
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study of the photoconduction and photomagnetism of the BPY[Ni(dmit)2]2 molecular crystal
Jhon
Zapata-Rivera
 a,
Rocío
Sánchez-de-Armas
b and
Carmen J.
Calzado
*b
a,
Rocío
Sánchez-de-Armas
b and
Carmen J.
Calzado
*b
aDepartamento de Química, Universidad de los Andes, Cra 1 No. 18A – 12, 111711 Bogotá, Colombia
bDepartamento de Química Física, Universidad de Sevilla, c/Profesor García González, s/n 41012 Sevilla, Spain. E-mail: calzado@us.es
First published on 26th August 2019
Abstract
The BPY[Ni(dmit)2]2 molecular crystal synthesized by Naito and coworkers (J. Am. Chem. Soc., 2012, 134, 18656) was characterized as a photo-magnetic-conductor. This crystal is a nonmagnetic semiconductor in the dark and becomes a magnetic conductor after UV irradiation. This work analyzes the key ingredients of the observed photomagnetism and photoconduction by means of wavefunction-based calculations on selected fragments and periodic calculations on the whole crystal. Our results demonstrate that UV-Vis light induces charge transfer processes between the closest [Ni(dmit)2]− and BPY2+ units, that introduce unpaired electrons on the unoccupied orbitals of the BPY cations. Since the conduction bands present a strong mixing of BPY and Ni(dmit)2, the optically activated anion–cation charge transfer enhances the conductivity. The photoinduced (BPY2+)* radicals can indeed interact with the close Ni(dmit)2 units, with non-negligible spin–spin magnetic couplings, which are responsible for the changes induced by the irradiation on the temperature dependence of the magnetic susceptibility.
Introduction
Among the large variety of multifunctional materials, the derivatives of metal–dithiolene complexes have been intensively studied due to their distinctive optical, magnetic and conducting properties,1 such as ferromagnetism,2 conductivity,3–5 superconductivity,6 chirality,7 electrocatalytic and photocatalytic water splitting ability,8,9 and their potential applications in photonic, spintronic and electronic devices.1,3,5,10–15In this family, the anion radicals of M(dmit)2, with M = Ni, Pd, Pt, Cu, Au,… and dmit2− = 1,3-dithiole-2-thione-4,5-dithiolate, have been distinguished as promising building blocks of magnetic and conducting molecular materials, the [Ni(dmit)2]− radical being one of the most studied radicals.1 The unpaired electron occupies a π-type orbital of the dmit ligands,16–18 and the interaction of these π electrons with adjacent complexes is responsible for the electronic conduction and the spin–spin interactions. The resulting properties are extremely sensitive to the packing patterns and the nature of the counter cations forming the salt.14 Hence, combined with a magnetic metal complex, it is possible to realize a hybrid molecular magnetic conductor.19,20 However, in most cases, the magnetic complexes occupy the voids of the Ni(dmit)2 network and remain isolated or exhibit weak interactions. If the spins of the magnetic cation and the radical anion have a strong interaction, it will result in a nonmagnetic system. At the same time, the presence of the cation in the conduction pathways of the Ni(dmit)2 units can affect the conducting behaviour to the point of turning the salt into an insulator.
Recently, a new strategy has been explored, consisting of an optical doping that introduces photocarriers by charge transfer (CT) excitation between the Ni(dmit)2 complex and the organic cations occurring in the UV-Vis region. This introduces unpaired electrons in the organic cations and significantly enhances the conductivity of the salts.21,22 Using this idea, several complexes have been reported, such as the [C8-Apy]2[Ni(dmit)2]3 molecular crystal,23 with C8-Apy+ = 4-amino-1-octylpyridinium, which behaves as a photoconductor under UV irradiation. Similarly, MV[Ni(dmit)2]2 and BPY[Ni(dmit)2]2 were classified as photomagnetic conductors21,22 (BPY2+ = N,N′-ethylene 2,2′-bypiridinium dication, MV2+ = methylviologen dication). They have been characterized as nonmagnetic insulators in the dark, and become conductors with magnetic behaviour under UV irradiation. In the case of the BPY[Ni(dmit)2]2 molecular crystal, a charge transfer between Ni(dmit)2 and BPY promoted by UV radiation has been invoked as the main factor responsible for the observed photomagnetism and photoconduction.21 The process is reversible and permits the optical switching between an ionic salt and a charge transfer complex, magnetic and conductor. The salt exhibits quite unique wavelength selectivity, being particularly reactive – with notable response in its electron spin resonance and conduction properties – when it is irradiated with a UV light source.
