
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and bonding in reduced boron and aluminium complexes with formazanate ligands†
Ranajit
Mondol
 and
Edwin
Otten
*
and
Edwin
Otten
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
First published on 25th July 2019
Abstract
Group 13 complexes of the type [(PhNNC(p-tol)NNPh)ZPh2]2− (Z = B, Al) containing a highly reduced, trianionic formazanate-derived ligand were studied and the differences in the structure, bonding and reactivity between the B and Al compounds were investigated. The increased ionic character in the bonding of the Al complex is evident from the enhanced charge delocalization onto the peripheral ligand substituents (N–Ph) via the π-framework, as shown by the rotation barrier around the N–C(Ph) bond. The electron-rich nature of these compounds allows facile benzylation at the ligand, and the structures of the products were analysed by X-ray crystallography. The products are inorganic analogues of 1-alkylated 1,2,3,4-tetrahydro-1,2,4,5-tetrazines (‘leucoverdazyls’). The six-membered heterocyclic cores of the B and Al compounds are shown to be different, having envelope- and boat-type conformations, respectively. Homolysis of the N–C(benzyl) bond in these compounds was studied by NMR spectroscopy under conditions that trap the organic radical as TEMPO-Bn. Analysis of the reaction kinetics affords activation parameters that approximate the N–C(benzyl) bond strength. The ionic Al compound has one of the weakest N–C bonds reported so far in this type of inorganic leucoverdazyl analogues.
Introduction
The direct involvement of ligands in redox reactions by coordination complexes bearing non-innocent ligands is an emerging research area.1–3 In this context, there is growing interest in designing new types of redox-active (non-innocent) ligands. In 2007, Hicks and coworkers observed reversible ligand-based reduction in a formazanate boron acetate compound.4 This demonstrated for the first time that the formazanate ligand can be considered as a redox-active analogue of the well-known β-diketiminate ligands.5–8 Taking inspiration from this work, our group has explored the coordination chemistry, redox behavior and reactivity of complexes with formazanate ligands.9–16 Along with our work, the Gilroy group17–26 and others27–29 have been involved in the synthesis and application of new molecular complexes with formazanate ligands. Previously, we showed that the ligands in boron and aluminum compounds with formazanate ligands could be sequentially reduced by 1- and 2-electrons at moderate reduction potentials (Scheme 1).30,31 The 2-electron reduced formazanate boron compound (12−) subsequently reacted with electrophiles (E+) such as benzyl bromide (BnBr) and water (H2O) to form ligand-benzylated and -protonated products (Bn1− and H1− in Scheme 1).32 In these compounds, the formazanate ligands are modified by the ‘storage’ of [2e−/E+], which could be converted to Bn˙ and H˙ radicals by the homolytic cleavage of the N–C(Bn) and N–H bonds, respectively (Scheme 1).32 These reactions occur readily, because the boron-containing radical that is generated (1˙−) is relatively stable due to the presence of a low-energy SOMO that is delocalized over all four N-atoms in the ligand backbone.30,32 It is anticipated that the basicity (nucleophilicity), radical stability and N–C/N–H bond strength of compounds bearing functionalized formazanate ligands could be tuned via either ligand substituent effects or incorporation of a different central element (main group or transition metal) in the formazanate chelate ring.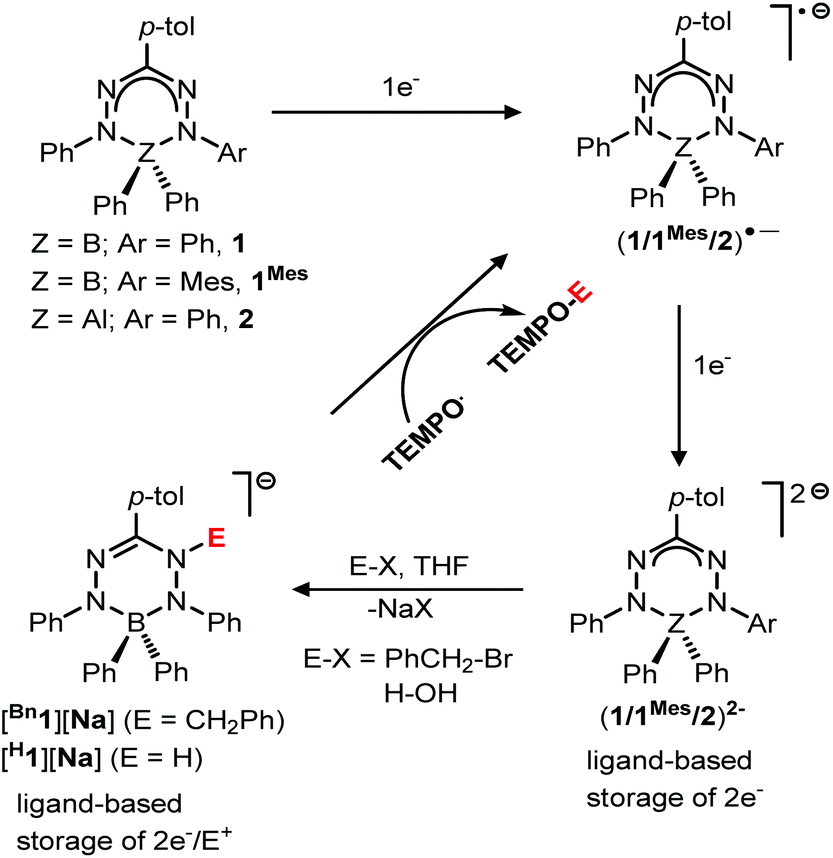 | ||
| Scheme 1 Ligand-based storage of [2e−/E+], and subsequent conversion to E˙ (Bn˙/H˙) radicals that can be trapped with TEMPO. | ||
In order to investigate the effects of changing the central element (boron) in 1 to the more electropositive aluminum, here we provide a detailed comparison of formazanate B and Al complexes with an identical ligand. The comparison includes an analysis of resonance delocalization in the two-electron reduced formazanate aluminum diphenyl compound (22−) via dynamic NMR spectroscopy. The synthesis of ligand-benzylated products Bn2− is described, and crystallographic and spectroscopic characterization data are provided. Furthermore, the kinetics of homolytic N–C(benzyl) cleavage is studied.
