
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbohydrate-functionalized N-heterocyclic carbene Ru(II) complexes: synthesis, characterization and catalytic transfer hydrogenation activity†
Joseph P.
Byrne
 ,
Pauline
Musembi
and
Martin
Albrecht
*
,
Pauline
Musembi
and
Martin
Albrecht
*
Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch
First published on 5th July 2019
Abstract
Three Ru complexes containing carbohydrate/N-heterocyclic carbene hybrid ligands were synthesized that were comprised of a triazolylidene coordination site and a directly linked per-acetylated glucosyl (5Glc) or galactosyl unit (5Gal), or a glycosyl unit linked through an ethylene spacer (6). Electrochemical and UV-vis analysis indicate only minor perturbation of the electronic configuration of the metal center upon carbohydrate installation. Deprotection of the carbohydrate was accomplished under basic conditions to afford complexes that were stable in solution over several hours, but decomposed in the solid state. Complexes 5 and 6 were used as pre-catalysts for transfer hydrogenation of ketones under basic conditions, i.e. conditions that lead to in situ deprotection of the carbohydrate entity. The carbohydrate directly influences the catalytic activity of the metal center. Remotely linked carbohydrates (complex 6) induce significantly lower catalytic activity than directly linked carbohydrates (complexes 5Glc, 5Gal), while unfunctionalized triazolylidenes are an order of magnitude more active. These observations and substrate variations strongly suggest that substrate bonding is rate-limiting for transfer hydrogenation in these hybrid carbohydrate/triazolylidene systems.
Introduction
N-Heterocyclic carbenes (NHCs) act as versatile ligands for various catalytic systems,1 as well as for biological and materials applications.2 Ruthenium complexes of NHCs, in particular, have shown a broad range of catalytic activity,3–6 with application in olefin-metathesis,7–9 transfer hydrogenation,10–18 as well as oxidation of alcohols and amines,19–23 and water.24 1,2,3-Triazolylidenes have emerged as a particularly promising subclass of NHC ligands that are stronger σ-donors than Arduengo-type imidazolylidenes and easily accessible through Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) ‘click’ chemistry.25 Because of these characteristics, they have found widespread applications in catalysis.26,27 CuAAC chemistry has excellent compatibility with most functional groups due to the mild reaction conditions28,29 and consequently triazoles have become ubiquitous linkers for molecular components in various domains of (bio)chemistry, including materials science,30–32 medicinal33 and supramolecular chemistry,34–37 as well as peptide/carbohydrate functionalisation.38–42 This CuAAC synthetic methodology allows the introduction of natural chiral pool motifs such as carbohydrates into triazoles, and thereby facilitates the decoration of 1,2,3-triazolylidene NHCs with functional wingtip substituents.26,43–45The introduction of carbohydrate substituents on the triazolylidene scaffold is particularly attractive as this approach introduces functional groups in close proximity to the metal active site. Such cooperation of ligand sites and the metal center has been demonstrated in so-called bifunctional catalysts, as introduced elegantly with Noyori-type catalysts containing an amide functionality,46–48 and Shvo's catalyst featuring a proximal oxygen functionality.49,50 Bifunctional NHC ligands have shown promise in a range of catalytic transformations including hydrogenations, giving rise to increased catalytic activities when compared to more classical analogs.13,17,23,51–55
The use of carbohydrate motifs in homogenous transition metal catalysts has attracted considerable attention, particularly for introducing chirality and water solubility. Several complexes with carbohydrate-based phosphine and phosphinite ligand scaffolds have shown excellent performance in asymmetric catalysis.56–58 Similar work with N-heterocyclic carbene ligands, however, is much more scarce.59,60 In pioneering work, Dyson and co-workers investigated the anticancer activity of Ru complexes of carbohydrate-functionalized NHC ligands,60,61 though only few examples have explored the catalytic activity of such carbohydrate–NHC hybrid complexes.8,22,62 Imidazolylidene systems functionalized with two carbohydrate wingtip groups, for example, induced up to 60% ee in asymmetric Rh(I)-catalyzed hydrosilylation of ketones.62 We have demonstrated that deprotected carbohydrate substituents in NHC–Ir(III) complexes are beneficial for base-free alcohol and amine oxidation.22 Based on these results, we became interested in exploiting this ligand design motif for transfer hydrogenation.
Herein we report three novel Ru–triazolylidene complexes that incorporate carbohydrate functionality, including their photophysical and electrochemical properties as well as their catalytic activity in transfer hydrogenation of ketones, which revealed that complexes are efficiently deprotected in situ under the basic catalytic conditions.