The aim of this work is to explore the electronic structure of this compound by means of a combined strategy of wavefunction-based calculations on isolated fragments and periodic DFT calculations on the whole crystal to elucidate the key ingredients of the observed photomagnetism and photoconduction. Our results indicate that the main features of the UV-vis spectra can be reproduced from the electronic states of the most interacting cation–anion dimer. While in the dark, the unpaired electrons of the Ni(dmit)2 radicals are antiferromagnetically coupled, forming strong dimers, UV-Vis irradiation converts some of the BPY cations into radicals, giving rise to a set of new spin–spin interactions among the photoexcited BPY cations and the neighbouring Ni(dmit)2 units, which can explain the changes observed on the EPR spectra and the magnetic susceptibility curve after UV irradiation. The position of the BPY2+ intercalated between the anions, with a large mixing in the conduction bands could be related with the observed enhancement of the electric conductivity once the cations are photoexcited.
Description of the system
The asymmetric unit of the BPY[Ni(dmit)2]2 crystal contains two Ni(dmit)2 molecules (Ni1 and Ni2) with a dihedral angle of ∼50°, and one crystallographically independent BPY molecule, distorted in such a way that the pyridine rings are parallel to each of the Ni(dmit)2 molecules (Fig. 1), with interplanar distances of ∼3.4–3.5 Å. The Ni1 and Ni2 units are connected through short S–S contacts, resulting in a 3D network. Fig. 1 and 2 show the main anion–anion and cation–anion interactions in the crystal, following the notation employed by Naito et al.21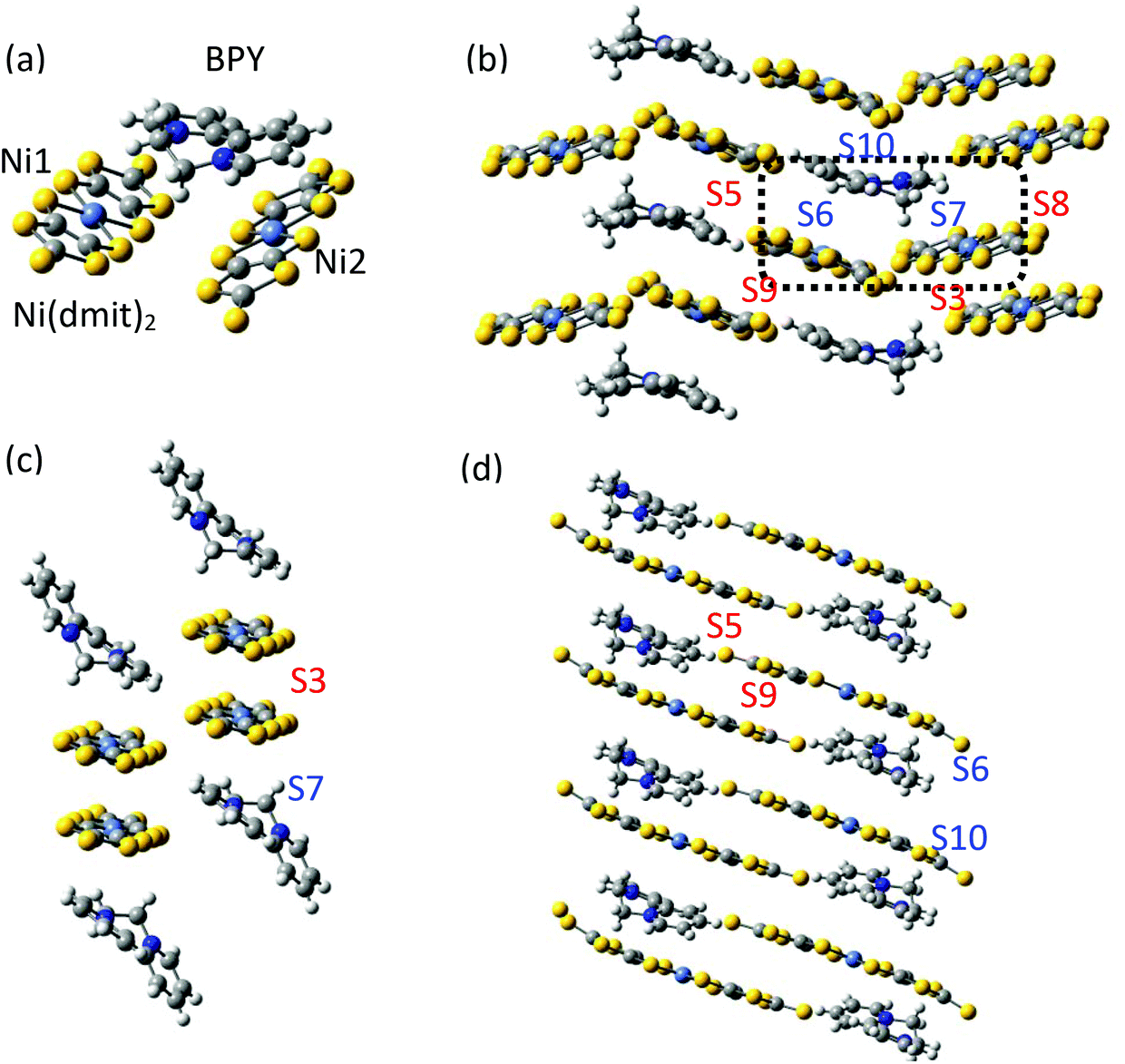 | ||
| Fig. 1 (a) Asymmetric unit of the BPY[Ni(dmit)2]2 crystal. (b) Main interactions on the crystal, following the notation by Naito et al.,21 with red labels for interactions between anions and blue labels for the cation–anion interactions. The black box contains the asymmetric unit. (c) Spatial distribution of the Ni1 units forming strong dimers S3. (d) One-dimensional chains of Ni2 units, with alternating S5 and S9 interactions. Yellow, grey, light blue, blue and white represent S, C, Ni, N and H atoms, respectively. | ||
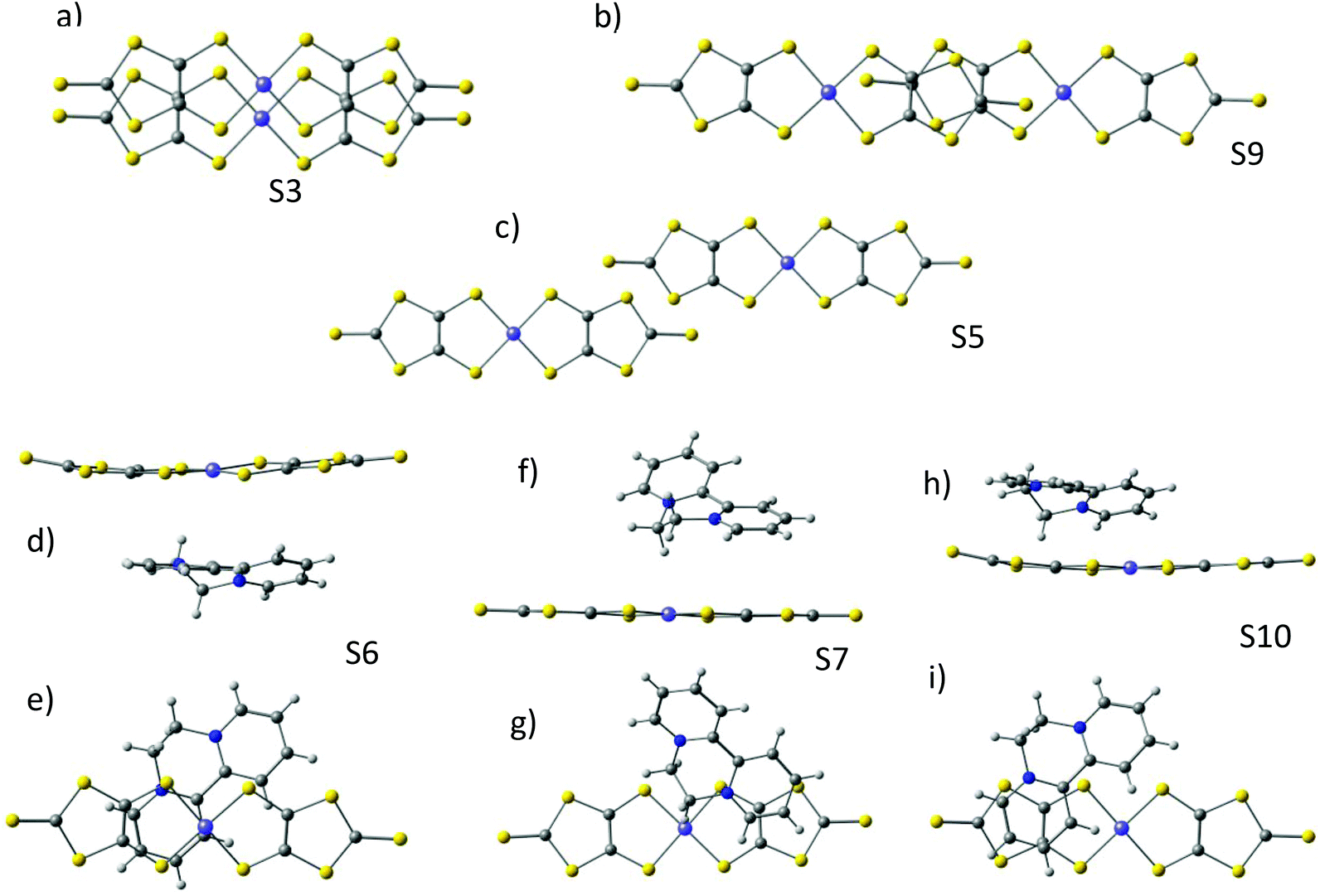 | ||
| Fig. 2 (a–c) Top views of the anion–anion S3, S5 and S9 dimers. Side and top views of the cation–anion dimers S6 (d and e), S7 (f and g) and S10 (h and i) used for the calculations. | ||
In the dark the BPY molecules have a formal charge +2, while the Ni(dmit)2 are radical anions, [Ni(dmit)2]−. The Ni1 anions form strong dimers (S3 in Fig. 1c), placed in between two BPY2+ units. These dimers are oriented in different directions and connected through short S–S contacts (3.6–3.7 Å). The Ni2 anions are arranged in one-dimensional chains, with two alternating weak dimers, S5 and S9 in Fig. 1d. The BPY cations are placed along this chain, occupying the voids, with one of the pyridine rings parallel to the molecular plane of the Ni2 monomer (S10 and S6 interactions, Fig. 1 and 2). Unlike the Ni1 dimers (S3) where the Ni(dmit)2 units are almost face-to-face, favoring strong π–π interactions (Fig. 2), the packing patterns for Ni2 anions present a noticeable slippage along the molecular plane that reduces the π overlap between the Ni2 units. This slippage is larger for dimer S5 than S9 (Fig. 2), and this should be related to the nature and amplitude of the intradimer magnetic interactions, as discussed below. Naito et al.21 suggested that the dominant interactions should be S3 among the anion–anion interactions and S10 for the cation–anion ones, based on the amplitude of the overlap and transfer integrals resulting from extended Hückel calculations in the crystal.
