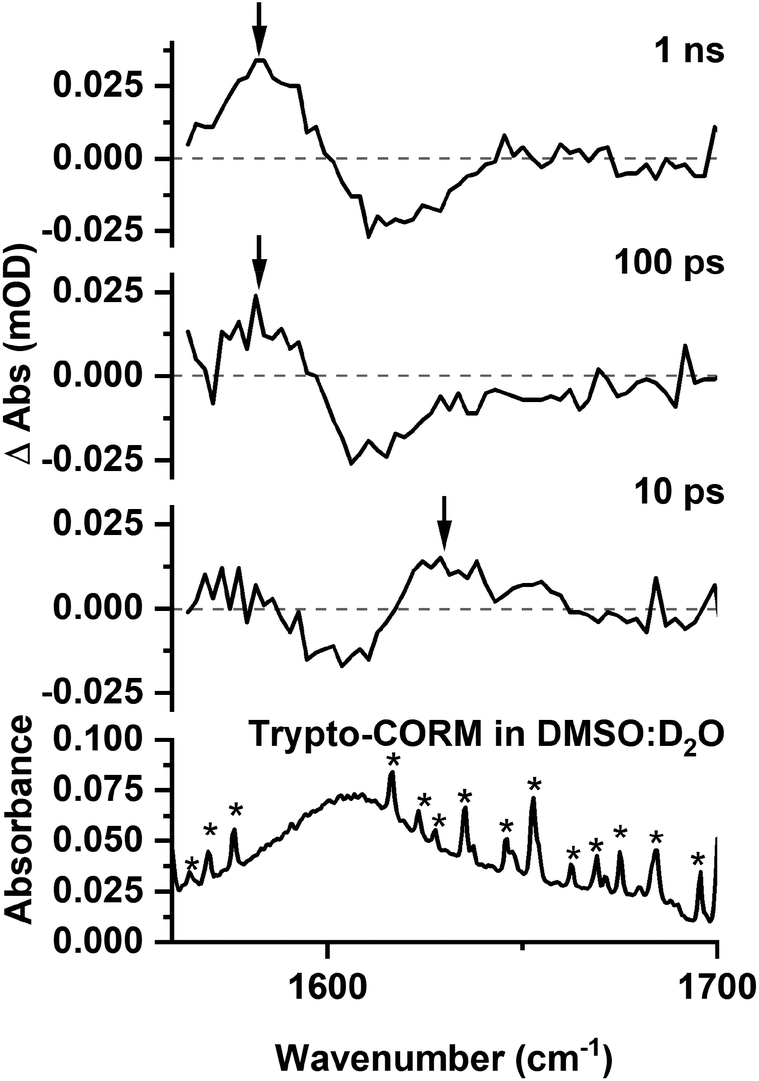 | ||
| Fig. 9 Bottom IR spectrum of trypto-CORM in DMSO:D2O (1:9 v:v) in the region 1560–1700 cm−1. Top time-resolved difference IR spectra for the photolysis (400 nm) of trypto-CORM in NCMe with pump–probe delays of 10 ps, 100 ps, and 1 ns, band positions marked with an arrow. * indicates bands due to H2O contamination. | ||
These observations demonstrate that the behaviour of trypto-CORM is similar in both media. Given that our previous data had indicated that the NCMe ligand in the complex is thermally labile, it was anticipated that speciation in a DMSO/D2O mixture was complicated. However, the shifts of the bands in the region 1800–1850 cm−1 to lower energy when compared to NCMe suggest that DMSO was coordinated to the metal.51
Density functional theory
The TRIR spectroscopy on trypto-CORM had demonstrated the formation of species A, B and C. In order to elucidate the potential structures of these states, a number of trial structures were probed by DFT. Geometries were optimised at the BP86/SV(P) level of theory, vibrational modes and subsequent free energy corrections were also calculated at this level. Single point energies were then calculated on these geometries at the D3BJ-PBE0/def2-TZVPP level with solvent correction using COSMO in NCMe.62 We have previously used this method on related studies to calculate reaction pathways for C–C bond formation in the coordination sphere of manganese.51,52 In these cases, there was a linear relationship between the calculated and experimental values for the reaction profiles studied and also between the calculated vibrational modes of the metal carbonyl ligands with those determined by time-resolved infra-red spectroscopy.The TRIR data had demonstrated that the products must contain two carbonyl ligands. Furthermore, as only small changes were observed in the position of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O mode of the coordinated tryptophanate, it was assumed that the binding mode of this ligand did not substantially change on photolysis.