Results and discussion
Synthesis and characterization
Triazole precursors 1 were synthesized in moderate yields by the CuAAC reaction from 1-hexyne and acetyl-protected anomeric azide derivatives of glucose and galactose (Scheme 1).63 HRMS analysis (ESI+) confirmed formation of 1Glc and 1Gal by characteristic signals at m/z = 456.1985 and 456.1980, respectively (m/z = 456.1977 calculated for [M + H]+). Also, 1H NMR analysis showed the triazolyl CH resonance for both compounds 1Glc and 1Gal at 7.5 ppm. Importantly, only a single anomeric proton resonance was observed for each triazole as a doublet at δH 5.85 and 5.81, respectively, each with a coupling constant of 9 Hz, which is consistent with stereoselective formation of β-anomers of the monosaccharide moiety. The remaining carbohydrate CH resonances differed between the glucose and galactose derivative, and the acetyl protecting groups appeared as four singlets between 1.7 and 1.3 ppm. | ||
| Scheme 1 Synthesis of complexes 5Glc, 5Gal and 6. | ||
Glycosidation of peracetylated glucopyranose with 1-bromoethanol and subsequent SN2 reaction with NaN3 according to literature procedures,64,65 yielded 1-azidoethyl-2,3,4,6-tetraacetylglucopyranoside, containing an ethylene spacer between the carbohydrate and azide functional groups. CuAAC of this azide with 1-hexyne (Scheme 1) yielded triazole 2, which gave a signal in HRMS analysis at m/z = 500.2236, corresponding to [M + H]+ (calculated m/z = 500.2239). The triazolyl CH resonance appeared in the 1H NMR spectrum at 7.21 ppm and the anomeric proton resonance was much less deshielded than in the spectra of 1, appearing as a doublet at 4.40 ppm, which overlaps with a multiplet from the ethylene spacer. The anomeric coupling constant of 7.9 Hz is again consistent with a β-conformation.
Carbohydrate-triazole compounds 1 and 2 were alkylated with Meerwein's reagent and isolated by precipitation from methanol to afford triazolium salts 3Glc, 3Gal and 4 in high to quantitative yields. A diagnostic downfield shift of ca. 1 ppm of the triazole CH resonance was observed upon alkylation (δH = 8.56, 8.50 and 8.17, respectively) along with a new singlet corresponding to the triazolium N-methyl group. The anomeric proton resonances also shifted 0.2–0.4 ppm downfield. HRMS analysis revealed a M+ ion [M − BF4]+ that is 14 amu higher than those of the corresponding triazole precursor [M + H]+ ions, indicative of successful methylation. These salts were used as ligand precursors without further purification.
Ruthenium(II) triazolylidene arene complexes 5 and 6 were synthesized from these triazolium salts via the well-established25,66 transmetalation procedure using Ag2O and [RuCl2(p-cym)]2. The silver(I) carbene intermediate was not isolated, but its formation was monitored by disappearance of the triazolium CH resonance in the 1H NMR spectrum and by HRMS analysis (m/z = 1045.3148 for [Ag(3Glc − H)2]+). Isolation of the transmetalated ruthenium(II) complexes by flash chromatography provided microanalytically pure complexes 5 in good yields (60–80%), yet 6 in a only moderate 30% yield. Successful ruthenation was confirmed by HRMS analysis, showing signals for the [M − Cl]+ ion at m/z = 740.1885, 740.1886 and 784.2153, for 5Glc, 5Gal and 6, respectively. Complex formation was further supported via NMR analysis by the absence of the downfield 1H resonance, corresponding to a triazolium CH group in the ligand precursor, and a characteristic14 carbenic 13C resonance at 167 ppm (5) or 163 ppm (6). The anomeric proton resonances in the spectra of 5 were further deshielded by 0.5–0.7 ppm when compared to the triazolium precursors and significantly broadened, appearing at δH 6.76 and 6.86 for 5Glc and 5Gal, respectively. This downfield shift indicates electronic perturbation at the anomeric position upon complexation when the carbohydrate is directly bound to the 1,2,3-triazolylidene ligand. In contrast, the shift of the anomeric resonance upon ruthenation is negligible for 6, which contains an ethylene spacer between the triazolylidene heterocycle and the carbohydrate unit. Moreover, complex 6 features two doublet resonances in the aromatic region due to the p-cymene ligand in the 1H NMR spectrum, suggesting Cs symmetry of the complex. In contrast, the p-cymene ligand is dissymmetric in complexes 5Glc and 5Gal revealing four distinct resonances for the aromatic protons and two non-identical isopropyl CH3 doublets at ca. 1.3 ppm for each complex. This splitting indicates restricted rotation about the Ru–CNHC bond and hence a more rigid second coordination sphere imparted by the directly linked carbohydrate units.
The photophysical and electrochemical properties of 5Glc, 5Gal and 6 in MeCN solution were probed. UV-vis absorption spectra of both the new carbohydrate-derived complexes 5 show a strong absorption band at λmax = 282 nm (ε 5700 M−1 cm−1) as well as a shoulder at 236 nm, both tentatively attributed to ligand-centered n–π*, π–π* transitions (Fig. 1a). In the spectrum of 6 an absorbance band at λmax = 272 nm (ε 4500 M−1 cm−1) was observed. Additionally, all complexes feature a broad and weak charge transfer band between 300 and 500 nm, giving rise to the yellow-orange colour of the complexes.