The UV-Vis and NIR spectra reproduced in Fig. 3 present much broader and slightly shifted peaks compared to those of BPY·Br2 and [n(C4H9)4N][Ni(dmit)2]. The overlapped broad peaks at 250–800 nm were tentatively assigned21 to a series of charge transfer transitions between BPY2+ and [Ni(dmit)2]−. The crystal has semiconducting behavior, with higher dark conductivity (σdark) than many other [Ni(dmit)2]− compounds with closed-shell organic cations.10 The photoconductivity (σph) examined at different temperatures and light intensities is singularly high (σph/σdark > 10). Indeed, the crystal exhibits a strong response to the conductivity as well as to the electron spin resonance spectrum when it is irradiated with light in the UV range, 250–450 nm, while the response is slighter with other wavelengths.21 This wavelength selectivity of the photoconductivity is a quite unique property, since most of the photoconductors are equally reactive to light, once it contains enough energy to generate the electron carriers.
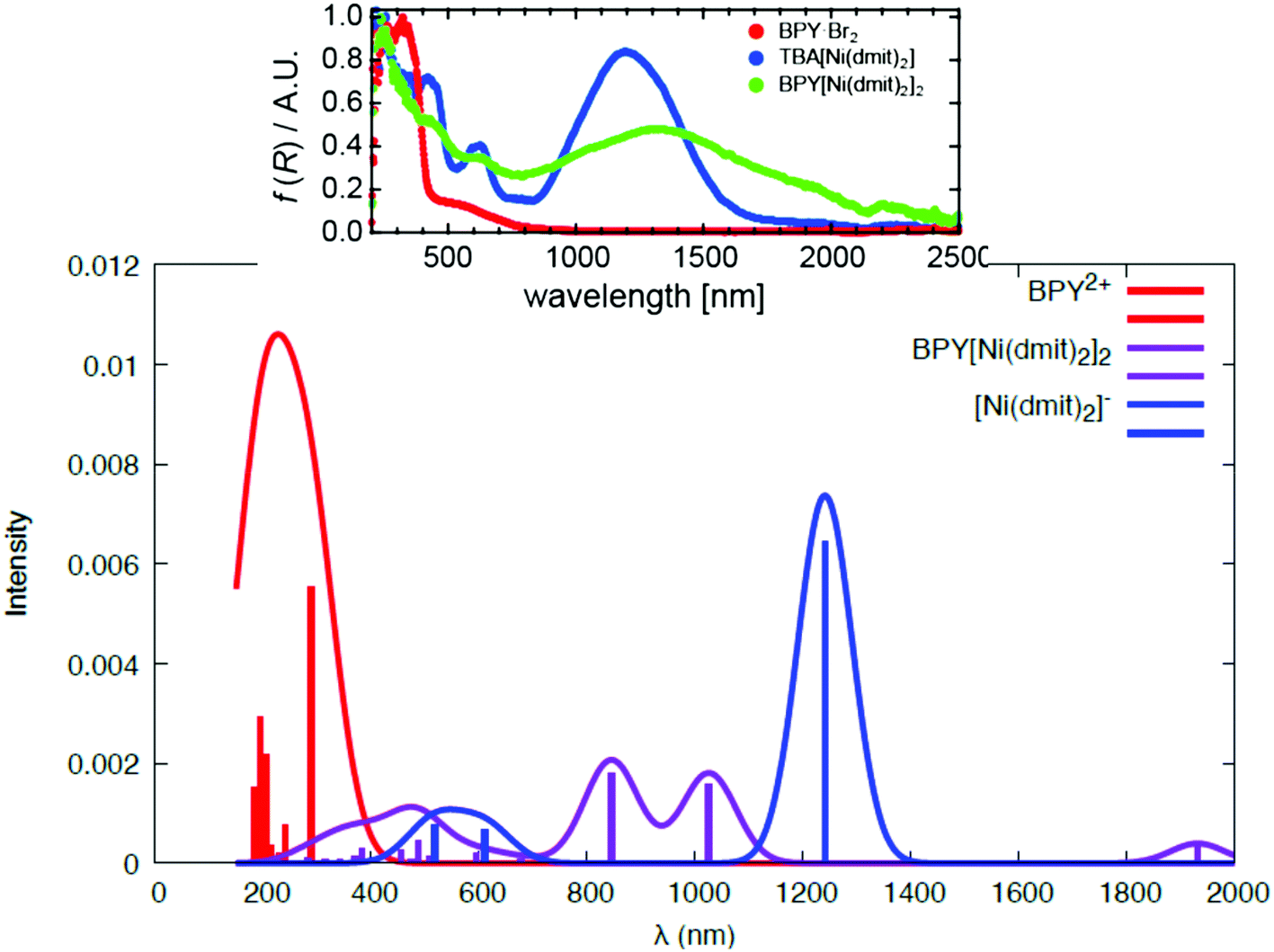 | ||
| Fig. 3 Calculated spectra of the isolated BPY2+ complex (red), the isolated [Ni(dmit)2]− (blue) and the cation–anion S10 dimer (violet). The inset corresponds to the experimental spectra, adapted with permission.21 Copyright 2012, American Chemical Society. | ||
The electron spin resonance spectra exhibit a single peak in the dark (g1 = 2.036), assigned to [Ni(dmit)2]− while under UV irradiation an additional signal appears, indicating the presence of two different types of spins (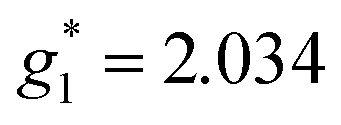 ,
, 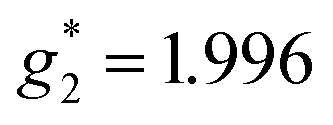 ). The signals were assigned to the unpaired electrons on the charge-transfer excited anion ([Ni(dmit)2]−)* and the charge-transfer excited cation (BPY2+)* with
). The signals were assigned to the unpaired electrons on the charge-transfer excited anion ([Ni(dmit)2]−)* and the charge-transfer excited cation (BPY2+)* with 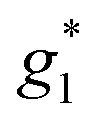 and
and 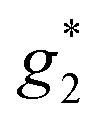 , respectively. The change in the ESR intensity gave a rough estimate of the amount of charge transfer induced by UV irradiation, about 10% decrease in the charge of the Ni(dmit)2 anions. Since there are eight anions per unit cell, this corresponds to approximately one charge transfer process per cell.
, respectively. The change in the ESR intensity gave a rough estimate of the amount of charge transfer induced by UV irradiation, about 10% decrease in the charge of the Ni(dmit)2 anions. Since there are eight anions per unit cell, this corresponds to approximately one charge transfer process per cell.
In the dark, the magnetic susceptibility χ is close to zero at all temperatures, except for T < 50 K where the observed increase of χ has been related to the presence of oxygen and lattice defects. Under UV irradiation, the material exhibits a qualitative different magnetic behavior. From 50 to 300 K, the difference in the magnetic susceptibility under the dark and irradiated conditions, Δχ, is nearly temperature independent. For T < 50 K, Δχ increases when the temperature decreases, until T ∼ 10 K, where a sharp decrease is observed. This has been related to an antiferromagnetic interaction between the photoexcited cation and anion.