Results and discussion
The two-electron reduced formazanate aluminium diphenyl compound [PhNNC(p-tol)NNPh]AlPh22− (22−) was synthesized as its disodium salt according to a previously published procedure.31 The product 22−, which has an electron-rich, formally trianionic formazanate ligand, is highly air-sensitive, but stable at room temperature under inert conditions. The 1H NMR spectrum of 22− in THF-d8 was shown to be temperature-dependent: at 233 K, the spectrum shows 5 inequivalent resonances due to the N–Ph groups. Increasing the temperature results in line broadening and ultimately coalescence of three distinct signals as expected for the ortho, meta and para-positions of a Ph group (Fig. 1).‡ These features are indicative of hindered rotation around the N–C(Ph) bond, which leads to inequivalent chemical environments for the two ortho- and meta-H positions, with exchange rates that are on the order of the NMR timescale.33,34 Lineshape analysis was carried out for the pairs of exchanging resonances in the temperature range 233–303 K, which gave the activation parameters for the exchange process of 22− as ΔH‡ = 54.1 ± 1.7 kJ mol−1 and ΔS‡ = −2.5 ± 6.0 J mol−1 K−1 (see the ESI† for details). The activation enthalpy reflects a substantial loss of the N-C(Ph) π-bonding character upon moving to the transition state, whereas there is little difference in entropy. A comparison with the related boron-containing compounds (12−) shows that the values are similar to those of the asymmetric derivative [MesNNC(p-tol)NNPh]BPh22− ([1Mes]2−; ΔH‡ = 57.4 ± 1.8 kJ mol−1 and ΔS‡ = 1 ± 6 J mol−1 K−1).30 On the other hand, the B-analogue 12−, which has the same (symmetrical) formazanate ligand as the Al complex 22− does not show evidence for dynamics on the NMR timescale:§ no line-broadening is observed down to 233 K.30 We interpret the difference between the B and Al complexes with an identical formazanate ligand (12− and 22−) as a reflection of the difference in bonding within the boron and aluminium heterocycles. In particular, the largely ionic character of the Al–N bonds in comparison with the more covalent B–N interactions is responsible for the increased accumulation of negative charge within the NNCNN framework in the Al compound. An increase in N–C(Ph) π-bonding due to resonance delocalization of the negative charge into the N–Ph group is also observed in the solid state structures obtained by X-ray crystallography: the N–C(Ph) bonds in 22− are 1.371(2)/1.375(3) Å,31 whereas those in 12− are marginally larger at 1.379(3)/1.385(3) Å.30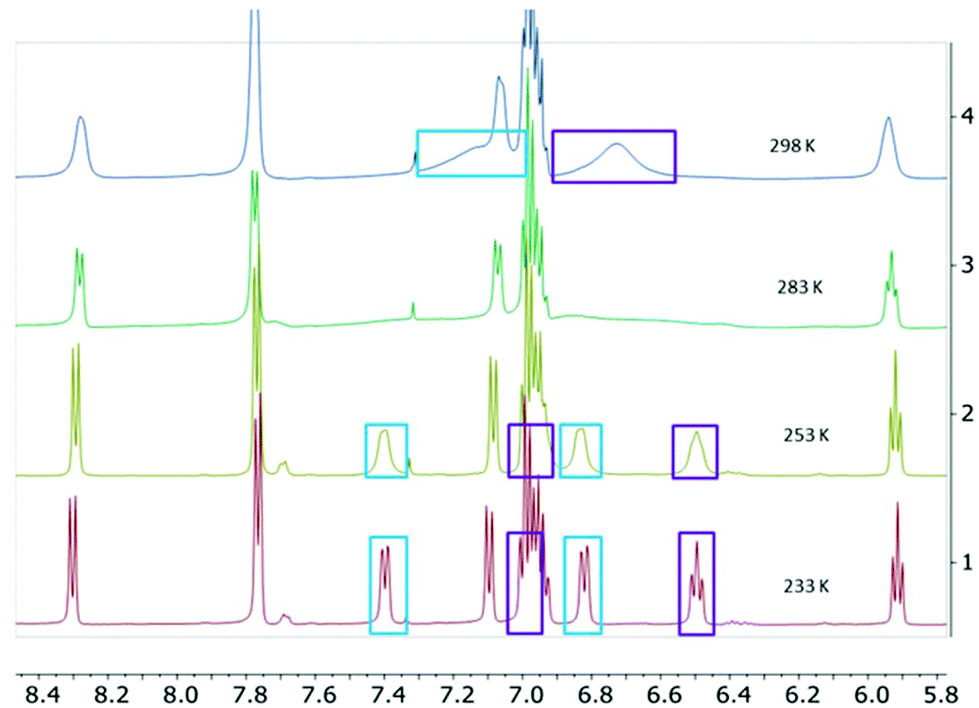 | ||
| Fig. 1 1H NMR spectra of 22− in THF-d8 at various temperatures. | ||
To further probe the bonding differences between formazanate boron and aluminium complexes, a DFT computational study was carried out (B3LYP functional and 6-311+G(d,p) basis set). The geometries were optimized in the gas phase starting from the crystallographic coordinates, with the Na(THF)x cations removed (the computational results for 12−calc were described previously).30 The metrical parameters of the optimized structures of 12−calc and 22−calc are in good agreement with the experimental structures, albeit the N–N bonds are somewhat shorter in the DFT models (see Table S2†). Visual inspection of the frontier orbitals shows that the HOMO is primarily π-antibonding between the N-atoms in the formazanate ligand, and in addition, evidences the presence of the π-bonding character in the N–C(Ph) fragment (Fig. S15†). Although the differences are small, the calculated Wiberg bond index35 for the N–C(Ph) bonds is larger in the Al compound (22−calc: 1.24) than in the B compound (12−calc: 1.22), corroborating the trend for the strength of π-bonding that was obtained from the NMR study. In addition, a higher Wiberg bond index is found for the B–N bonds in 12− (0.68) in comparison with the Al–N bonds in 22− (0.40) and the natural charges indicate that the formazanate ligand bears a significantly higher negative charge in the Al complex (−2.40e) than in the B analogue (−1.99e).