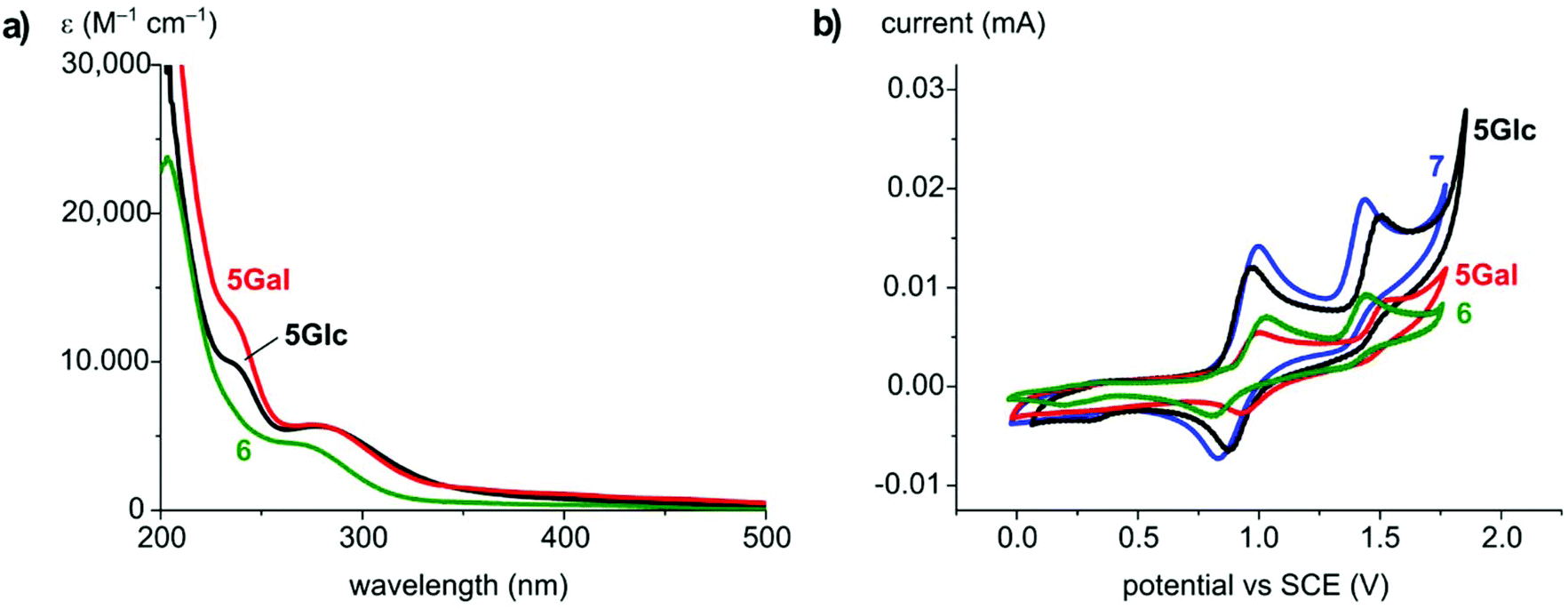 | ||
| Fig. 1 (a) UV-Vis absorption spectra of 5Glc, 5Gal and 6 at room temperature in MeCN; and (b) cyclic voltammetry of 5Glc, 5Gal, 6, and 7 in MeCN solution, scan rate 100 mV s−1, [NBu4]PF6 as the supporting electrolyte (0.1 mM), complexes at 0.5 mM except 5Gal (0.25 mM). | ||
Electrochemical analysis using cyclic voltammetry showed two oxidations for complex 5Glc, a quasi-reversible and presumably metal-based Ru2+/3+ process at E1/2 = +0.91 V vs. SCE and an irreversible oxidation at Epa = +1.50 V (Fig. 1b, Table 1). 5Gal and 6 showed very similar behavior with only slight shifts of the oxidation potentials. These redox features are reminiscent to those of the known14,67 ruthenium complex [RuCl2(cym)(trzBuBu)] 7 containing two n-butyl wingtip groups as triazolylidene substituents (E1/2 = 0.91 V, Epa = 1.4 V vs. SCE), indicating that the carbohydrate unit has no significant influence on the electron density at the ruthenium center, and that this unit is not redox active at these potentials.