Results and discussion
Electronic structure of isolated BPY2+ and [Ni(dmit)2]−
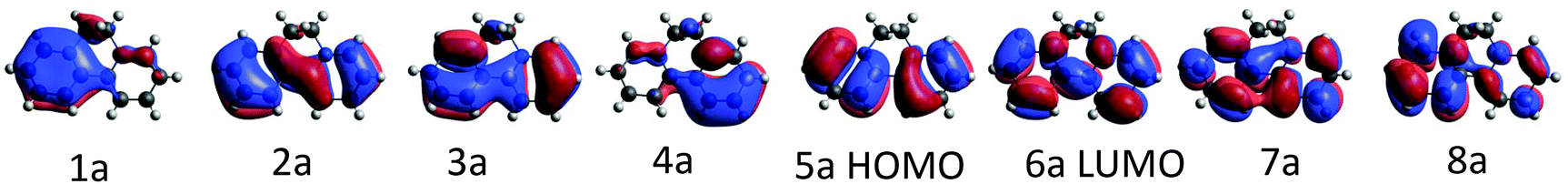
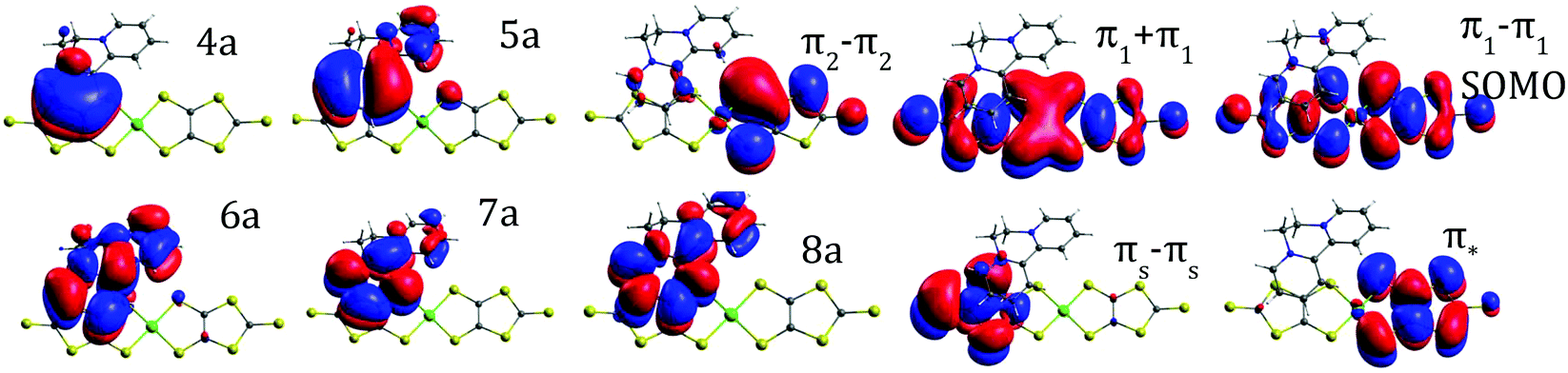
The ground state of the BPY cation is mainly dominated by a single determinant; the HOMO and LUMO are represented in Table 1. The excited states are placed at 4.3–6.8 eV above the ground state (Table 1). All the transitions show non-negligible values of the oscillator strength; the higher value corresponds to the single excitation from the HOMO to the LUMO, with λ = 289 nm. The next in intensity should be the transitions to the excited states placed at about 6.0–6.7 eV, which are related to a broad peak at about 200 nm. These states are multideterminantal in nature, with different single excitations playing a noticeable role in the description of the wavefunction, as shown in Table 1, most of them involving the HOMO or the LUMO. The calculated spectrum, shown in Fig. 3, is in general agreement with the diffuse reflectance spectra of BPY·Br2 dispersed in KBr.22| State | ΔE (eV) | λ (nm) | f | Dominant contributions to the wavefunction |
|---|---|---|---|---|
| 11A | 0.0 | — | — | |1a22a23a24a25a2| |
| 21A | 4.29 | 288.7 | 0.792 | HOMO → LUMO (77%) |
| 31A | 5.15 | 240.9 | 0.109 | HOMO → 8a (50%); 3a → LUMO (20%) |
| 41A | 5.37 | 230.9 | 0.030 | 5a → LUMO (27%); 3a → LUMO (25%) |
| 51A | 5.78 | 214.4 | 0.053 | 2a → LUMO (49%); 3a → LUMO (12%) |
| 61A | 6.04 | 205.4 | 0.310 | 4a → LUMO (36%); HOMO → 7a (21%) |
| 71A | 6.38 | 194.2 | 0.419 | 2a → 8a (35%); HOMO → 7a (17%) |
| 81A | 6.71 | 184.8 | 0.216 | HOMO → 7a(21%); 2a → 8a (14%); 4a → LUMO (13%) |
|
||||
In the case of the isolated radical [Ni(dmit)2]− the doublet ground state is also dominated by a single determinant (Table 2), where the SOMO corresponds to the antibonding combination of the π1 orbital of the dmit ligands (Table 2), in line with previous theoretical studies on this radical.18,24 The excited states are distributed in two groups, about 1.0 and 2.4 eV above the ground state. The first excited state at about 1.0 eV results from the single excitation from the (HOMO−1) to the SOMO. The transition between the ground and this excited state, with an associated wavelength of 1224 nm, can be assigned to the intense band at 1200 nm observed in the diffuse reflectance spectra of [n-(C4H9)4N][Ni(dmit)2].21 The peak at 600 nm, with half of the intensity of the fundamental transition, can be related to the transitions between the ground state and the second and third excited states, both involving the SOMO orbital. Our calculations then reproduce correctly both the energy and relative intensity of the main excitations of the isolated anion, and are in agreement with previous studies based on TD-DFT.21
| State | ΔE (eV) | λ (nm) | f | δ Ni | Dominant contributions to the wavefunction |
|---|---|---|---|---|---|
| 12A | 0.0 | — | — | 0.0088 | |(π2 − π2)2(π1 + π1)2(π1 −π 1)1| |
| 22A | 1.00 | 1241.4 | 0.924 | 0.0289 | (π1 + π1) → SOMO (98%) |
| 32A | 2.03 | 610.6 | 0.095 | 0.0003 | (π2 − π2) → SOMO (99%) |
| 42A | 2.39 | 517.9 | 0.110 | 0.0048 | SOMO → LUMO (68%) |
| 52A | 2.41 | 514.4 | 1 × 10−5 | 0.0053 | SOMO → (πS − πS) (66%) |
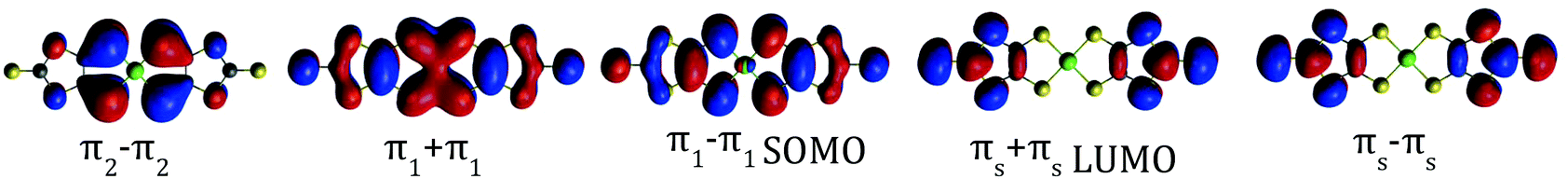
|
|||||
Charge transfer between the cation and anion units
To analyze the photoinduced charge transfer, we have calculated the UV-Vis spectrum of the S10 dimer (Fig. 2) that contains the cation and anion with the largest overlap and transfer integrals.21 The electronic spectrum resulting from the thirty lowest doublet states is represented in Fig. 3 and the relative energy of the excited states, those for which the transition from the ground state has an oscillator strength larger than 0.03, are collected in Table 3.| State | ΔE (eV) | λ (nm) | f | Nature of the dominant excitations |
|---|---|---|---|---|
| 12A | 0 | 0 | — | 88% |(4a-BPY)2 (5a-BPY)2(π1 + π1)2(π2 − π2)2 (π1 −π 1)1| |
| 22A | 0.71 | 1736.8 | 3 × 10−4 | 73% CT SOMO Ni(dmit)2 → BPY LUMO |
| 32A | 1.19 | 1044.7 | 0.17 | 53% CT Ni(dmit)2 → BPY; 31% intra Ni(dmit)2 (π1 + π1) → SOMO |
| 42A | 1.50 | 826.5 | 0.40 | 45% intra Ni(dmit)2 (π1 + π1) → SOMO; 40% CT Ni(dmit)2 → BPY |
| 52A | 1.66 | 744.7 | 0.05 | 64% CT Ni(dmit)2 → BPY |
| 62A | 2.25 | 550.1 | 0.04 | 73% CT Ni(dmit)2 → BPY |
| 72A | 2.38 | 521.8 | 0.03 | Intra Ni(dmit)2 (π2 − π2) → SOMO |
| 102A | 2.93 | 422.4 | 0.03 | Intra Ni(dmit)2 SOMO → LUMO |
| 122A | 3.45 | 359.4 | 0.03 | Intra Ni(dmit)2 |
| 132A | 3.53 | 351.5 | 0.13 | Intra Ni(dmit)2 |
| 172A | 3.94 | 315.0 | 0.12 | Intra Ni(dmit)2 |
|
||||