Computational evaluation of the barrier to rotation around the N–C(Ph) bond in 22−calc was carried out by scanning the dihedral angle between the Ph ring and the ligand backbone. As expected, a maximum was found at a dihedral angle of ca. 90°, and at this geometry a transition state optimization was carried out to arrive at a saddle point (22−TS, Nimag = 1). The computed barrier for N–C(Ph) bond rotation was found to be 55.9 kJ mol−1, which is in good agreement with the experimental value (∼54.9 kJ mol−1 at 298 K). A comparison between the ground and transition states shows that π-bonding is lost in the N–Ph ring that is rotated (Wiberg bond index = 1.04), whereas a small increase is observed in the other N–C(Ph) bond (1.26). This is also reflected in the different N–C(Ph) bond lengths that are calculated for 22−TS (1.411 Å and 1.365 Å; cf. 1.370 Å in 22−calc).
We subsequently evaluated the reactivity of compound 22− towards the electrophile benzyl bromide. Treatment of an orange THF-d8 solution of 22− with 1 equivalent of BnBr resulted in an immediate colour change from orange to yellowish-green. The 1H NMR spectrum of the reaction mixture at room temperature (400 MHz, THF-d8) reveals the diagnostic resonances for the diastereotopic protons of the benzyl–CH2 group at δ 4.29 and 4.45 ppm with a geminal coupling constant of 2JHH = 12.5 Hz. Also, the 1H NMR spectrum shows two inequivalent resonances for the para-protons of the N–Ph rings at δ 6.09 and 6.34 ppm due to a descent in symmetry from the C2v-symmetric precursor 22−. The NMR data of the reaction product are similar to those of the boron analogue,32 and suggest that it contains a benzyl group that is attached to a N-atom of the ligand backbone. Consequently, we formulate the product as an anionic, ligand-benzylated complex [PhNN(Bz)C(p-tol)NNPh]AlPh2− (Bn2−, Scheme 2). On a preparative scale, the reaction between 22− and BnBr allowed isolation of the sodium salt [Bn2][Na] as a solid material in 52% yield. Crystals of [Bn2][Na] were obtained by slow diffusion of hexane into the THF solution at −30 °C. Although the previously reported boron analogue [Bn1][Na] also crystallizes under these conditions, these crystals quickly melt at room temperature, thus thwarting structure determination.32 Treatment of both these sodium salts with Bu4NBr resulted in cation exchange and formation of the tetrabutyl ammonium salts [Bn1][NBu4] and [Bn2][NBu4] in yields of ca. 75%. Gratifyingly, crystals of [Bn1][NBu4] had a much higher melting point, and structure determination using X-ray was successfully completed for both B- and Al-containing products. The molecular structures of [Bn1][NBu4] and [Bn2][Na] show that in both of these compounds, the benzyl group is attached to one of the internal N-atoms to retain the 6-membered chelate ring structures of the dianionic precursors (Fig. 2, pertinent metrical data in Table 1). A closer inspection of the central six-membered heterocyclic rings in these compounds reveals some noteworthy differences. Compound [Bn1][NBu4] adopts an envelope conformation which is similar to the precursor 12−,30 with the formazanate backbone atoms (NNCNN) nearly coplanar and the B atom residing 0.568 Å above that plane. In contrast, the six-membered ring in the Al compound [Bn2][Na] is found to be present in a boat conformation in which N1, Al1, N3 and C7 are coplanar, whereas N2 and N4-atoms are displaced out from that plane by 0.502 and 0.507 Å, respectively (Fig. 2). This is markedly different from the nearly planar conformation in the precursor 22−.31 A Cremer–Pople puckering analysis36 was carried out using PLATON,37 which allowed a more quantitative distinction between the divergent geometrical parameters within the core of [Bn1][NBu4]vs.[Bn2][Na]. In this analysis, a combination of the puckering amplitude (Q) and two polar coordinates (θ and φ) are used to map the conformations onto a sphere. The following puckering parameters are obtained for these compounds: for [Bn1][NBu4] (Q = 0.44 Å; θ = 57.0°; φ = 332.2°) and [Bn2][Na] (Q = 0.58 Å; θ = 89.8°; φ = 300.2°). Of these parameters, the θ angles in six-membered rings distinguish among the chair, envelope and boat conformations, whereas φ defines the pseudorotation pathways that transverse twisted conformations as well. The angles θ found for both compounds are indicative of either an envelope (idealized: 54.7°; [Bn1][NBu4]: 57.0°) or boat conformation (idealized: 90°; [Bn2][Na]: 89.8°).
