
Unsymmetrical nonplanar ‘push–pull’ β-octasubstituted porphyrins: facile synthesis, structural, photophysical and electrochemical redox properties†
 b
and
Muniappan
Sankar
*a
b
and
Muniappan
Sankar
*a
Abstract
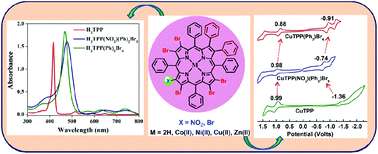
Mixed substitution at the β-position of porphyrins influences their photophysical and electrochemical redox properties. Two new series of asymmetrically mixed β-octasubstituted porphyrins viz. MTPP(Ph)2Br5X (X = NO2 or Br and M = 2H, Co(II), Ni(II), Cu(II), and Zn(II)) have been synthesized and characterized by various spectroscopic techniques. The single crystal X-ray structure of H2TPP(NO2)(Ph)2Br5 showed a nonplanar saddle shape conformation of the macrocyclic core. Furthermore, the fully optimized geometries confirmed the saddle shape conformation of H2TPP(Ph)2Br5X (X = NO2 or Br). Electronic spectra revealed a significant bathochromic shift by appending both electron donor and acceptor substituents at the β-position of the meso-tetraphenylporphyrin skeleton, which reflects the following order H2TPP < H2TPP(NO2) < H2TPP(NO2)(Ph)2 < H2TPP(Ph)2Br6 < H2TPP(NO2)(Ph)2Br5. H2TPP(Ph)2Br5X (X = NO2 or Br) exhibited a significant bathochromic shift (Δλmax = 53–61 nm) in the Soret band and (Δλmax = 90–95 nm) in the longest wavelength Qx(0,0) band as compared to H2TPP. Nonplanar conformations and electron withdrawing β-substituents induce higher protonation and deprotonation constants for H2TPP(NO2)(Ph)2Br5 and H2TPP(Ph)2Br6 as compared to precursor porphyrins viz. H2TPP, H2TPP(NO2) and H2TPP(NO2)(Ph)2. The electronic spectral properties and redox potentials of MTPP(Ph)2Br5X (X = NO2 or Br and M = 2H, Co, Ni, Cu and Zn) are affected by β-substituents at the periphery of the porphyrin core. Redox tunability was achieved by appending push–pull substituents at the β-position of the MTPP (M = 2H, CoII, NiII, CuII, and ZnII) skeleton of the macrocycle. CuTPP(Ph)2Br6 and CuTPP(NO2)(Ph)2Br5 exhibited a dramatically reduced HOMO–LUMO gap with a difference of 0.55 V and 0.62 V, respectively as compared to CuTPP due to the push–pull effect of β-substituents and nonplanarity of the porphyrin core.
 Please wait while we load your content...
Please wait while we load your content...