The calculations indicate that most of the excitations involving the charge transfer (CT) from the Ni(dmit)2 MOs to the BPY cation are concentrated in the low-lying part of the spectrum, in the region between 550 and 1050 nm. The first excited state is at only 0.71 eV above the ground state and results from the charge transfer between the Ni(dmit)2 SOMO and the BPY LUMO, with a λ = 1737 nm. With such a weak oscillator strength, it can be assigned to the low intensity broad absorption in the NIR region (1500–2000 nm). The 32A and 42A excited states are placed at 1.19 and 1.50 eV above the ground state. Both with a noticeable multideterminantal nature contain a significant percentage of anion → cation CT, together with intramolecular Ni(dmit)2 excitations, in line with the excitation at 1.0 eV obtained in the isolated anion calculations. The 52A and 62A excited states are strongly characterized by the charge transfer, while the rest of the explored states correspond to local excitations on the Ni(dmit)2 unit. In particular, the states 72A and 102A at 2.38 and 2.93 eV above the ground state are represented by the excitations (π2–π2) → SOMO and SOMO → LUMO, respectively, in concordance with the excited states of the isolated Ni(dmit)2, with relative energies of 2.03 and 2.39 eV. The differences found in the energies when comparing with the isolated units can be due to limitations in our calculations, and also due to the physical effects related to the mutual influence of the cation and anion fragments on the distribution of their molecular orbitals. In fact, the experimental spectra of BPY[Ni(dmit)2]2 show slightly shifted bands21 with respect to those of the BPY·Br2 and [n(C4H9)4N][Ni(dmit)2], as do our calculations.
Fig. 3 shows the electronic spectrum resulting from the excitations in dimer S10. The calculated spectrum for dimer S10 reproduces correctly the main features of the experimental one. The local excitations of the cation and anion are well separated in the UV-Vis region, and the charge transfer excitations are placed in between, in the 500–1000 nm region, giving rise to a broad band. It is important to mention that we have inspected only one of the cation–anion interactions in the crystal, and then the comparison with the experimental spectrum should be done assuming that this dimer should be the most representative, but there exists additional contributions coming from the rest of the crystals.
The projected density-of-states (DOS) resulting from the periodic B3LYP calculations are shown in Fig. 4. Our calculation predicts a small bandgap of only 0.2 eV for the ground state (NUPDOWN = 0), in line with the semiconducting properties of the system. If instead, PBE + U is used, the calculation fails in predicting the semiconductor nature of the crystal, in fact the valence and conduction bands touch each other and since there is no energy gap, the resulting DOS is closer to a semimetal than to a semiconductor. This underestimation of the energy band gap is a well-known drawback of the Kohn–Sham implementation of the DFT formalism,25–27 and here it persists despite the use of the +U correction. In this system, the evaluation is indeed particularly difficult since a rather small band gap is expected, as it indicates the separation of only 0.7 eV between the ground and first excited states in the calculations of the cation–anion S10 dimer (Table 3), and corroborates the small bandgap resulting from the periodic B3LYP calculation.
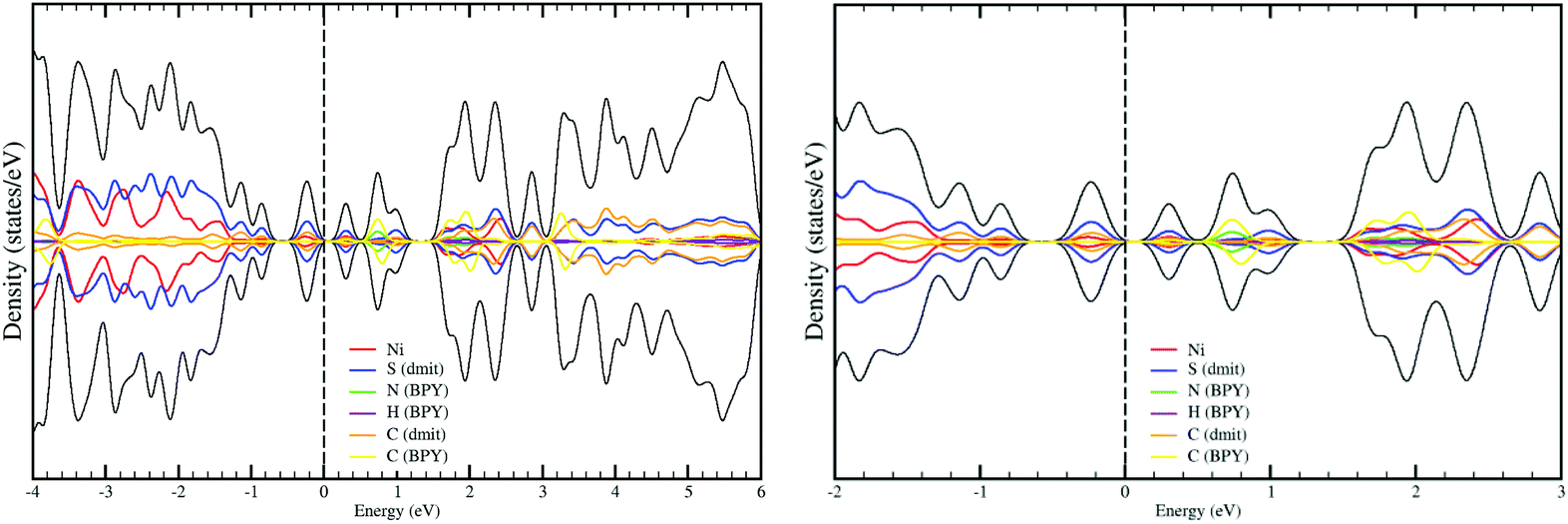 | ||
| Fig. 4 Density of electronic states (DOS) (black line) and projected DOS on the different atoms of the crystal (color lines) for the ground state (NUPDOWN = 0) at the B3LYP level. Zoomed view of the Fermi region is shown on the right part. | ||
The DOS gives additional insight into the UV-vis absorption and the photoconduction. Unlike the calculations on the S10 dimer, the calculation on the crystal takes into account all the interactions and provides a complete picture of the distribution of the energy levels in the crystal. The bands near the Fermi level are markedly of Ni(dmit)2 in nature for the valence band, while the conduction bands in the range of 3 eV above the Fermi level present a strong mixing of BPY and Ni(dmit)2. These results indicate the presence of accessible states that can be populated after UV-Vis-NIR irradiation. The conduction in the dark is then essentially related to the 3D packing of the Ni(dmit)2 units, but the population of the conduction BPY bands, after illumination, should produce an enhancement of the conducting properties, since the BPY cations are placed in the voids of the Ni2 columns, and once photoexcited, they enhance the conduction pathways. The excitations from the valence band to these conduction bands correspond to charge-transfer and local excitations to the Ni(dmit)2 (or between anions), energetically in the Vis-NIR region, in agreement with the results obtained from the MS-CASPT2 calculations. The excitations between cations (or/and local to the cations) are much higher in energy, the separation between the occupied and unoccupied BPY bands is larger than 3 eV and it corresponds to bands with λ < 400 nm in the UV region.