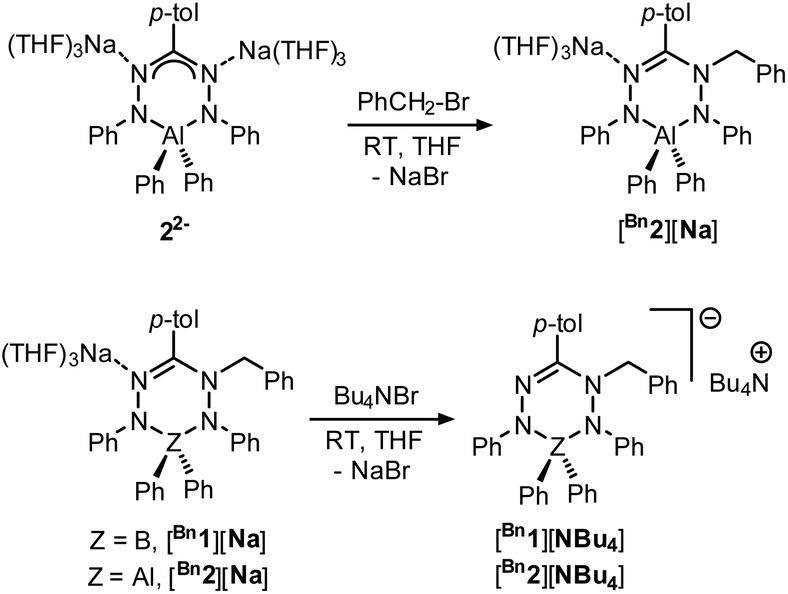 | ||
| Scheme 2 Synthesis of ligand-benzylated products [Bn2][Na], [Bn2][NBu4] and [Bn1][NBu4]. | ||
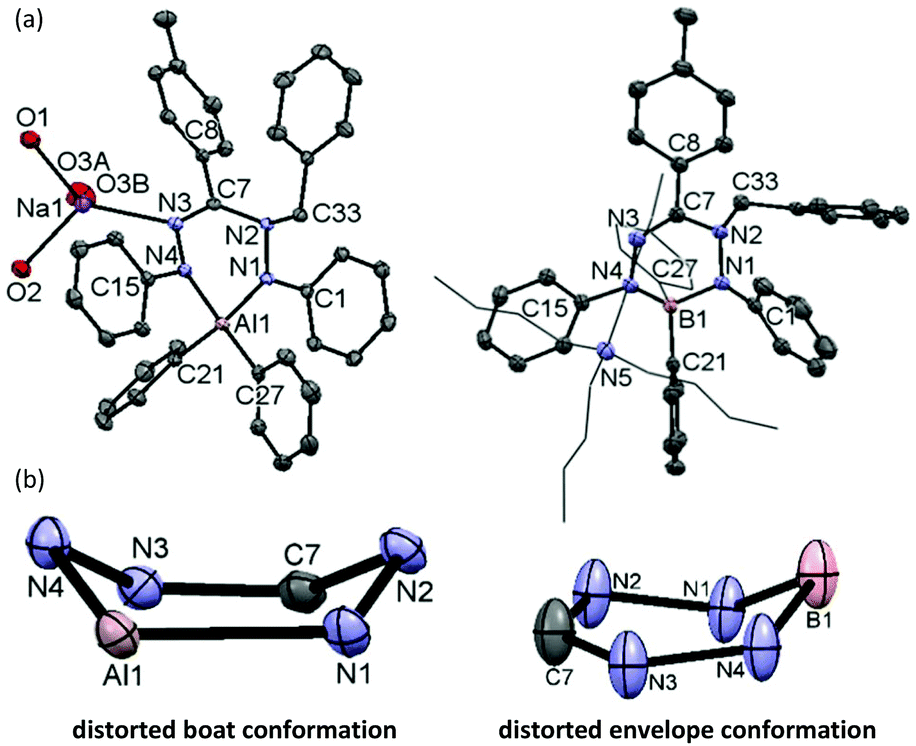 | ||
| Fig. 2 (a) Molecular structures of [Bn2][Na] (top, left) and [Bn1][NBu4] (top, right), showing 50% probability ellipsoids. Hydrogen atoms for [Bn2][Na] and [Bn1][NBu4] are omitted for clarity. THF molecules (except for the O atoms bonded to Na) for [Bn2][Na] are omitted for clarity. (b) Six-membered chelate rings for [Bn2][Na] (bottom, left) and for [Bn1][NBu4] (bottom, right). | ||
| [Bn1a][NBu4] | [Bn2][Na] | |
|---|---|---|
| N1–N2 | 1.428(2) | 1.446(1) |
| N3–N4 | 1.393(2) | 1.402(1) |
| N2–C7 | 1.396(2) | 1.437(1) |
| C7–N3 | 1.289(2) | 1.292(1) |
| Al1–N1 | 1.867(1) | |
| Al1–N4 | 1.914(1) | |
| B1–N1 | 1.575(2) | |
| B1–N4 | 1.584(2) | |
| N1–C1 | 1.414(2) | 1.387(1) |
| N4–C15 | 1.392(2) | 1.399(1) |
| N2–C33 | 1.468(2) | 1.486(1) |
| N1–B1–N4 | 105.78(1) | |
| N1–Al1–N4 | 95.24(4) | |
| N1–N2–C7 | 115.41(1) | 112.27(8) |
| N1–N2–C33 | 113.94(1) | 110.33(8) |
| C7–N2–C33 | 118.79(1) | 112.13(8) |
In the B and Al dianions (12− and 22−) the bonding in the six-membered core is delocalized, as shown by the equivalent N–N and C–N bond lengths, whereas ligand benzylation leads to a more localized bonding picture. For example, the C–N bonds in the ligand backbone change from 1.332(2)/1.328(2) Å in 22− to 1.437(1)/1.292(1) Å in [Bn2][Na]. Similar changes in bond lengths occur in the boron analogue (see Tables S2/S3† for a full comparison of metrical data). In both compounds, the benzylated N-atom is pyramidal as indicated by the sum of angles around N(2) being much smaller than 360° (Σ∠(N(2)) = 348.1° and 334.7° for [Bn1][NBu4] and [Bn2][Na], respectively). In [Bn2][Na] the Ph-substituted N-atoms are planar (sp2-hybridized) with Σ∠(N) = 359.5° for N(1) and 354.5° for N(4). Conversely, in the boron analogue [Bn1][NBu4] the N(4) atom is almost planar (Σ∠(N) = 357.5°), while N(1) is pyramidalized ((Σ∠(N) = 347.8°). It appears that these differences reflect a larger degree of directionality (covalency) in the B–N bonds in comparison with the more ionic Al congener.
