
Effects of the preparation method of Pt/g-C3N4 photocatalysts on their efficiency for visible-light hydrogen production†
 a
Yan
Xing,
*a
a
Yan
Xing,
*a
Abstract
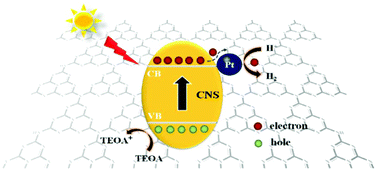
As a two-dimensional (2D) nanomaterial, bulk g-C3N4 (CNB) has a low specific surface area and weak electron transport ability, which limit its application in photocatalysis. In this paper, ultrathin porous g-C3N4 nanosheets (CNS) have been synthesized by thermal oxidation etching of CNB. Compared with CNB, CNS possess a larger surface area of 234.65 m2 g−1, good dispersity in water and a high electron transfer rate. As a co-catalyst, ultra-small Pt nanoparticles (NPs) with high dispersity are successfully loaded on the surface of CNS. It is found that changing the loading method of Pt NPs in the preparation step remarkably alters the efficiency for hydrogen production. The Pt/CNS-CR photocatalyst fabricated by the chemical reduction (CR) method shows a much higher efficiency for H2 evolution from water splitting, compared to the Pt/CNS-PR photocatalyst obtained by the loading of Pt NPs by the traditional photo-reduction (PR) method. When triethanolamine (TEOA) is used as a hole sacrificial agent, the hydrogen production rate of 2.0%-Pt/CNS-CR is 7862.5 μmol g−1 h−1, which is 6.92 times higher than that of 2.0%-Pt/CNS-PR (1136.8 μmol g−1 h−1). The valence states of the Pt element in the Pt/CNS-CR and Pt/CNS-PR nanocomposites have been analyzed by X-ray photoelectron spectroscopy, respectively. At the same time, the effects of the loading amount of Pt and different sacrificial reagents on the photocatalytic H2 generation activity have also been systematically investigated.
 Please wait while we load your content...
Please wait while we load your content...