Magnetic coupling constants
The magnetic coupling constants between the Ni(dmit)2 anions in the unit cell have been evaluated at the DDCI level and reported in Table 4 for dimers S3, S5 and S9. The Ni1 units form strong dimers, with a remarkable antiferromagnetic interaction, J(S3) = −2315 cm−1, so large that even at room temperature the S3 dimers retain the singlet ground state, and then their contribution to the molar susceptibility is null in all the range of temperatures. The amplitude of this interaction is in line with the strong π–π overlap of the active orbitals of the dimer (Fig. 5).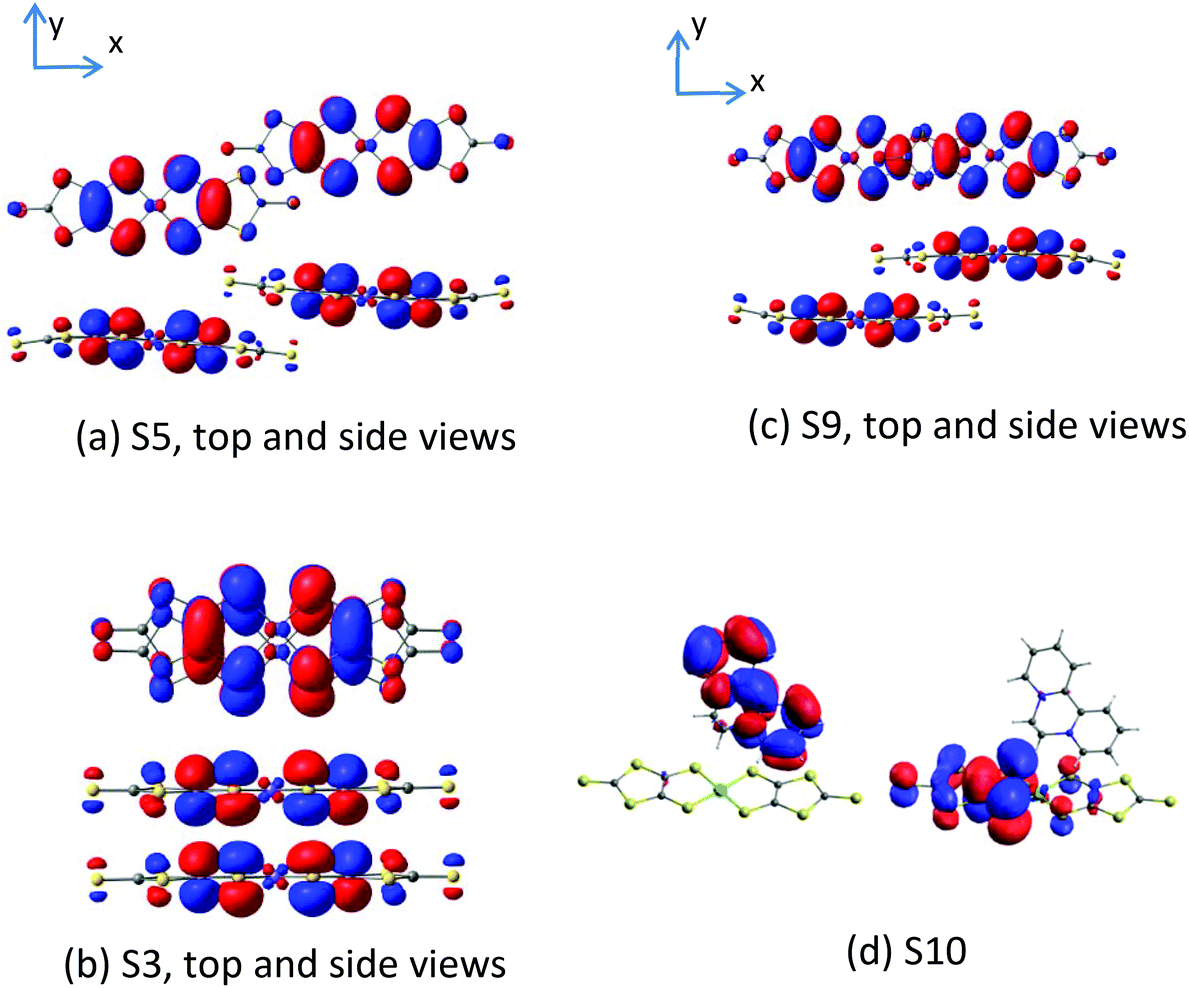 | ||
| Fig. 5 Singly occupied molecular orbitals (SOMOs) on anion–anion dimers S3, S5 and S9 (left) and the cation–anion S10 dimer (right). The side views show the different relative slippage for each dimer, and how it does affect the π–π overlap. | ||
| Anion–anion | Anion–(cation)* | ||||||
|---|---|---|---|---|---|---|---|
| S3 | S5 | S9 | S7 | S6 | S10 | S15 | |
| J/cm−1 | −2315 | −1.6 | −73.9 | −36.3 | +17.4 | −264.8 | 0.0 |
The Ni2 units form an alternating 1D chain, with two AF interaction parameters J(S9) = −73.9 cm−1 and J(S5) = −1.6 cm−1. As for S3, the amplitude of the interactions can also be related to the overlap of the SOMOs. The lateral slippage in S5 prevents a large overlap, which is much more efficient in the case of the S9 dimer, where the Ni(dmit)2 units are slipped along the molecular x axis (Fig. 5). The S5 interaction is so weak that it is possible to alternatively describe these chains as columns of isolated S9 dimers. The amplitude of the interaction in S9 dimers is in agreement with that obtained for salts of the [Ni(dmit)2]− radicals with similar packing patterns.18 In addition to these interactions, there exists the possibility of interactions between the Ni1 and Ni2 units in the crystal through the S–S contacts, but due to their relative orientation and large distances, it is expected that they result in rather weak coupling constants, as illustrated in our previous study of the magnetic behaviour of [Ni(dmit)2]− radicals.18
The photoinduced charge transfer promotes one electron from an occupied orbital of the Ni(dmit)2 anion to an unoccupied orbital of the BPY2+. We have just analysed the charge transfer process inside the cation–anion S10 dimer, i.e., between the D unit in Fig. 6 and the blue highlighted BPY cation. The resulting excited (BPY2+)* unit carries one unpaired electron that can now interact with the spin orbital on the neighbouring Ni(dmit)2 molecules. We have considered the magnetic interactions in three dimers containing the excited (BPY2+)* and the closest Ni1 (dimer S7) and Ni2 (dimers S6 and S15) anions (Fig. 6).