The anions Bn1− and Bn2− were also evaluated by DFT calculations. Optimized geometries of Bn1−calc and Bn2−calc were obtained starting from the crystallographic coordinates (counter cations were removed) and were shown to be in good agreement with the empirical structures. Importantly, the dissimilarities in pyramidalization at the N-atoms of both the B and Al compounds are reproduced in the computational models, suggesting that these are intrinsic and are not due to packing or ion pairing effects. In both the experimental structures and the DFT models, there is a small but noticeable elongation of the N–C(Bn) bond in the Al compound Bn2− (X-ray: 1.486(1) Å; DFT: 1.492 Å) compared to that in the B congener Bn1− (X-ray: 1.468(2) Å; DFT: 1.472 Å). An NBO analysis indicated that the bonding within the NNCNN ligand backbone is similar in both B and Al compounds and can be explained by a normal σ-bonding framework with an additional localized N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π-bond (Fig. 3A). The hybridization of the benzyl-substituted nitrogen atom is intermediate between sp2 and sp3, with a lone pair that has a small but non-negligible amount of the s-character (10% for Bn1−calc; 16% for Bn2−calc). Importantly, the NBO analysis shows that the N–B and N–Al interactions in both the compounds are quite different. For the boron compound Bn1−, we find two natural bond orbitals that represent the relatively covalent B–N σ-bonds (ca. 77% contribution from N; 23% from B). For the Al analogue Bn2−, on the other hand, no 2-center Al–N bonds are obtained and, instead, the NBO analysis indicates that the N-based lone pairs interact with an empty Al orbital (Fig. 3). Similarly, the natural charges indicate that the Al complex is much more ionic (NPA charge for Al in Bn2−calc: 1.82; B in Bn1−: 0.74).
C π-bond (Fig. 3A). The hybridization of the benzyl-substituted nitrogen atom is intermediate between sp2 and sp3, with a lone pair that has a small but non-negligible amount of the s-character (10% for Bn1−calc; 16% for Bn2−calc). Importantly, the NBO analysis shows that the N–B and N–Al interactions in both the compounds are quite different. For the boron compound Bn1−, we find two natural bond orbitals that represent the relatively covalent B–N σ-bonds (ca. 77% contribution from N; 23% from B). For the Al analogue Bn2−, on the other hand, no 2-center Al–N bonds are obtained and, instead, the NBO analysis indicates that the N-based lone pairs interact with an empty Al orbital (Fig. 3). Similarly, the natural charges indicate that the Al complex is much more ionic (NPA charge for Al in Bn2−calc: 1.82; B in Bn1−: 0.74).
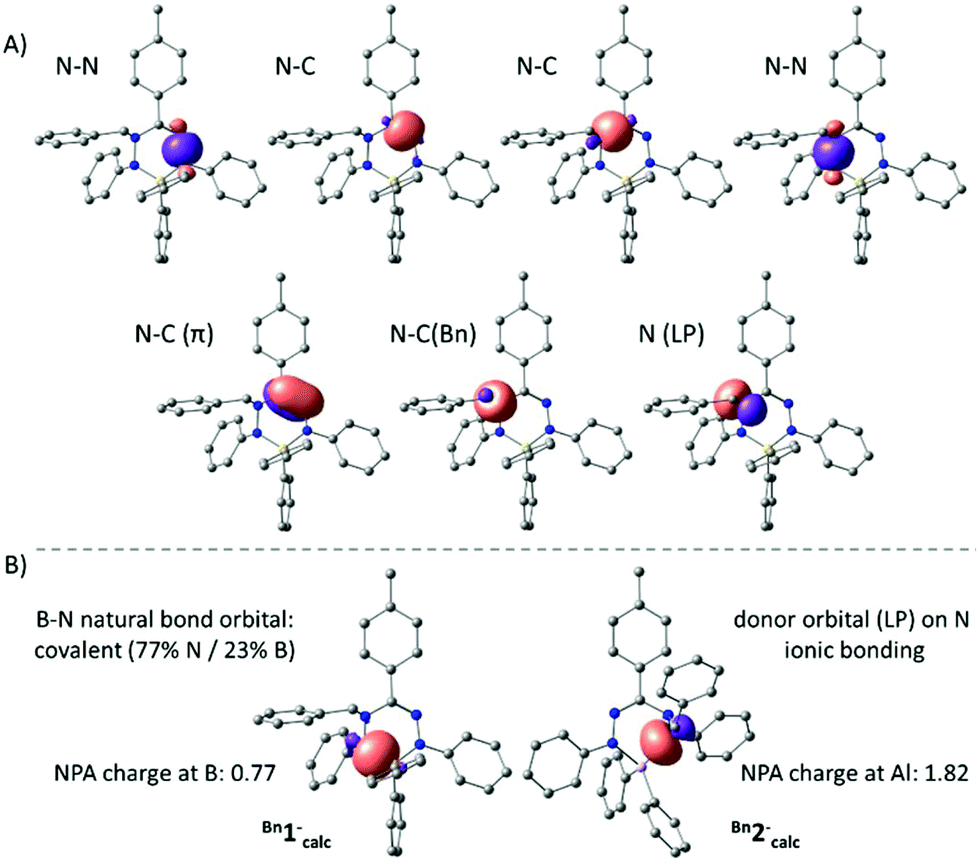 | ||
| Fig. 3 (A) Selected natural bond orbitals for the σ-framework in the ligand backbone (top); the N–C π-bond and those around the N–benzyl atom (bottom). (B) Comparison of the B–N (covalent) vs. Al–N (ionic) bonding interactions. | ||
A comparison of the UV/Vis spectra of the Na+ and Bu4N+ salts of Bn1− and Bn2− shows that the absorption maxima in dilute THF solution are independent of the nature of the cation. The boron compounds Bn1− and the aluminium compounds Bn2− absorb at 395 and 389 nm, respectively (Fig. S1†). These absorbance maxima are most likely due to a π–π* transition of the localized NC bonds present in these compounds, as shown by the crystallographic data (vide supra), and are blue-shifted compared to the delocalized precursors 12− and 22− (λmax = 486 nm).¶,31
The compounds Bn1− and Bn2− are inorganic, anionic analogues of purely organic 1-alkylated tetrahydro-1,2,4,5-tetrazines (‘leucoverdazyls’).38 Leucoverdazyls (with N–H bonds) and the alkylated (N–C) derivatives are known to have weak N–H/N–C bonds because homolytic cleavage generates a stable verdazyl radical.38–41 The stability of this type of radical extends to inorganic systems.4,9,10,14,17,26,30,31,42 In order to examine the effect on the N–C(Bn) bond dissociation energy (BDE) due to the replacement of B to the more electropositive Al in [Bn2][Na], the cleavage of the N–C bond in [Bn2][Na] was investigated. The N–C(Bn) bond dissociation enthalpy in [Bn2][Na] was obtained experimentally by NMR spectroscopic monitoring of the kinetics of benzyl radical transfer from [Bn2][Na] to TEMPO (present in excess) in the temperature range of 65–85 °C (Scheme 3).