O mode of the coordinated tryptophanate, it was assumed that the binding mode of this ligand did not substantially change on photolysis.
Two general classes of photoproducts were considered on this basis. The first were based on an 18-electron [Mn(tryp)(CO)2(NCMe)2], [1], framework. Four possible isomers (Fig. 10) are possible for this composition. The resulting geometry optimisations predicted, as expected, that all four of the complexes formed pseudo-octahedral geometries. In order to evaluate the relative Gibbs free energies, [1a] was taken as the reference state. Complex [1b] was found to lie at +14 kJ mol−1, [1c] at 3 kJ mol−1 whereas [1d] with two mutually trans carbonyl ligands is at a much higher energy (+74 kJ mol−1).
 | ||
| Fig. 10 Potential structures for complexes A and C studied by DFT. | ||
The second class of potential photoproducts considered were 16-electron complexes [Mn(tryp)(CO)2(NCMe)], [2], in which a CO ligand had dissociated leaving a formally vacant coordination site at the metal. Both singlet 1[Mn(tryp)(CO)2(NCMe)], 1[2], and triplet 3[Mn(tryp)(CO)2(NCMe)], 3[2] spin states were calculated. Three geometric isomers were located for 1[2] which possessed geometries intermediate between a trigonal bipyramid and a square-based pyramid with the largest angle in the equatorial plane being ca. 150° in all cases. This is in marked contrast to the related 16-electron complex fac-[Mn(ppy)(CO)3] which is calculated to have a square-based pyramidal shape (angles in the basal plane 168° and 170°).51 This species is proposed to be generated from the photochemically induced loss of CO from [Mn(ppy)(CO)4], however, solvation occurs within 1 ps. The difference in geometry between the complexes 1[2] and fac-[Mn(ppy)(CO)3] may be assigned to the presence of the coordinated ester group in the tryptophante ligand. This is a good π-donor group and may provide electron density for the formally vacant d-orbital in a d6 16-electron system.63 The significant shortening of the Mn–O bond length in all three isomers of 1[Mn(tryp)(CO)2(NCMe)] (all less than 1.93 Å) when compared to trypto-CORM (calculated 2.020 Å) and the four isomers of [Mn(tryp)(CO)2(NCMe)2] (all longer than 2 Å) presumably reflect this effect. Complex 1[2c], in which the two carbonyl ligands and the oxygen of the tryptophanate ligand occupy the three equatorial sites, is the lowest energy (−17 kJ mol−1 relative to 1[2a]) of the three isomers investigated.
Two minima were located for 3[Mn(tryp)(CO)2(NCMe)] despite using a number of different starting geometries. These only differ in the orientation of the indole substituent with respect to the NCMe ligand. In 3[2a] the indole is on the opposite face to the NCMe, in 3[2b] it is on the same face. Despite only a small conformational difference, the DFT calculations indicated a remarkable difference in energy. Complex 3[2a] has an energy of −16 kJ mol−1 relative to 1[2a] whereas 3[2b] is at −117 kJ mol−1. A further analysis of the calculations indicated this difference in energy is almost entirely due to the empirical dispersion correction. If this correction is not applied, then the relative energies of the two isomers are −18 kJ mol−1 (3[2a]) and −8 kJ mol−1 (3[2b]). This significant dispersion effect is assigned to an interaction between the methyl group of the NCMe ligand and the π-system of the indole which is present in 3[2b], but not 3[2a]. However, significant care must be taken in the interpretation of such an effect as the current model is only accounting for intramolecular interactions. It is highly likely that the indole in 3[2a] (and indeed in all of the other structures examined that do not shown this effect) is interacting with the NCMe solvent in a similar fashion. It is argued that the D3-correction is, in this instance, significantly over stabilising 3[2b].
The calculated vibrational modes for the metal carbonyl groups at the BP86/SV(P) level for all the structures explored are presented in Fig. 11. The values have been scaled using the linear empirical relationship from our previous study on related manganese carbonyl complexes.52 As shown in Fig. 11, using this method also give a good approximation to the experimental values of trypto-CORM.
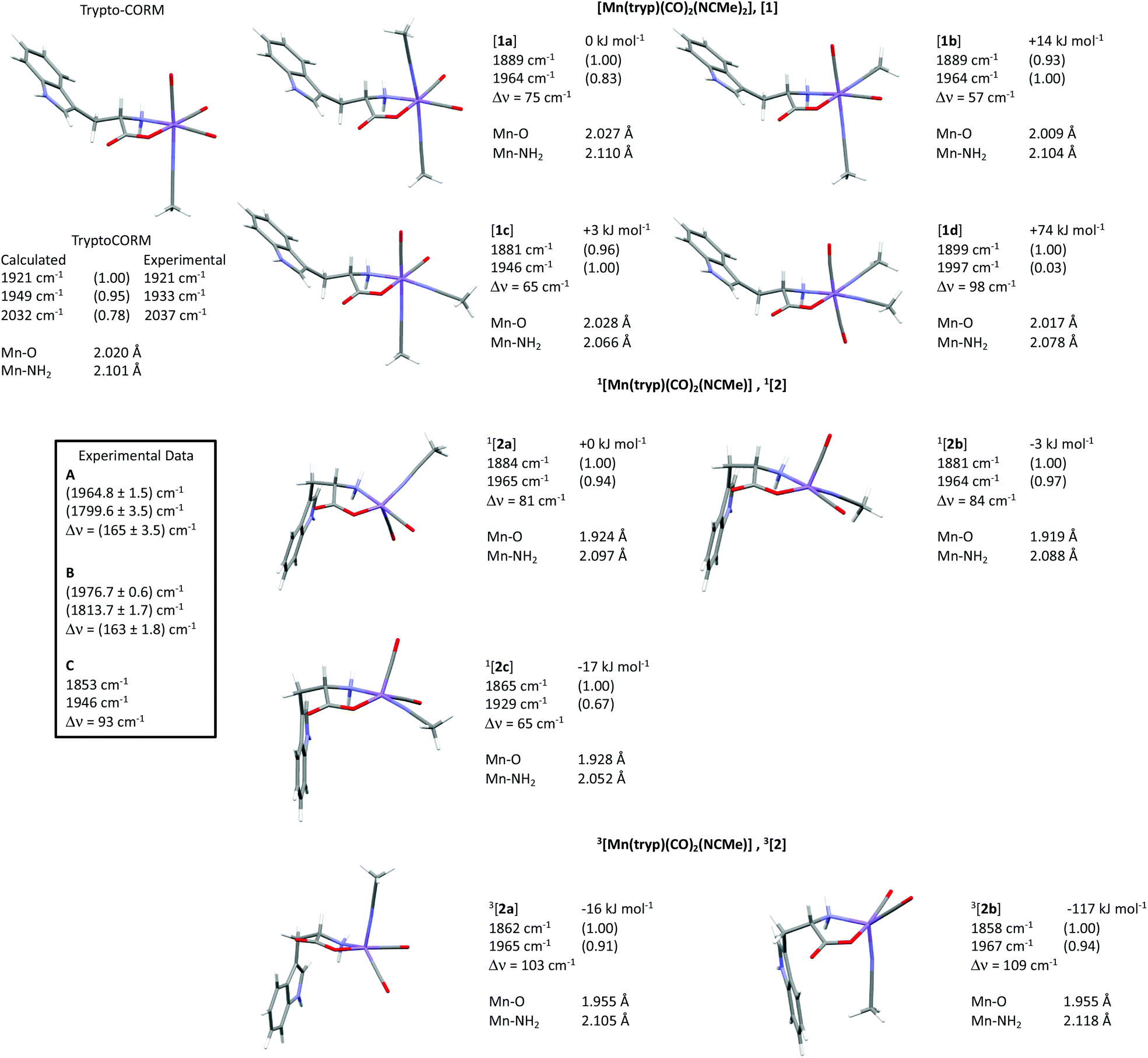 | ||
| Fig. 11 Calculated structures, scaled infra-red modes for the metal carbonyl stretches (with normalised predicted intensities), relative free energies at 298 K in kJ mol−1 and Mn–O and Mn–NH2 bond lengths, of trypto-CORM and isomers of [1], 1[2] and 3[2] at the D3-PBE0/def2-TZVPP/BP86/SV(P) level of theory. Manganese is shown in purple, carbon grey, hydrogen white, oxygen red and nitrogen blue. The relative energies of complexes [1] are relative to [1a], those for [2] relative to 1[2a]. Δν refers to the difference in frequency between the two carbonyl modes. | ||
Structural assignment and mechanistic discussion
The TRIR data indicate that photolysis of trypto-CORM results in the process A → B → C. Focussing firstly on the identity of species C, the DFT-predicted bands for complex [1c] are the closest match to the experimentally determined values (Fig. 11). The predicted bands for [1a] and [1b] are similar, but are all at higher energy than [1c]. Across all the compounds investigated, the asymmetric stretching mode is predicted to occur at higher energy than the experiment, but for [1c] the symmetric stretch is a close match to experiment. Complex [1d] can be excluded as the two mutually trans carbonyl ligands would entail that the symmetric stretching mode would be at high energy with a very low intensity when compared to the experimental values.The assignment of the short-lived species observed in the TRIR experiments, A and B, is more complex. As shown in Table 1, these species show unusually low frequency bands for the asymmetric stretch with a large difference in energy between the two bands, Δν. The fact that only two metal-carbonyl bands are observed for A and B supports an assignment to complexes containing two mutually cis carbonyl ligands. This indicates that the CO-loss event must occur in less than 3 ps: the data are not consistent with the formation of a tricarbonyl complex which then undergoes CO loss on a ps timescale to give C,¶ as is observed in chromium arene complexes.64 An examination of the data in Fig. 11 reveals that only the two isomers of 3[2] are predicted to have a large (>100 cm−1) value of Δν. Complexes 3[2] are also predicted to have one of the lowest energy bands for the asymmetric stretching mode. Therefore, states A and B are both assigned to 3[2c]. One important spectroscopic feature of note is the extremely low frequency of the asymmetric stretch for both A and B. This is in a region more typical of bridging carbonyl ligands, however, the mass spectrum of trypto-CORM indicates that the complex is monomeric, precluding this assignment.
It is proposed that vibrational cooling is responsible for the process A → B. As the energy of the photon which results in photodissociation is greater than that needed to break the M–CO bond, 3[2c] is generated in a vibrationally excited state (v > 0).65 Due to the anharmonic nature of the vibrational energy well, cooling to leads to a blue shift in energy of these modes, as observed for A → B. Indeed, the difference in frequency between the bands in A and B (ca. 14 cm−1) is consistent with the former being the v = 1 → 2 transition of 3[2c], whereas the latter is v = 0 → 1.66
A mechanistic picture is proposed, Scheme 1, in which the ultra-fast photodissociation of CO from trypto-CORM results in the formation of 3[2] in a vibrationally excited state (A) which undergoes cooling to v = 0 to give B. A change in spin with coordination of the solvent gives [1c] (C). Our calculations indicate that the binding of the solvent to 3[2] would be unfavourable as attempts to calculate a triplet spin isomer of [1] were unsuccessful. As expected for a d6 18-electron octahedral complex, 3[1] is metal–ligand dissociative and the geometry optimisation resulted in loss of an NCMe ligand with the metal adopting a structure related to 3[2]. Hence solvent coordination is predicted to only occur to the single state. The lifetime for the formation of C ([1c]) is ≈20 ps and solvation of unsaturated d6 metal complexes by NCMe can occur in 1 ps,67 hence the change in spin-state is proposed to be rate controlling. Similar behaviour has been reported for photochemically generated 3[Fe(CO)4] and 3[Co(η5-C5H5)(CO)].68 Recent studies on 3[Mn(η5-C5H5)(CO)2] have indicated that the nature of the solvent effects the lifetime of the triplet state which may indicate a solvent-promoted change in spin as an additional mechanistic pathway.69
 | ||
| Scheme 1 Proposed mechanism for the photochemically induced loss of CO from trypto-CORM. | ||
Conclusions
Photolysis of trypto-CORM at 400 nm results in ultrafast CO dissociation and formation of 3[Mn(tryp)(CO)2(NCMe)] in a vibrationally hot state which then undergoes cooling. This triplet state subsequently forms a long-lived species, assigned to all-cis-[Mn(tryp)(CO)2(NCMe)2], [1c], with a lifetime of ≈20 ps. This species remains unchanged for the duration of the experiment (800 μs). Previous studies have demonstrated that trypto-CORM is capable of releasing between two and three molecules of CO38 and these data indicate that any subsequent thermal loss of CO must be slower than 800 μs.An alternative pathway involving the light-induced loss of NCMe may also be considered. However, loss of the coordinated NCMe in the same solvent would, on re-coordination, regenerate trypto-CORM. If such a process is occurring then it must take place in under 3 ps as no evidence for a tricarbonyl photoproduct, [Mn(tryp)(CO)3], was obtained in this study.
The experiments demonstrate that TRIR spectroscopy, coupled with DFT, can provide detailed information about the nature of the ultra-fast processes underpinning photochemical CO-release, including in model systems for an aqueous environment.
Experimental
Trypto-CORM was prepared a described previously.38 Time-resolved infra-red spectra were recorded on the LIFEtime instrument in the ULTRA facility at the Rutherford Appleton. Details of the experiment have been described previously,60 however, in brief, the pump source was the output of a Yb:KGW amplifier providing 15 W, 260 fs pulses at 1030 nm with a 100 kHz repetition rate (Pharos). This was used to drive a BBO-based 515 nm pumped optical parametric amplifier (OPA) to deliver pulses at 400 nm. The pump beam was collimated, travelled over a computer programmable 0–16 ns optical delay (1200 mm long, double pass), and focused onto the sample. The pump energy at the sample was attenuated down to 500 nJ and focused down to a 120 × 120 μm2 spot. The probe source was the output of a Yb:KGW amplifier providing 6 W, 180 fs pulses at 1030 nm with a 100 kHz repetition rate (Pharos). This drove two 3 W BBO/KTA based OPAs. The two Pharos sources share a common 80 MHz oscillator to allow for pump–probe delay steps of 12.5 ns. The probe beam was split to provide probe and reference pulses. To go beyond pump–probe delays of 12.5 ns, subsequent seed pulses were selected from the 80 MHz oscillator. Data were collected using pump–probe delays ranging from 1 ps to 988.5 μs. The probe beams were collimated, synchronised by a fixed optical delay, and focused by a gold parabolic mirror onto the sample. The three beams were overlapped on the sample using a 50 μm pinhole. The probe beams were measured by two separate 128-element detectors. To cover the full spectroscopic window required, data from a number of different detector positions were combined to generate the required spectra.For the experiments in NCMe, trypto-CORM ca. 30 mg was dissolved in 20 ml anhydrous NCMe and placed in a Duran flask. The experiments in DMSO/D2O were performed by dissolving ca. 13 mg of trypto-CORM in 2 ml DMSO before adding D2O to make the total volume 20 ml. The solutions were then flowed through a Harrick cell fitted with a 100 μm Teflon spacer using a peristaltic pump for the duration of the experiment.
Data were initially visualised in the ULTRA View version 2 software,70 where baseline-correction was undertaken. The resulting spectra were then exported as comma-separated values files into Origin 2019.71 The spectra in the metal carbonyl region were calibrated against the GSB of an authentic sample of trypto-CORM allowing for detector pixels to be allocated to specific frequencies. The region between 1860 and 1500 cm−1 was calibrated against a 190 μm polystyrene standard. Kinetic data were analysed using the expdec and expgrowdec functions in Origin 2019 and errors are presented as 95% confidence limits. Subtraction of the GSB from the difference spectrum in Fig. 5 was performed by scaling the difference spectrum and a spectrum of authentic trypto-CORM recorded in NCMe on an FTIR instrument to the peak at 2013 cm−1. The subtraction was then performed using the Subtract Reference Data feature in Origin 2019.
DFT calculations were performed using the TURBOMOLE V6.4 package using the resolution of identity (RI) approximation.72–78 Initial optimisations were performed at the (RI-)BP86/SV(P) level, followed by frequency calculations at the same level. All minima were confirmed as such by the absence of imaginary frequencies. Single-point calculations on the (RI-)BP86/SV(P) optimised geometries were performed using the hybrid PBE0 functional and the flexible def2-TZVPP basis set. The (RI-)PBE0/def2-TZVPP SCF energies were corrected for their zero-point energies, thermal energies and entropies (obtained from the (RI-)BP86/SV(P)-level frequency calculations). Vibrational frequencies were scaled using the empirical relationship developed in our previous work [νexpt = (νcalc − 561.7)/0.7369].52 No symmetry constraints were applied during optimisations. Triplet states were optimised using a spin unrestricted formalism. Solvent corrections were applied with the COSMO62 dielectric continuum model and dispersion effects modelled with Grimme's D3 method.79,80 Energies, xyz coordinates and the first 50 lines of the vibrational spectra are presented in the electronic ESI.‡
Additional TD-DFT calculations were performed using the Gaussian 16 Rev. A.03 Win6481 at the PBE0/DGDZVP/def2tzv level, with NCMe as the implicit solvent using CPCM. The first 10 states calculated are collated in the ESI.‡
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the EPSRC (studentship to B. J. A., EP/M506680/1 an iCASE award with Syngenta, EP/N509413/1 studentship to L. A. H. and for the computational equipment used in this study, grants EP/H011455/1 and EP/K031589/1) the STFC (programme access to the ULTRA facility grant 1813) and the University of York (studentship to J. B. E.) for funding.Notes and references
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- R. Foresti, M. G. Bani-Hani and R. Motterlini, Intensive Care Med., 2008, 34, 649–658 CrossRef CAS PubMed.
- F. Zobi, Future Med. Chem., 2013, 5, 175–188 CrossRef CAS PubMed.
- R. Alberto and R. Motterlini, Dalton Trans., 2007, 1651–1660 RSC.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, e2–e8 CrossRef CAS.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, E17–E24 CrossRef CAS.
- I. J. S. Fairlamb and J. M. Lynam, in Advances in Bioorganometallic Chemistry, ed. T. Hirao and T. Moriuchi, Elsevier, 2019, pp. 137–154 Search PubMed.
- A. R. Marques, L. Kromer, D. J. Gallo, N. Penacho, S. S. Rodrigues, J. D. Seixas, G. J. L. Bernardes, P. M. Reis, S. L. Otterbein, R. A. Ruggieri, A. S. G. Goncalves, A. M. L. Goncalves, M. N. De Matos, I. Bento, L. E. Otterbein, W. A. Blattler and C. C. Romao, Organometallics, 2012, 31, 5810–5822 CrossRef CAS.
- X. Ji, K. Damera, Y. Zheng, B. Yu, L. E. Otterbein and B. Wang, J. Pharm. Sci., 2016, 105, 406–416 CrossRef CAS PubMed.
- S. Romanski, E. Stamellou, J. T. Jaraba, D. Storz, B. K. Krämer, M. Hafner, S. Amslinger, H. G. Schmalz and B. A. Yard, Free Radical Biol. Med., 2013, 65, 78–88 CrossRef CAS PubMed.
- S. Romanski, B. Kraus, M. Guttentag, W. Schlundt, H. Rucker, A. Adler, J.-M. Neudorfl, R. Alberto, S. Amslinger and H.-G. Schmalz, Dalton Trans., 2012, 41, 13862–13875 RSC.
- S. Romanski, B. Kraus, U. Schatzschneider, J.-M. Neudörfl, S. Amslinger and H.-G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- I. J. S. Fairlamb, A. K. Duhme-Klair, J. M. Lynam, B. E. Moulton, C. T. O'Brien, P. Sawle, J. Hammad and R. Motterlini, Bioorg. Med. Chem. Lett., 2006, 16, 995–998 CrossRef CAS PubMed.
- I. J. S. Fairlamb, J. M. Lynam, B. E. Moulton, I. E. Taylor, A. K. Duhme-Klair, P. Sawle and R. Motterlini, Dalton Trans., 2007, 3603–3605 RSC.
- A. J. Atkin, S. Williams, P. Sawle, R. Motterlini, J. M. Lynam, I. J. S. Fairlamb, A. J. Atkin, S. Williams, P. Sawle, R. Motterlini, J. M. Lynam and I. J. S. Fairlamb, Dalton Trans., 2009, 38, 3653–3656 RSC.
- W.-Q. Zhang, A. J. Atkin, R. J. Thatcher, A. C. Whitwood, I. J. S. Fairlamb and J. M. Lynam, Dalton Trans., 2009, 38, 4351–4358 RSC.
- W.-Q. Zhang, A. C. Whitwood, I. J. S. Fairlamb and J. M. Lynam, Inorg. Chem., 2010, 49, 8941–8952 CrossRef CAS PubMed.
- S. McMahon, S. Amirjalayer, W. J. Buma, Y. Halpin, C. Long, A. D. Rooney, S. Woutersen and M. T. Pryce, Dalton Trans., 2015, 44, 15424–15434 RSC.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- M. A. Wright and J. A. Wright, Dalton Trans., 2016, 45, 6801–6811 RSC.
- I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- P. C. Ford, Coord. Chem. Rev., 2018, 376, 548–564 CrossRef CAS.
- M. Tinajero-Trejo, N. Rana, C. Nagel, H. E. Jesse, T. W. Smith, L. K. Wareham, M. Hippler, U. Schatzschneider and R. K. Poole, Antioxid. Redox Signal., 2016, 24, 765–780 CrossRef CAS PubMed.
- C. Nagel, S. McLean, R. K. Poole, H. Braunschweig, T. Kramer and U. Schatzschneider, Dalton Trans., 2014, 43, 9986–9997 RSC.
- H. Pfeiffer, T. Sowik and U. Schatzschneider, J. Organomet. Chem., 2013, 734, 17–24 CrossRef CAS.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- S. J. Carrington, I. Chakraborty and P. K. Mascharak, Chem. Commun., 2013, 49, 11254–11256 RSC.
- F. Mohr, J. Niesel, U. Schatzschneider and C. W. Lehmann, Z. Anorg. Allg. Chem., 2012, 638, 543–546 CrossRef CAS.
- J. Niesel, A. Pinto, H. W. Peindy, N.'D. K. Merz, I. Ott, R. Gust and U. Schatzschneider, Chem. Commun., 2008, 1798–1800 RSC.
- S. H. C. Askes, G. U. Reddy, R. Wyrwa, S. Bonnet and A. Schiller, J. Am. Chem. Soc., 2017, 139, 15292–15295 CrossRef CAS PubMed.
- Q. Jiang, Y. Xia, J. Barrett, A. Mikhailovsky, G. Wu, D. Wang, P. Shi and P. C. Ford, Inorg. Chem., 2019, 58, 11066–11075 CrossRef CAS PubMed.
- N. Rana, H. E. Jesse, M. Tinajero-Trejo, J. A. Butler, J. D. Tarlit, M. L. U. zur Muhlen, C. Nagel, U. Schatzschneider and R. K. Poole, Microbiology, 2017, 163, 1477–1489 CrossRef CAS.
- J. Betts, C. Nagel, U. Schatzschneider, R. Poole and R. M. La Ragione, PLoS One, 2017, 12, e186359 CrossRef PubMed.
- W. Huber, R. Linder, J. Niesel, U. Schatzschneider, B. Spingler and P. C. Kunz, Eur. J. Inorg. Chem., 2012, 2012, 3140–3146 CrossRef CAS.
- E. Kottelat, A. Ruggi and F. Zobi, Dalton Trans., 2016, 45, 6920–6927 RSC.
- J. S. Ward, J. M. Lynam, J. Moir and I. J. S. Fairlamb, Chem. – Eur. J., 2014, 20, 15061 CrossRef CAS PubMed.
- J. S. Ward, R. Morgan, J. M. Lynam, I. J. S. Fairlamb and J. W. B. Moir, MedChemComm, 2017, 8, 346–352 RSC.
- R. N. Perutz, O. Torres and A. Vlcek, in Comprehensive Inorganic Chemistry II, Elsevier Press, 2013, vol. 8, pp. 229–253 Search PubMed.
- R. N. Perutz and J. J. Turner, J. Am. Chem. Soc., 1975, 97, 4791–4800 CrossRef CAS.
- J. A. Calladine, S. B. Duckett, M. W. George, S. L. Matthews, R. N. Perutz, O. Torres and Q. V. Khuong, J. Am. Chem. Soc., 2011, 133, 2303–2310 CrossRef CAS PubMed.
- O. Torres, J. A. Calladine, S. B. Duckett, M. W. George and R. N. Perutz, Chem. Sci., 2015, 6, 418–424 RSC.
- J. M. Butler, M. W. George, J. R. Schoonover, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2007, 251, 492–514 CrossRef CAS.
- A. J. Cowan and M. W. George, Coord. Chem. Rev., 2008, 252, 2504–2511 CrossRef CAS.
- O. Torres, J. A. Calladine, S. B. Duckett, M. W. George and R. N. Perutz, Chem. Sci., 2015, 6, 418–424 RSC.
- J. Guan, A. Wriglesworth, X. Z. Sun, E. N. Brothers, S. D. Zarić, M. E. Evans, W. D. Jones, M. Towrie, M. B. Hall and M. W. George, J. Am. Chem. Soc., 2018, 140, 1842–1854 CrossRef CAS PubMed.
- J. A. Calladine, S. B. Duckett, M. W. George, S. L. Matthews, R. N. Perutz, O. Torres and K. Q. Vuong, J. Am. Chem. Soc., 2011, 133, 2303–2310 CrossRef CAS PubMed.
- S. A. Bartlett, N. A. Besley, A. J. Dent, S. Diaz-Moreno, J. Evans, M. L. Hamilton, M. W. D. Hanson-Heine, R. Horvath, V. Manici, X.-Z. Sun, M. Towrie, L. Wu, X. Zhang and M. W. George, J. Am. Chem. Soc., 2019, 141, 11471–11480 CrossRef CAS PubMed.
- M. Fumanal, Y. Harabuchi, E. Gindensperger, S. Maeda and C. Daniel, J. Comput. Chem., 2019, 40, 72–81 CrossRef CAS PubMed.
- B. J. Aucott, A.-K. Duhme-Klair, B. E. Moulton, I. P. Clark, I. V. Sazanovich, M. Towrie, L. A. Hammarback, I. J. S. Fairlamb and J. M. Lynam, Organometallics, 2019, 38, 2391–2401 CrossRef CAS.
- L. A. Hammarback, I. P. Clark, I. V. Sazanovich, M. Towrie, A. Robinson, F. Clarke, S. Meyer, I. J. S. Fairlamb and J. M. Lynam, Nat. Catal., 2018, 1, 830–840 CrossRef CAS.