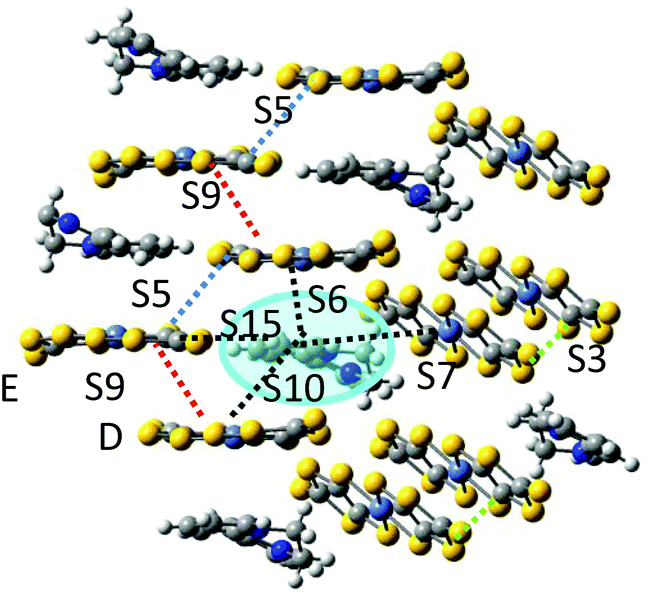 | ||
| Fig. 6 A charge transfer between the Ni(dmit)2 and BPY2+ in dimer S10 generates a photoexcited (BPY2+)* radical (in the blue circle). This (BPY2+)* radical can now interact with the spin orbital on the neighbouring Ni(dmit)2 complexes through interactions S6, S7 and S15. | ||
The interactions are ferromagnetic for S6, J(S6) = +17.4 cm−1, antiferromagnetic for dimer S7, J(S7) = −36.3 cm−1 and negligible for S15 (Table 4), and then the Ni(dmit)2 of this cation–anion unit (anion E in Fig. 6) remains almost magnetically isolated. But if the transfer integral between D and E units is significant, as indicated by the antiferromagnetic interaction between them (J(S9) = −73.9 cm−1), the unpaired electron can transfer from E to D, i.e. within the S9 dimer, and a new magnetic interaction with the (BPY2+)* radical can now be activated, corresponding to dimer S10. The coupling in this dimer is strongly antiferromagnetic, with J(S10) = −264.8 cm−1.
These results indicate that the photoexcitation of the BPY cations introduces a new set of spin–spin interactions that could explain the changes observed experimentally. The simulation of the temperature dependence of the magnetic susceptibility has not been attempted due to the low quality of the reported χ vs. T curve and the presence of impurities that make difficult a reliable analysis of this curve.
Conclusions
The photoconduction and photomagnetism of the BPY[Ni(dmit)2]2 molecular crystal have been investigated employing a combined strategy of wave-function based calculations on selected fragments and periodic calculations on the whole crystal. Despite the complexity of the system and the process itself, our calculations provide some clues about the origin of the main changes observed after UV irradiation. In this system, the BPY cations occupy the voids of the Ni2 chains. Their close proximity allows a charge transfer process between the Ni(dmit)2 anion and the BPY cation. This process introduces unpaired electrons on the cation, populating the BPY conduction band. The calculations indicate that the valence band is markedly Ni(dmit)2 in nature, while the conduction bands show a strong cation–anion mixing. A narrow band gap favors the photoconduction. Hence, the UV-Vis irradiation populates the conduction BPY bands and introduces alternative conduction pathways through the photoexcited BPY bands. As a result, an enhancement of the conducting properties is observed.The electronic spectrum of the crystal can be considered as the superposition of local excitations on each component, slightly modified by the presence of the counterpart, and the photoinduced charge transfer excitations between the cation and anion units. The excited states involved in the charge transfer are relatively close in energy to the ground state, in the region between 550 and 1050 nm, most of them with a noticeable multideterminantal nature, which points to the need for multiconfigurational approaches. The unpaired electrons on the photoexcited BPY cations introduce additional spin–spin interactions with the neighbouring Ni(dmit)2 radicals, which are responsible for the changes observed on the temperature dependence of the magnetic susceptibility after irradiation.
Computational details
The geometries of all the models employed in our calculations were directly obtained from the resolved X-ray structure of the crystal at 120 K under dark conditions.21 It is worth mentioning that no geometrical modification has been observed in the irradiated structure.21Wavefunction-based calculations
The study of the electronic states of the BPY2+ cation has been carried out with an active space of 8 molecular orbitals (MO), all π in nature, and ten electrons CAS(10,8), represented in Table 1. The fifteen lowest roots are included in the state-average approach, all with the same weight. For [Ni(dmit)2]−, the CAS contains five electrons in five orbitals, shown in Table 2, all (mainly) localized on the dmit ligands, with a small contribution of the Ni 3d orbitals. The five lowest doublet states are evaluated from state-average calculations, all with the same weight.
To study the photoinduced charge transfer between BPY2+ and [Ni(dmit)2]− molecules, the cation–anion dimer S10 model has been employed, since it exhibits the largest overlap and transfer integral according to extended Hückel calculations.21 We have carried out SA-CASSCF(9,10)/MS-CASPT2 on the thirty lowest doublet states, where the CAS includes occupied and virtual orbitals of both units (Table 3).
In all the models considered the radicals present only through-space interactions, which can be described by an isotropic Heisenberg Hamiltonian, following the notation:
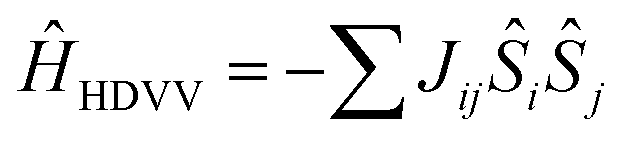
 and
and  correspond to the spin operators of the two interacting sites i and j, and Jij is the magnetic coupling constant between these sites. In this notation, for only two interacting sites the coupling constant J equals the energy difference between the singlet and triplet states, being negative for a singlet ground state (antiferromagnetic coupling) and positive for a ferromagnetic interaction (triplet ground state). The term “site” is used here in a broad sense, since it refers to the π orbitals of the BPY and dmit ligands.
correspond to the spin operators of the two interacting sites i and j, and Jij is the magnetic coupling constant between these sites. In this notation, for only two interacting sites the coupling constant J equals the energy difference between the singlet and triplet states, being negative for a singlet ground state (antiferromagnetic coupling) and positive for a ferromagnetic interaction (triplet ground state). The term “site” is used here in a broad sense, since it refers to the π orbitals of the BPY and dmit ligands.
The energies of the singlet and triplet states on each [Ni(dmit)2]2 dimer have been evaluated following the same strategy as in our previous work on the salts of the [Ni(dmit)2]− radicals with supramolecular-cations.18 As starting orbitals, we employed those resulting from a CASSCF(4,4) calculation on the singlet ground state of the neutral dimers. The CAS contains the (π1 + π1) bonding and (π1 − π1) antibonding combinations of the dmit ligand orbitals of each Ni(dmit)2 unit. The orbitals remain symmetrically distributed in the two sides of the [Ni(dmit)2] unit. These orbitals are now required to be optimized in the presence of two extra electrons for the subsequent calculations on the anionic dimers. The main delocalization effects can be introduced through the interaction of the CAS with the singly excited determinants (i.e. CAS + S calculations). The natural orbitals determined from the diagonalization of the average density matrix of the singlet and triplet CAS + S wavefunctions (Fig. 5) are used to finally evaluate the magnetic coupling constants in an additional run at the DDCI(2,2) level. The difference-dedicated configuration interaction (DDCI) calculation,30,31 considered as the reference method in the field, includes all the active double-excited determinants in the CI space (i.e, those double excitations involving at least one active orbital). These determinants take into account the effects of the dynamic electronic correlation, and their contribution could significantly modify (20–40%) the J coupling.32,33
In the case of the cation–anion magnetic interaction, the active space contains just two electrons on two molecular orbitals, the SOMO of the Ni(dmit)2 anion and the LUMO of the BPY2+ cation (Fig. 4). The cation–anion magnetic coupling constants have been determined from DDCI(2,2) calculations on the singlet and triplet states of the dimers, on the basis of the CASSCF(2,2) MOs.
In all the wavefunction-based calculations, ANO-RCC basis sets have been used for all the atoms with contractions 6s5p4d1f for Ni, 5s4p1d for S, 4s3p1d for C and 2s1p for H atoms.34,35 All CASSCF calculations have been performed using the MOLCAS@UU package.36 The CASDI code37 has been used for CI calculations, combined with the Lewis program38 to carry out the localization of the molecular orbitals.