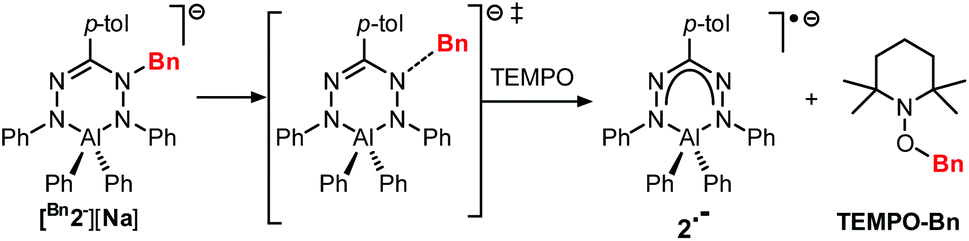 | ||
| Scheme 3 Benzyl group transfer from [Bn2][Na] to TEMPO. | ||
Clean exponential decay of the starting material and concomitant appearance of TEMPO-Bn were observed, which allowed the rate constants to be determined (Fig. S8†). An Eyring analysis provided the activation parameters ΔH‡ and ΔS‡ of 107 ± 4 kJ mol−1 and 17 ± 11 J mol−1 K−1, respectively, for the benzyl transfer (Fig. 4; see the ESI† for details). Both these values are somewhat smaller than those found for the boron analogue [Bn1][Na] (ΔH‡ = 121 ± 5 kJ mol−1; ΔS‡ = 77 ± 14 J mol−1 K−1).32 A likely explanation for these differences is the ground-state destabilization of the N–C(Bn) bond which is indicated by the somewhat larger N–C(Bn) distance found by both experiment and theory (vide supra).
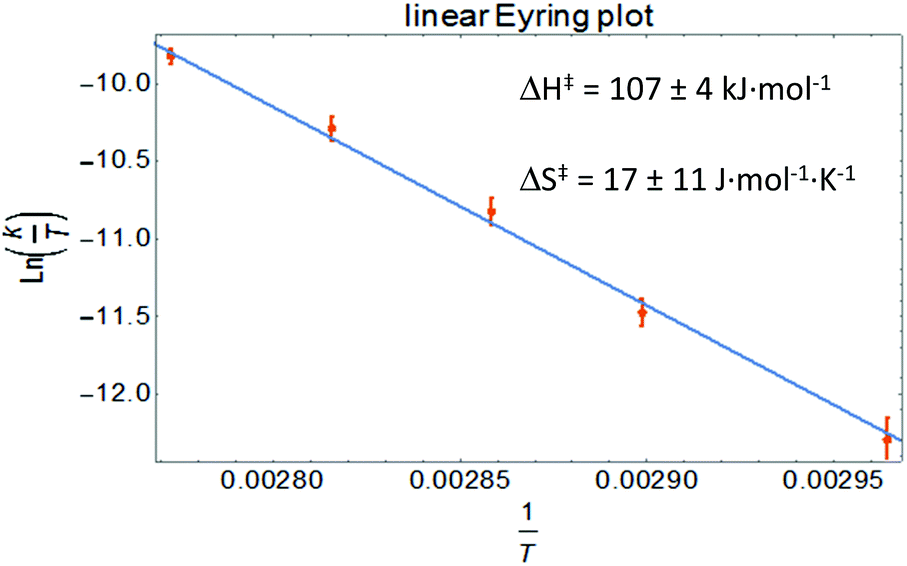 | ||
| Fig. 4 Eyring analysis for benzyl transfer from [Bn2][Na] to TEMPO. | ||
To investigate a possible influence of ion-pairing on the N–C(Bn) bond dissociation energy, the kinetics of benzyl transfer were also measured for the tetrabutyl ammonium salts [Bn1][NBu4] and [Bn2][NBu4] using the same methodology. The resulting rate constants are in good agreement with those of the sodium salts (see the ESI† for details). Thus, even though in the solid state the sodium cation is bound to the ligand backbone, this weak interaction is likely broken in the solution. This is further supported by the observation that the UV/Vis spectra of the solution do not depend on the nature of the cation.
These data indicate that N–C(Bn) bond homolysis is modulated by the central element in these heterocyclic leucoverdazyl analogues: on going from relatively covalent, C-based parent structures (i.e., 1-benzyl-substituted 1,2,3,4-tetrahydro-1,2,4,5-tetrazines), the N–C(Bn) bond strength progressively decreases with an increase in the electropositive nature of the central element (i.e., C > B > Al).
Conclusions
In conclusion, this work addresses the differences in the structure and bonding between dianionic formazanate boron (12−) and aluminum (22−) complexes. Experimental (NMR, UV/Vis spectroscopy, and X-ray crystallography) and computational studies (Wiberg bond indices and NBO analysis) reveal that the increased ionic character of the Al compounds results in a higher degree of resonance delocalization of the ligand negative charge into the periphery of the ligand (i.e., the N–Ph substituents), which is reflected in the rotation barrier around the N–C(Ph) bond. For both the compounds, facile ligand benzylation occurs upon reaction with benzyl bromide to form Bn1− and Bn2− as anionic analogues of carbon-based leucoverdazyls. X-ray diffraction studies of Bn2− and the boron congener Bn1− are reported and a comparison shows distinct differences in the solid state structures between these complexes which can be related to a different degree of the ionic character in the bonding. The kinetics of benzyl transfer show that the N–C(Bn) bond homolysis is modulated by the nature of the central element present in the six-membered heterocyclic rings of Bn1− and Bn2−, with the ionic Al-based compound Bn2− having the weakest N–benzyl bond.Experimental section
General considerations
All manipulations were carried out under a nitrogen atmosphere using standard glovebox, Schlenk, and vacuum-line techniques. THF and hexane (Aldrich, anhydrous, 99.8%) were passed over columns of Al2O3 (Fluka). The compounds [Bn1][NBu4], [Bn2][Na] and [Bn2][NBu4] are highly air-sensitive, and the solvents (THF and hexane) used for their preparation and characterization were additionally dried on a Na/K alloy and subsequently vacuum transferred and stored under nitrogen. All solvents were degassed prior to use and stored under nitrogen. THF-d8 (Sigma-Aldrich) was vacuum transferred from the Na/K alloy and stored under nitrogen. The compounds [Bn1][Na]32 and 22− (as its disodium salt, [(PhNNC(p-tol)NNPh)AlPh2][Na2(DME)4])31 were synthesized according to published procedures. NMR spectra were recorded on a Varian Mercury 400 or Inova 500 spectrometer. The 1H and 13C NMR spectra were referenced internally using the residual solvent resonances and reported in ppm relative to TMS (0 ppm); J is reported in Hz. Assignments of NMR resonances were aided by COSY, NOESY, HSQC and HMBC experiments using standard pulse sequences. UV-Vis spectra were recorded in THF solution (∼10−3 M) in a quartz cuvette using an AVANTES AvaSpec-2048. Samples for elemental analyses were sent to Kolbe Microanalytical Laboratory (Mülheim an der Ruhr, Germany). However, despite our best efforts, no satisfactory analysis data could be obtained for these compounds, which is likely due to their highly air-sensitive nature.X-ray crystallography
Suitable crystals of compounds [Bn1][NBu4] and [Bn2][Na] were mounted on top of a cryoloop and transferred into the cold (100 K) nitrogen stream of a Bruker D8 Venture diffractometer. Data collection and reduction were performed using the Bruker software suite APEX2.43 Data collection was carried out at 100 K using Cu radiation (1.54178 Å) (for [Bn1][NBu4]) and Mo radiation (0.71073 Å) (for [Bn2][Na]). The final unit cell was obtained from the xyz centroids of 9929 ([Bn1][NBu4]) and 3515 ([Bn2][Na]) reflections after integration. A multiscan absorption correction was applied, based on the intensities of symmetry-related reflections measured at different angular settings (SADABS).43 The structures were solved by intrinsic phasing methods using SHELXT,44 and refinement of the structures was performed using SHELXL.45 For [Bn1][NBu4], the refinement indicated the presence of smeared out electron-density due to a disordered THF molecule that could not be modelled in a satisfactory manner. The contribution of this electron-density was removed using the PLATON/SQUEEZE routine,46 giving 4 solvent-accessible volumes in the unit cell, each containing 37 electrons (in agreement with THF). For [Bn2][Na], refinement was frustrated by a disorder problem: two of the THF molecules bound to the Na+ cation showed unrealistic displacement parameters when refined freely. For one of these, a two-site occupancy model was applied for all the atoms of the THF molecule and the site occupancy factor was refined (major fraction: 0.82). A SAME instruction was applied for the two disorder components, such that the two disordered THF molecules were restrained to have a similar geometry to that found for the THF molecule without disorder. Some atoms in the disordered THF molecule showed non-positive definite displacement parameters when refined freely, and RIGU/DELU instructions were applied. One of the carbon atoms of the remaining Na-THF fragment was split into two disorder components, which refined to an s.o.f. of 0.73 for the major component. The hydrogen atoms were generated by geometrical considerations, constrained to idealized geometries and allowed to ride on their carrier atoms with an isotropic displacement parameter related to the equivalent displacement parameter of their carrier atoms. Crystal data and details on data collection and refinement are presented in Table S1.†Computational studies
Calculations were performed with the Gaussian 09 program47 using density functional theory (DFT) in the gas phase. The geometries of the anions were fully optimized (after removing the counter cation from the crystallographically determined structure) using the B3LYP exchange–correlation functional with the 6-311+G(d,p) basis set. Optimization was performed without (symmetry) constraints, and the resulting structures were confirmed to be minimal on the potential energy surface by frequency calculations (number of imaginary frequencies = 0). The stationary points found for 22−calc, Bn1−calc and Bn2−calc closely resemble their crystallographically determined structures, respectively. For the NBO analysis, single point calculations were performed on the final optimized geometries of 12−calc,3022−calc, Bn1−calc and Bn2−calc by including keywords for NBO calculation.To explore the chemical exchange process of 22− due to the rotation around the N–C(Ph) bond, we scanned the dihedral angle between the two adjacent N atoms in the ligand and the ipso- and ortho-carbons of the Ph group in steps of 10 degrees (all other degrees of freedom were optimized). Starting from the highest energy points along the scan coordinate, the transition state was optimized and confirmed by frequency analysis (number of imaginary frequencies for 22−calc-ts = 1).
GaussView 5.0.948 was used to visualize the computed structures and molecular orbitals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Netherlands Organisation for Scientific Research (NWO) (VIDI grant to EO) is gratefully acknowledged. We thank Dr. Remco Havenith for suggestions regarding computational chemistry. We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster.Notes and references
- V. Lyaskovskyy and B. De Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- D. L. J. Broere, R. Plessius and J. I. Van Der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- J. B. Gilroy, M. J. Ferguson, R. McDonald, B. O. Patrick and R. G. Hicks, Chem. Commun., 2007, 126–128 RSC.
