
A family of readily synthesised phosphorescent platinum(ii) complexes based on tridentate N^N^O-coordinating Schiff-base ligands†

Abstract
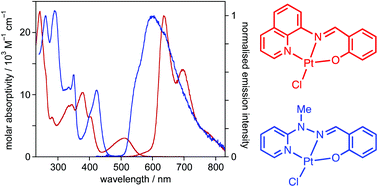
The synthesis and photophysical properties of 22 platinum(II) complexes featuring N^N^O-coordinating ligands are described. The complexes have the form Pt(N^N^O-Ln)Cl (n = 1 to 20). The tridentate ligands comprise lateral pyridine and phenolate rings, offering the metal N and O coordination respectively, linked via an imine or hydrazone unit that provides a further, central N atom for coordination. The proligands HLn, some of which have previously been reported for the coordination of 1st row transition metal ions in other contexts, are Schiff bases that are readily synthesised by condensation of salicylaldehydes either with 8-aminoquinoline (to generate imine-based ligands HL1–4) or with 2-hydrazinopyridines (to generate hydrazone-based proligands HL5–20). The Pt(II) complexes are prepared under mild conditions upon treatment of the proligands with simple Pt(II) salts. Metathesis of the chloride ligand by an acetylide is possible, as exemplified by the preparation of two further complexes of the form Pt(N^N^O-Ln)(–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ar), where Ar = 3,5-bis(trifluoromethyl)phenyl. Nine of the complexes have been characterised in the solid state by X-ray diffraction. The imine-based complexes have intense low-energy absorption bands around 520 nm attributed to charge-transfer transitions. They display deep red phosphorescence in solution at ambient temperature, with λmax in the range 635–735 nm, quantum yields up to 4.6% and lifetimes in the microsecond range. The hydrazone complexes that feature a py–NH–N
C–Ar), where Ar = 3,5-bis(trifluoromethyl)phenyl. Nine of the complexes have been characterised in the solid state by X-ray diffraction. The imine-based complexes have intense low-energy absorption bands around 520 nm attributed to charge-transfer transitions. They display deep red phosphorescence in solution at ambient temperature, with λmax in the range 635–735 nm, quantum yields up to 4.6% and lifetimes in the microsecond range. The hydrazone complexes that feature a py–NH–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–Ar linker display pH-dependent absorption spectra owing to the acidity of the hydrazone NH: these complexes have poor photostability in solution. In contrast, their N-methylated analogues (i.e., py–NMe–NC–Ar) show no evidence of photodecomposition. They are phosphorescent in solution at room temperature in the 600–640 nm region, the emission maximum being influenced by substituents in the phenolate ring. The results show how simply prepared tridentate Schiff base ligands – which offer the metal a combination of 5- and 6-membered chelate rings – can provide access to phosphorescent Pt(II) complexes that have superior emissive properties to those of terpyridines, for example.
C–Ar linker display pH-dependent absorption spectra owing to the acidity of the hydrazone NH: these complexes have poor photostability in solution. In contrast, their N-methylated analogues (i.e., py–NMe–NC–Ar) show no evidence of photodecomposition. They are phosphorescent in solution at room temperature in the 600–640 nm region, the emission maximum being influenced by substituents in the phenolate ring. The results show how simply prepared tridentate Schiff base ligands – which offer the metal a combination of 5- and 6-membered chelate rings – can provide access to phosphorescent Pt(II) complexes that have superior emissive properties to those of terpyridines, for example.
 Please wait while we load your content...
Please wait while we load your content...