Periodic calculations on the BPY[Ni(dmit)2]2 molecular crystal
The crystals have been studied with density functional theory (DFT) using the Vienna ab initio simulation package (VASP) code,39–42 employing the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof exchange–correlation functional43,44 and projector-augmented wave (PAW) potentials.45,46 Effective Hubbard corrections of 8 eV have been used to describe the localized Ni 3d orbitals, and 4 eV has been used to describe C 2p and S 3p orbitals, using Dudarev's approach.47 The PBE + U method is considered as a practical alternative to hybrid methods when plane-wave basis sets are used, where the evaluation of the Fock exchange terms is computationally prohibitive.48–50 Valence electrons are described using a plane-wave basis set with a cutoff of 500 eV and a Γ-centred grid of k-points is used for integrations in the reciprocal space, where the smallest allowed spacing between k-points is set at 0.2 Å−1.51 van der Waals interactions were taken into account through the Tkatchenko–Scheffler method.52We have considered the experimental structure of the crystal, and single-point calculations have been done for different magnetic solutions. The NUPDOWN option is used, which forces the difference between the number of electrons in up and down spin channels Nα-Nβ to be equal to a certain value (NUPDOWN = 0 and 8). Relaxation has been performed until the change in the total energy between two consecutive steps is smaller than 10−6 eV. For the most stable magnetic solution (NUPDOWN = 0) we performed single-point calculations with the B3LYP hybrid functional53–55 in order to improve the description of the electronic structure. As this calculation is computationally prohibitive at the same level of precision, we performed it at the Γ point of the Brillouin zone. Valence electrons are described using a plane-wave basis set with a cutoff of 500 eV and relaxation has been performed until the change in the total energy between two consecutive steps is smaller than 10−6 eV.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support provided by the Ministerio de Economía y Competitividad (Spain) and the funds from the FEDER through Project CTQ-2015-69019-P (MINECO/FEDER). J. Z. R. acknowledges the financial support provided by COLCIENCIAS through the project 1411712-51515. R. Sánchez-de-Armas thanks VPPI-US for the financial support. The technical support of the Supercomputing Team of the Centro Informático Científico de Andalucía (CICA) and the access to the computational facilities of the “Centro de Servicios de Informática y Redes de Comunicaciones” (CSIRC, Universidad de Granada, Spain) are also acknowledged.References
- T. Kusamoto and H. Nishihara, Coord. Chem. Rev., 2019, 380, 419–439 CrossRef CAS.
- A. T. Coomber, D. Beljonne, R. H. Friend, J. L. Brédas, A. Charlton, N. Robertson, A. E. Underbill, M. Kurmoo and P. Day, Nature, 1996, 380, 144 CrossRef CAS.
- R. Kato, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed.
- A. Kobayashi, E. Fujiwara and H. Kobayashi, Chem. Rev., 2004, 104, 5243–5264 CrossRef CAS PubMed.
- N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93–127 CrossRef CAS.
- H. Kobayashi, E. Fujiwara, H. Fujiwara, H. Tanaka, T. Otsuka, A. Kobayashi, M. Tokumoto and P. Cassoux, Mol. Cryst. Liq. Cryst., 2002, 380, 139–144 CrossRef CAS.
- F. Pop and N. Avarvari, Coord. Chem. Rev., 2017, 346, 20–31 CrossRef CAS.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef CAS PubMed.
- A. Zarkadoulas, E. Koutsouri and C. A. Mitsopoulou, Coord. Chem. Rev., 2012, 256, 2424–2434 CrossRef CAS.
- P. Cassoux, L. Valade, H. Kobayashi, A. Kobayashi, R. A. Clark and A. E. Underhill, Coord. Chem. Rev., 1991, 110, 115–160 CrossRef CAS.
- L. Valade, D. de Caro, C. Faulmann and K. Jacob, Coord. Chem. Rev., 2016, 308, 433–444 CrossRef CAS.
- B. Garreau-de Bonneval, K. I. Moineau-Chane Ching, F. Alary, T.-T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- M. L. Mercuri, P. Deplano, L. Pilia, A. Serpe and F. Artizzu, Coord. Chem. Rev., 2010, 254, 1419–1433 CrossRef CAS.
- R. Kato, Bull. Chem. Soc. Jpn., 2014, 87, 355–374 CrossRef CAS.
- F. Alary, J.-L. Heully, A. Scemama, B. Garreau-de Bonneval, K. I. Chane Ching and M. Caffarel, Theor. Chem. Acc., 2010, 126, 243–255 Search PubMed.
- K. Ray, T. Petrenko, K. Wieghardt and F. Neese, Dalton Trans., 2007, 1552–1566 RSC.
- J. Zapata-Rivera and C. J. Calzado, Molecules, 2019, 24, 1088 CrossRef CAS PubMed.
- J. Zapata-Rivera, D. Maynau and C. J. Calzado, Chem. Mater., 2017, 29, 4317–4329 CrossRef CAS.
- Y. Kosaka, H. M. Yamamoto, A. Nakao, M. Tamura and R. Kato, J. Am. Chem. Soc., 2007, 129, 3054–3055 CrossRef CAS PubMed.
- T. Kusamoto, H. M. Yamamoto, N. Tajima, Y. Oshima, S. Yamashita and R. Kato, Inorg. Chem., 2013, 52, 4759–4761 CrossRef CAS PubMed.
- T. Naito, T. Karasudani, S. Mori, K. Ohara, K. Konishi, T. Takano, Y. Takahashi, T. Inabe, S. Nishihara and K. Inoue, J. Am. Chem. Soc., 2012, 134, 18656–18666 CrossRef CAS PubMed.
- T. Naito, T. Karasudani, K. Ohara, T. Takano, Y. Takahashi, T. Inabe, K. Furukawa and T. Nakamura, Adv. Mater., 2012, 24, 6153–6157 CrossRef CAS PubMed.
- H. Yang, J.-L. Liu, L.-C. Zhou and X.-M. Ren, Inorg. Chem. Front., 2014, 1, 426–433 RSC.
- K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2005, 44, 5345–5360 CrossRef CAS PubMed.
- C. S. Wang and W. E. Pickett, Phys. Rev. Lett., 1983, 51, 597–600 CrossRef CAS.
- Á. Morales-García, R. Valero and F. Illas, J. Phys. Chem. C, 2017, 121, 18862–18866 CrossRef.
- W. Li, C. F. J. Walther, A. Kuc and T. Heine, J. Chem. Theory Comput., 2013, 9, 2950–2958 CrossRef CAS PubMed.
- P.-A. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- J. Miralles, J.-P. Daudey and R. Caballol, Chem. Phys. Lett., 1992, 198, 555–562 CrossRef CAS.
- J. Miralles, O. Castell, R. Caballol and J.-P. Malrieu, Chem. Phys., 1993, 172, 33–43 CrossRef CAS.
- C. J. Calzado, J. Cabrero, J. P. Malrieu and R. Caballol, J. Chem. Phys., 2002, 116, 2728–2747 CrossRef CAS.
- J. P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihery, Chem. Rev., 2014, 114, 429–492 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, P. A. Malmqvist, V. Veryazov and P. O. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed.
- B. O. Roos, R. Lindh, P. A. Malmqvist, V. Veryazov and P. O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. F. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- D. Maynau , unpublished work.
- L. Tenti, D. Maynau, C. Angeli and C. J. Calzado, Phys. Chem. Chem. Phys., 2016, 18, 18365–18380 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- P. Verma, R. Maurice and D. G. Truhlar, J. Phys. Chem. C, 2016, 120, 9933–9948 CrossRef CAS.
- C. Franchini, R. Podloucky, J. Paier, M. Marsman and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195128 CrossRef.
- A. Sorkin, M. A. Iron and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 307–315 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |