
Highly efficient room-temperature phosphorescence achieved by gadolinium complexes†
Zhiwei
Liu,  a
Zuqiang
Bian
*a
and
a
Zuqiang
Bian
*a
and
a
Zuqiang
Bian
*a
and
Abstract
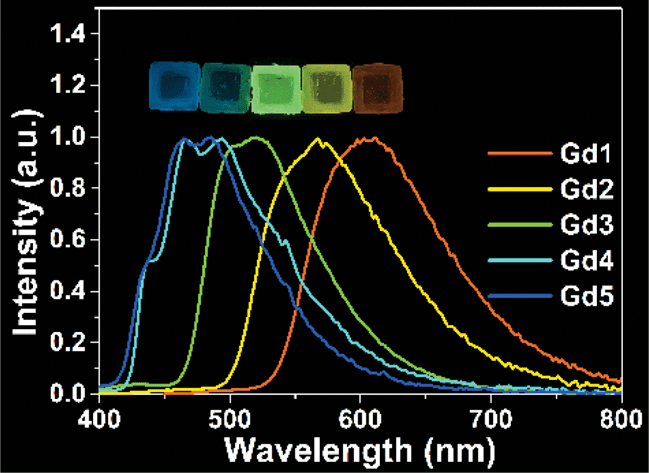
A new family of room temperature phosphorescent materials with emission lifetimes in microseconds has been reported in this work. Phosphorescence of gadolinium complexes with emission color from blue to orange has been obtained at room temperature with a maximum photoluminescence quantum yield of 66%, benefiting from appropriate molecular structures and favorable encapsulation methods.
 Please wait while we load your content...
Please wait while we load your content...