| Complex | λ max (nm) [ε (M−1 cm−1)] | E 1/2 (V) [ΔEp (mV)]b | E pa (V) |
|---|---|---|---|
| a All measurements in MeCN, sh = shoulder. b All potentials vs. SCE using Fc+/Fc as the internal standard (E1/2 = +0.40 V, ΔEp = 72 mV), scan rate 100 mV s−1, [NBu4]PF6 as the supporting electrolyte (0.1 mM). c Peak potential of irreversible oxidation. | |||
| 5Glc | 282 [5700], 236 (sh) [10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000] 000] |
0.91 [89] | 1.50 |
| 5Gal | 282 [5700], 236 (sh) [13000] |
0.96 [68] | 1.52 |
| 6 | 272 [4500] | 0.92 [226] | 1.45 |
| 7 | 272 [4000] | 0.91 [178] | 1.44 |
In situ deprotection of 5Glc
Deprotection of the carbohydrate unit in complexes 5 and 6 is essential for exploiting the potential of the carbohydrate wingtip group for reductive catalytic processes and for promoting potentially bifunctional interactions involving the carbohydrate hydroxyl groups and the metal center. Such deprotection has been achieved only in a few specific cases.59,68,69 Recent results from our laboratory have demonstrated the deacetylation of carbohydrate wingtip groups of iridium complexes in methanolic HCl.22 The ruthenium complexes 5 and 6, however, were not stable under these conditions and although deprotection was observed, simultaneous formation of significant amounts of free triazolium salt due to metal dissociation occurred. Acid-lability of Ru–triazolylidene bonds has been established by Grubbs, Bertrand, and co-workers in olefin methathesis catalysis.7 The Ru triazolylidene complexes were also not stable to conventional Zemplén deprotection conditions using methanolic NaOMe.70 However, stability tests of complex 5Glc under typical transfer hydrogenation conditions, i.e. KOH in iPrOH (20 mM) revealed rapid deprotection, which was accompanied by a color change of the reaction solution from orange to yellow (Scheme 2). Deprotection and formation of 8Glc was also accomplished in D2O using 10 equiv. KOH and was confirmed by a HRMS (ESI+) signal at m/z = 536.1684, corresponding to [M − 2Cl − H]+. Moreover, 1H NMR analysis in D2O showed the coalescence of the four distinct acetate signals of 5Glc into a single resonance consistent with the formation of KOAc (see ESI, Fig. S19†). In addition, the C1–C5 carbohydrate ring proton resonances are shifted upfield upon deprotection. The asymmetry of the p-cymene ligand is retained, as indicated by two distinct isopropyl methyl signals at 1.13 and 1.29 ppm. Analogous results were observed for 5Gal, suggesting wider applicability of this method. | ||
| Scheme 2 Synthesis of 8Glc by base-mediated deprotection of 5Glc. | ||
The deprotected complex 8Glc was not isolated as a pure solid since it displayed instability over the course of several hours upon drying. In addition, substantial amounts of degradation products were detected by 1H NMR spectroscopy over the course of 24 h when kept in either D2O or (CD3)2CDOD solution. Consequently, the more robust protected complexes 5 were used as pre-catalysts and complexes 8 were generated by in situ deprotection in the course of transfer hydrogenation catalysis.
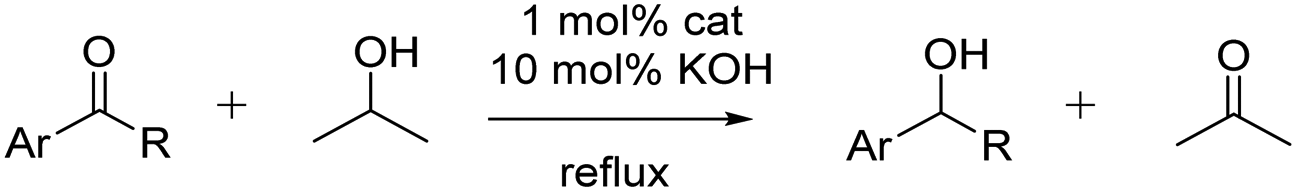
Transfer hydrogenation catalysis