- J. S. Ward, J. T. W. Bray, B. J. Aucott, C. Wagner, N. E. Pridmore, A. C. Whitwood, J. W. B. Moir, J. M. Lynam and I. J. S. Fairlamb, Eur. J. Inorg. Chem., 2016, 5044–5051 CrossRef.
- J. S. Ward, J. M. Lynam, J. W. B. Moir, D. E. Sanin, A. P. Mountford and I. J. S. Fairlamb, Dalton Trans., 2012, 41, 10514–10517 RSC.
- B. J. Aucott, J. S. Ward, S. G. Andrew, J. Milani, A. C. Whitwood, J. M. Lynam, A. Parkin and I. J. S. Fairlamb, Inorg. Chem., 2017, 56, 5431–5440 CrossRef CAS PubMed.
- N. P. Yahaya, K. M. Appleby, M. Teh, C. Wagner, E. Troschke, J. T. W. Bray, S. B. Duckett, L. A. Hammarback, J. S. Ward, J. Milani, N. E. Pridmore, A. C. Whitwood, J. M. Lynam and I. J. S. Fairlamb, Angew. Chem., Int. Ed., 2016, 55, 12455–12459 CrossRef CAS PubMed.
- Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS PubMed.
- L. A. Hammarback, A. Robinson, J. M. Lynam and I. J. S. Fairlamb, Chem. Commun., 2019, 55, 3211–3214 RSC.
- L. A. Hammarback, A. Robinson, J. M. Lynam and I. J. S. Fairlamb, J. Am. Chem. Soc., 2019, 141, 2316–2328 CrossRef CAS PubMed.
- G. M. Greetham, P. M. Donaldson, C. Nation, I. V. Sazanovich, I. P. Clark, D. J. Shaw, A. W. Parker and M. Towrie, Appl. Spectrosc., 2016, 70, 645–653 CrossRef CAS PubMed.
- G. M. Greetham, D. Sole, I. P. Clark, A. W. Parker, M. R. Pollard and M. Towrie, Rev. Sci. Instrum., 2012, 83, 103107 CrossRef CAS PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. F. Riehl, Y. Jean, O. Eisenstein and M. Pelissier, Organometallics, 1992, 11, 729–737 CrossRef CAS.
- I. P. Clark, M. W. George, G. M. Greetham, E. C. Harvey, C. Long, J. C. Manton, H. McArdle and M. T. Pryce, J. Phys. Chem. A, 2012, 116, 962–969 CrossRef CAS PubMed.
- J. E. Shanoski, C. K. Payne, M. F. Kling, E. A. Glascoe and C. B. Harris, Organometallics, 2005, 24, 1852–1859 CrossRef CAS.
- T. P. Dougherty and E. J. Heilweil, J. Chem. Phys., 1994, 100, 4006–4009 CrossRef CAS.
- L. Zhu, S. Saha, Y. Wang, D. A. Keszler and C. Fang, J. Phys. Chem. B, 2016, 120, 13161–13168 CrossRef CAS PubMed.
- J. P. Lomont, S. C. Nguyen and C. B. Harris, Acc. Chem. Res., 2014, 47, 1634–1642 CrossRef CAS PubMed.
- X. Wu, Z. Liu, T. S. Murphey, X. Z. Sun, M. W. D. Hanson-Heine, M. Towrie, J. N. Harvey and M. W. George, Faraday Discuss., 2019 10.1039/C9FD00067D.
- M. R. Pollard and G. M. Greetham, ULTRA View Data Analysis, STFC Search PubMed.
- OriginPro, OriginLab Corporation, Northampton, MA, USA, 2019 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- M. von Arnim and R. Ahlrichs, J. Chem. Phys., 1999, 111, 9183–9190 CrossRef CAS.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- P. Deglmann, K. May, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2004, 384, 103–107 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, Wallingford, CT Search PubMed.
Footnotes |
| † This manuscript is dedicated to Professor Robin Perutz, an inspirational scientist, colleague and mentor on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Details xyz coordinates and collated energies for DFT calculations. See DOI: 10.1039/c9dt03343b |
| § These authors contributed equally. |
| ¶ We are grateful to a referee who suggested the possibility that complexes A and B may be singlet excited states of trypto-CORM which then undergo CO loss to give dicarbonyl C. This possibility had been discounted on the basis that A and B exhibit two bands in the difference spectrum (Fig. 3). However, we cannot completely discount the presence of a weak feature obscured by the GSB for trypto-CORM. |
| This journal is © The Royal Society of Chemistry 2019 |