- C. Chen, S. M. Bellows and P. L. Holland, Dalton Trans., 2015, 44, 16654–16670 RSC.
- C. Camp and J. Arnold, Dalton Trans., 2016, 45, 14462–14498 RSC.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS.
- M. C. Chang, T. Dann, D. P. Day, M. Lutz, G. G. Wildgoose and E. Otten, Angew. Chem., Int. Ed., 2014, 53, 4118–4122 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431–7433 RSC.
- R. Travieso-Puente, M. C. Chang and E. Otten, Dalton Trans., 2014, 43, 18035–18041 RSC.
- R. Travieso-Puente, J. O. P. Broekman, M. C. Chang, S. Demeshko, F. Meyer and E. Otten, J. Am. Chem. Soc., 2016, 138, 5503–5506 CrossRef CAS PubMed.
- M. C. Chang and E. Otten, Organometallics, 2016, 35, 534–542 CrossRef CAS.
- M. C. Chang, A. Chantzis, D. Jacquemin and E. Otten, Dalton Trans., 2016, 45, 9477–9484 RSC.
- M. C. Chang, P. Roewen, R. Travieso-Puente, M. Lutz and E. Otten, Inorg. Chem., 2015, 54, 379–388 CrossRef CAS PubMed.
- M.-C. Chang and E. Otten, Inorg. Chem., 2015, 54, 8656–8664 CrossRef CAS PubMed.
- S. M. Barbon, J. T. Price, P. A. Reinkeluers and J. B. Gilroy, Inorg. Chem., 2014, 53, 10585–10593 CrossRef CAS PubMed.
- M. Hesari, S. M. Barbon, V. N. Staroverov, Z. Ding and J. B. Gilroy, Chem. Commun., 2015, 51, 3766–3769 RSC.
- S. M. Barbon, J. T. Price, U. Yogarajah and J. B. Gilroy, RSC Adv., 2015, 5, 56316–56324 RSC.
- S. M. Barbon, J. V. Buddingh, R. R. Maar and J. B. Gilroy, Inorg. Chem., 2017, 56, 12003–12011 CrossRef CAS PubMed.
- S. Novoa and J. B. Gilroy, Polym. Chem., 2017, 8, 5388–5395 RSC.
- S. M. Barbon, S. Novoa, D. Bender, H. Groom, L. G. Luyt and J. B. Gilroy, Org. Chem. Front., 2017, 4, 178–190 RSC.
- S. M. Barbon, V. N. Staroverov and J. B. Gilroy, Angew. Chem., 2017, 129, 8285–8289 CrossRef.
- R. R. Maar, A. Rabiee Kenaree, R. Zhang, Y. Tao, B. D. Katzman, V. N. Staroverov, Z. Ding and J. B. Gilroy, Inorg. Chem., 2017, 56, 12436–12447 CrossRef CAS PubMed.
- J. S. Dhindsa, R. R. Maar, S. M. Barbon, M. Olivia Avilés, Z. K. Powell, F. Lagugné-Labarthet and J. B. Gilroy, Chem. Commun., 2018, 54, 6899–6902 RSC.
- A. Van Belois, R. R. Maar, M. S. Workentin and J. B. Gilroy, Inorg. Chem., 2019, 58, 834–843 CrossRef CAS PubMed.
- A. Mandal, B. Schwederski, J. Fiedler, W. Kaim and G. K. Lahiri, Inorg. Chem., 2015, 54, 8126–8135 CrossRef CAS PubMed.
- G. Mu, L. Cong, Z. Wen, J. I. C. Wu, K. M. Kadish and T. S. Teets, Inorg. Chem., 2018, 57, 9468–9477 CrossRef CAS PubMed.
- E. Kabir, D. Patel, K. Clark and T. S. Teets, Inorg. Chem., 2018, 57, 10906–10917 CrossRef CAS PubMed.
- R. Mondol, D. A. Snoeken, M.-C. Chang and E. Otten, Chem. Commun., 2017, 53, 513–516 RSC.
- R. Mondol and E. Otten, Inorg. Chem., 2019, 58, 6344–6355 CrossRef CAS PubMed.
- R. Mondol and E. Otten, Inorg. Chem., 2018, 57, 9720–9727 CrossRef CAS PubMed.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS.
- A. D. Bain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- I. Polumbrik and O. Ryabokon, Zh. Org. Khim., 1978, 14, 1332 Search PubMed.
- A. Misyura, O. Polumbrik and L. Markovskii, Zh. Org. Khim., 1989, 25, 424 CAS.
- C. W. Johnston, T. R. Schwantje, M. J. Ferguson, R. McDonald and R. G. Hicks, Chem. Commun., 2014, 50, 12542–12544 RSC.
- J. L. Lukkarila, PhD thesis, University of Toronto, 2009.
- R. R. Maar, S. D. Catingan, V. N. Staroverov and J. B. Gilroy, Angew. Chem., Int. Ed., 2018, 57, 9870–9874 CrossRef CAS PubMed.
- Bruker, APEX2 (v2012.4-3), SAINT (Version 8.18C) and SADABS (Version 2012/1), Bruker AXS Inc., Madison, Wisconsin, 2012 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1938708 and 1938709. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02831e |
| ‡ The additional broadening observed above 283 K is attributed to the presence of a small amount of radical species (the paramagnetic monoanion 2˙−) that engages in electron transfer with 22−. |
| § The estimated upper limit of the barrier for N–C(Ph) bond rotation is ca. 40 kJ mol−1. See ESI† for details. |
| ¶ The UV/Vis spectrum of 12− reported in the ESI† of ref. 30 is incorrect, likely due to the decomposition of this highly sensitive compound. |
| This journal is © The Royal Society of Chemistry 2019 |