To assess the suitability of the carbohydrate-functionalized carbene ruthenium complexes as pre-catalysts for base-promoted transfer hydrogenation of ketones, a model reaction with benzophenone was carried out under standard conditions,43i.e., refluxing iPrOH as hydrogen source, KOH as activator, 100:10:1 substrate/base/complex ratio. For both 8Glc and 8Gal, both generated in situ from the acetate-protected precursors 5Glc and 5Gal, respectively, the reaction proceeded to completion within 8 h, forming diphenylmethanol as the exclusive product (Table 2, entries 1 and 2). The results for both glucose- and galactose-derivatives were essentially identical, suggesting little influence of the remote carbohydrate C4 configuration and hence no relevance of a chair conformation of the pyranose ring.71 Complex 6 featuring an ethylene spacer between the triazolylidene and carbohydrate units showed decreased activity as pre-catalyst for the model reaction, when compared to those complexes with the carbohydrate directly linked to the triazolylidene, reaching only 57% within 8 h and incomplete conversion even after 24 h (84%, entry 3). In contrast, the unfunctionalized complex 7 generates a considerably faster catalyst and reaches completion in less than 2 h (TOF50 = 310 h−1). These differences point to a direct catalytic relevance of the carbohydrate functionality, either through chelation or through stabilization of substrates or intermediates within the catalytic cycle. The location of the carbohydrate is critical and proximal oxo functionalities are less inhibiting than the more remote functional group in complex 6.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex | Substrate | Conversionb (%) | TOF50c (h−1) | σ p | |
| 8 h | 24 h | |||||
| a General reaction conditions: Ru complex (0.01 mmol), substrate (1.0 mmol), KOH (0.05 mL, 2 M, 0.1 mmol), 2-propanol (5 mL) at reflux. b Determined by 1H NMR analysis. c TOF50 (mol product)/(mol pre-catalyst) at 50% conversion. d Hammett parameter of aryl para-substituent. e Elemental Hg (ca. 1 mmol) added after 2 h. | ||||||
| 1 | 5Glc |
|
98 | >98 | 20 | 0 |
| 2 | 5Gal | >98 | >98 | 22 | 0 | |
| 3 | 6 | 57 | 84 | 9 | 0 | |
| 4 | 7 | >98 | >98 | 310 | 0 | |
| 5 | 5Glc | 62 | >98 | 8 | 0 | |
| 6 | 5Glc |
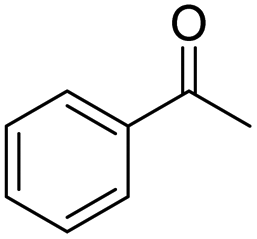
|
66 | 98 | 10 | 0 |
| 7 | 5Glc |
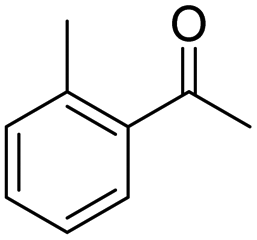
|
74 | 98 | 13 | — |
| 8 | 5Glc |
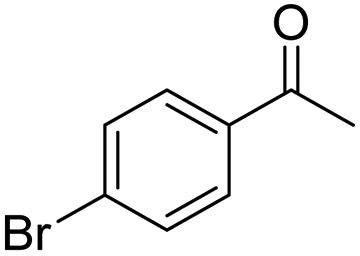
|
56 | 75 | 8 | +0.23 |
| 9 | 5Glc |
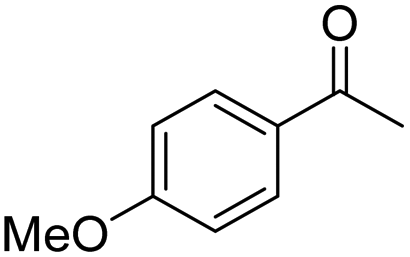
|
51 | 77 | 7 | −0.27 |
| 10 | 5Glc |
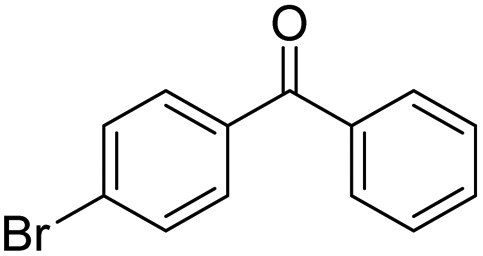
|
62 | 74 | 11 | +0.23 |
| 11 | 5Glc |
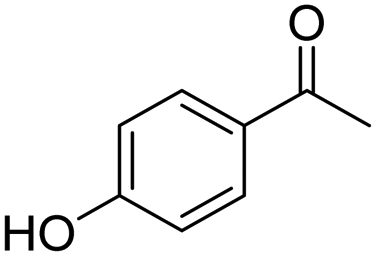
|
10 | 16 | — | −0.36 |
| 12 | 5Glc |
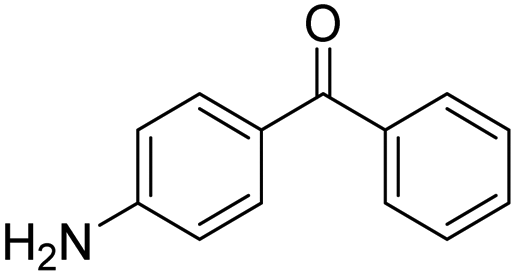
|
15 | 31 | — | −0.66 |
| 13 | 5Glc |
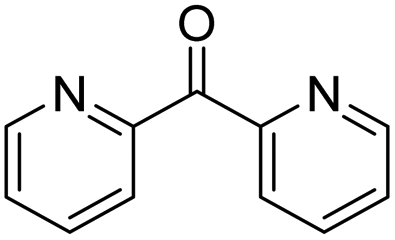
|
97 | >98 | 60 | — |
Transfer hydrogenation of benzophenone by 5Glc was not impeded by elemental mercury. Thus, addition of Hg (0.2 g, ca. 1 mmol, 100 molequiv relative to Ru) after 2 h when the reaction has reached 34% substrate conversion proceeded to full conversion (entry 5). The initial decrease of activity was attributed to mass transfer limitations induced by the mercury drop. The preservation of catalytic activity supports a homogeneous catalyst as active species rather than significant dissociation of the triazolylidene ligand and formation of a heterogeneous layer as catalytically active phase.
Since the triazolylidene ligand is robustly coordinated to the ruthenium center, transfer hydrogenation allows for evaluating the asymmetric induction of the stereochemically well-defined carbohydrate functionality by using prochiral ketones. Similar carbohydrate-based ligands have previously been shown to be effective in asymmetric catalysis,58,62,72–75 including hydrosilylation of ketones.62 The enantioselectivity of 5Glc was probed with acetophenone as prochiral substrate for transfer hydrogenation (entry 6). The reaction proceeded at a lower rate than with benzophenone (TOF50 = 10 vs. 20 h−1), reaching 66% conversion after 8 h and full conversion after 24 h. Product analysis by chiral gas chromatography revealed negligible enantioselectivity (<2%, see ESI, Fig. S28†), indicating that chiral information from the carbohydrate wingtip group is not transferred to the substrate under these conditions. Modification of the chiral pocket and enhanced enantioselectivity may be induced through variation of the carbohydrate, drawing on the vast pool of pyranose and ribose structures present in nature.
In order to investigate electronic and steric influences on substrate reactivity as well as functional group tolerance, a small substrate scope was carried out with derivatives of benzophenone and acetophenone using 5Glc as pre-catalyst (entries 7–13). Using sterically more demanding 2-methylacetophenone as substrate in place of acetophenone led to a modest increase in activity (TOF50 = 13, entry 7). Very little variation of activity was noted when using electronically distinct substrates. For example, conversion of electron-withdrawing 4-bromoacetophenone (σp = +0.23, TOF50 = 8 h−1) and electron-donating 4-methoxyacetophenone (σp = −0.27, TOF50 = 7 h−1) was essentially identical (entries 8 and 9). Notably, neither substrate achieved full conversion after 24 h and thus proved less suitable than unsubstituted acetophenone. 4-Bromobenzophenone showed similar activity (TOF50 = 11, entry 10), implying a markedly decreased performance with respect to the unsubstituted benzophenone. Under the same conditions, benzaldehyde was not converted to the corresponding alcohol. Alcohol and amine substituents markedly inhibited catalytic activity and ketones with these functional groups reached conversions of only 10 and 15%, respectively after 8 h (entries 11 and 12). In contrast, di(2-pyridyl)ketone was converted much faster than benzophenone (TOF50 = 60 vs. 20 h−1, entry 13). This substrate scan suggests a few features of the catalytic transfer hydrogenation induced by 8Glc. First, the lack of influence of electronic substituent effects on the turnover frequency indicates that the rate-limiting step is not associated with substrate conversion. Moreover, the decrease of the rate for the conversion of substituted substrates, in particular in para position, hints to a steric bias and the requirement of sufficient space around the metal center for turnover to proceed. This observation renders β-hydrogen elimination from coordinated isopropoxide unlikely as rate-limiting step, as this step would be substrate-independent and lead to equal rates. Instead, these data suggest instead that substrate coordination is rate-limiting, a step that is obviously affected by the steric demand and the potential (hydrogen bond) interactions of the carbohydrate with the substrate. Substrate coordination may also involve decoordination of potentially chelating carbohydrate hydroxy groups from the ruthenium center. This decoordination is accelerated with soft pyridyl coordinating groups (entry 13), which may rationalize the enhanced conversion rate observed with this substrate. In contrast, phenol and aniline functional groups are presumably too acidic and deprotonated under the basic reaction conditions, leading to a strongly coordinating ligand which is even more difficult to be displaced by the carbonyl group for catalytic turnover (entries 11 and 12).
Conclusions
We have successfully synthesized and characterized new carbohydrate–NHC Ru complexes. Deprotection of acetylated carbohydrate unit on the ruthenium complex was achieved in situ under basic conditions in protic solvents without affecting the Ru–triazolylidene bond and affording the first example of an unprotected carbohydrate–NHC system with Ru. This method provides access to hydroxy-functionalized carbene complexes, even though their isolation is compromised by stability issues. The carbohydrate functionality has a profound impact on catalytic activity in transfer hydrogenation of ketones. Directly triazolylidene-bound glucose substantially enhances turnover rates compared to more remote carbohydrate functionalization, though it reduces activity compared to unfunctionalized carbene complexes. This prominent role of the carbohydrate substituent provides opportunities for further catalyst engineering towards other transformations involving hydrogen transfer such as oxidations or hydrogen borrowing processes, as well as by modulation of the carbohydrate entity using other pyranoses or furanoses to exploit their beneficial properties in catalysis.Experimental section
General
Unless otherwise stated, all reagents were obtained from commercial suppliers and used as received. Carbohydrate azide precursors were prepared according to modified literature procedures.63–65 Ag2O was used after regeneration by heating to >160 °C under vacuum. Dry, degassed solvents were obtained by filtration over columns of dried aluminium oxide under a positive pressure of argon. NMR spectra were recorded on Bruker spectrometer operating at room temperature. Chemical shifts (δ in ppm, J in Hz) were referenced to residual solvent resonances and are reported downfield from SiMe4. High resolution mass spectrometry and elemental analysis were performed by the Analytical Research Services at University of Bern.General synthesis of triazoles
Acetylated pyranosyl azide (1.000 g, 2.68 mmol) and 1-hexyne (0.31 mL, 2.7 mmol) were reacted in the presence of CuSO4·5H2O (0.270 g, 1.07) and sodium ascorbate (0.530 g, 2.68 mmol) in tert-butanol–water mixture (1:1, 20 mL), at room temperature for 3 days. The reaction mixture was diluted with EDTA (20 mL, 0.2 M in 1.0 M NH4OH(aq) solution) and extracted into CH2Cl2 (3 × 20 mL). The organic layers were combined, washed with brine, dried over MgSO4 and filtered through Celite, yielding a white solid (1) or yellow oil (2).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). FT-IR (ATR, cm−1): 3070 (w), 2928 (w), 2357, 1746 (s, CO), 1436, 1366, 1216 (s), 1093, 1041 (s), 928.
O); FT-IR (ATR, cm−1): 2958 (w), 2929 (w), 1743 (s, CO), 1369, 1218 (s), 1044 (s), 923.
:5) was used to obtain the product as an off-white hygroscopic solid. HRMS (m/z) (ESI+): Calculated for C22H34N3O10+m/z = 500.2239 [M + H]+; found m/z = 500.2236. 1H NMR (300 MHz, CDCl3): δ = 0.92 (t, 3H, 3JH,H = 7.3 Hz, butyl CH3), 1.31–1.48 (m, 2H, butyl CH2), 1.57–1.72 (m, 2H, butyl CH2), 1.93, 1.98, 2.00, 2.07 (4 × s, 3H, C(O)CH3), 2.56–2.80 (m, 2H, butyl CH2), 3.68 (ddd, 1H, 3JH,H = 10.0, 4.8, 2.4 Hz, glucosyl C5H), 3.90 (ddd, 1H, 2JH,H = 10.4 Hz, 3JH,H = 8.5, 3.4 Hz, ethylene CHH), 4.06–4.29 (m, 3H, ethylene CHH + glucosyl C6H2), 4.39–4.59 (m, 2H, ethylene CH2), 4.46 (d, 1H, 3JH,H = 7.9 Hz, glucosyl C1H), 4.98 (dd, 1H, 3JH,H = 9.5, 7.9 Hz, glucosyl C2H), 5.05 (dd, 1H, 3JH,H = 10.0, 9.5 Hz, glucosyl C4H), 5.16 (t, 1H, 3JH,H = 9.5 Hz, glucosyl C3H), 7.31 (s, 1H, CtrzH). 13C{1H} NMR (75 MHz, CDCl3): δ = 13.8 (butyl CH3), 20.5, 20.5, 20.7, 20.7, (4 × C(O)CH3), 22.3, 25.4, 31.5 (3 × butyl CH2), 49.8 (CH2), 61.8 (CH2), 68.0 (CH2), 68.3, 71.0, 72.0, 72.5 (glucosyl C2–5H, 100.6 (glucosyl C1H), 122.0 (CtrzH), 148.3 (Ctrz–Bu), 169.2, 169.4, 170.1, 170.5 (4 × CO); FT-IR (ATR, cm−1): 2935 (w), 1744 (s, CO), 1432, 1365, 1211 (s), 1032 (s), 907.
O). FT-IR (ATR, cm−1): 3070 (w), 2928 (w), 2357, 1746 (s, CO), 1436, 1366, 1216 (s), 1093, 1041 (s), 928.
O); FT-IR (ATR, cm−1): 2958 (w), 2929 (w), 1743 (s, CO), 1369, 1218 (s), 1044 (s), 923.
:5) was used to obtain the product as an off-white hygroscopic solid. HRMS (m/z) (ESI+): Calculated for C22H34N3O10+m/z = 500.2239 [M + H]+; found m/z = 500.2236. 1H NMR (300 MHz, CDCl3): δ = 0.92 (t, 3H, 3JH,H = 7.3 Hz, butyl CH3), 1.31–1.48 (m, 2H, butyl CH2), 1.57–1.72 (m, 2H, butyl CH2), 1.93, 1.98, 2.00, 2.07 (4 × s, 3H, C(O)CH3), 2.56–2.80 (m, 2H, butyl CH2), 3.68 (ddd, 1H, 3JH,H = 10.0, 4.8, 2.4 Hz, glucosyl C5H), 3.90 (ddd, 1H, 2JH,H = 10.4 Hz, 3JH,H = 8.5, 3.4 Hz, ethylene CHH), 4.06–4.29 (m, 3H, ethylene CHH + glucosyl C6H2), 4.39–4.59 (m, 2H, ethylene CH2), 4.46 (d, 1H, 3JH,H = 7.9 Hz, glucosyl C1H), 4.98 (dd, 1H, 3JH,H = 9.5, 7.9 Hz, glucosyl C2H), 5.05 (dd, 1H, 3JH,H = 10.0, 9.5 Hz, glucosyl C4H), 5.16 (t, 1H, 3JH,H = 9.5 Hz, glucosyl C3H), 7.31 (s, 1H, CtrzH). 13C{1H} NMR (75 MHz, CDCl3): δ = 13.8 (butyl CH3), 20.5, 20.5, 20.7, 20.7, (4 × C(O)CH3), 22.3, 25.4, 31.5 (3 × butyl CH2), 49.8 (CH2), 61.8 (CH2), 68.0 (CH2), 68.3, 71.0, 72.0, 72.5 (glucosyl C2–5H, 100.6 (glucosyl C1H), 122.0 (CtrzH), 148.3 (Ctrz–Bu), 169.2, 169.4, 170.1, 170.5 (4 × CO); FT-IR (ATR, cm−1): 2935 (w), 1744 (s, CO), 1432, 1365, 1211 (s), 1032 (s), 907.
General synthesis of triazolium salts
The triazole compound (0.44 mmol) and Me3OBF4 (0.067 g, 0.45 mmol) were dissolved in dry CH2Cl2 (30 mL) and stirred at room temperature for 16 h. Then MeOH (0.5 mL) was added and stirring continued for 30 min. All volatiles were evaporated under reduced pressure. The residue was dissolved in a minimum of MeOH and stored at −20 °C, yielding the triazolium salt as a white solid. 3Gal and 4 were hygroscopic and were therefore used without further purification.O); FT-IR (ATR, cm−1): 2968 (w), 2359, 1747 (s, CO), 1434, 1371 (s), 1215 (s), 1024 (br, s, BF4), 927.
O); FT-IR (ATR, cm−1): 3121 (w), 2962 (w), 1745 (s, CO), 1370, 1210 (s), 1034 (s, br, BF4), 922, 496.
O); FT-IR (ATR, cm−1): 2959 (w), 2848(w), 1744 (s, CO), 1435, 1369, 1218 (s), 1034 (s, br, BF4), 908.
General synthesis of Ru(II) complexes
The triazolium salt (0.43 mmol), Ag2O (0.060 g, 0.26 mmol) and Me4NCl (0.057 g, 0.52 mmol) were suspended in dry CH3CN (50 mL) and stirred in the absence of light at room temperature for 5 h. The reaction mixture was diluted with CH2Cl2 (25 mL), filtered through Celite and concentrated under reduced pressure. CH2Cl2 (20 mL) and [Ru(p-cym)Cl2]2 (0.099 g, 0.16 mmol) were added and the mixture was stirred for 3 h. The reaction mixture was cooled in an ice bath, filtered through Celite, and evaporated to dryness. The red residue was purified by gradient flash chromatography (SiO2; CH2Cl2 to CH2Cl2/acetone 9:1).
Ru complex 5Glc
From 3Glc (0.240 g) according to the general procedure, 5Glc was obtained (0.189 g, 80%). Anal. calcd for C31H45N3O9RuCl2 (775.683 g mol−1): C 48.00, H 5.85, N 5.42%; found C 48.06, H 5.39, N 5.84%. HRMS (m/z) (ESI+): Calculated for C31H45N3O9RuCl+m/z = 740.1882 [M − Cl]+; found m/z = 740.1885. 1H NMR (400 MHz, CDCl3): δ = 0.95 (t, 3H, 3JH,H = 7.3 Hz, butyl CH3), 1.32 (d, 3H, 3JH,H = 6.9 Hz, CHCH3), 1.34 (d, 3H, 3JH,H = 6.9 Hz, CHCH3), 1.46 (quintet, 2H, 3JH,H = 7.3 Hz, butyl CH2), 1.54–1.64 (br s, 2H, butyl CH2), 1.93 (s, 3H, C(O)CH3), 1.97 (s, 3H, cym–CH3), 2.02, 2.04, 2.06 (3 × s, 3H, C(O)CH3), 2.88–3.08 (m, 3H, butyl CH2 + CHMe2), 3.97–4.09 (m, 4H, NCH3 + glucosyl C5H), 4.12–4.27 (m, 2H, glucosyl C6H2), 5.00 (d, 1H, 3JH,H = 5.8 Hz, CcymH), 5.14–5.26 (m, 2H, CcymH + glucosyl C4H), 5.30–5.43 (m, 2H, CcymH + glucosyl C3H), 5.53 (d, 1H, 3JH,H = 5.8 Hz, CcymH), 6.01 (t, 1H, 3JH,H = 9.4 Hz, glucosyl C2H), 6.76 (br s, 1H, glucosyl C1H). 13C{1H} NMR (100 MHz, CDCl3): δ = 14.0 (butyl CH3), 18.8 (cym–CH3), 20.71, 20.74, 20.97, 21.12 (4 × C(O)CH3), 22.7, 23.0 (2 × CH–CH3), 23.2 (butyl CH2), 26.3 (butyl CH2), 30.8 (CHMe2), 31.8 (butyl CH2), 37.3 (NCH3), 62.3 (glucosyl C6H2), 68.3 (glucosyl C4H), 70.1 (glucosyl C2H), 74.1 (glucosyl C3H), 74.5 (glucosyl C5H), 81.1, 81.9, 86.3, 86.8 (4 × CcymH), 87.1 (glucosyl C1H), 97.6 (Ccym), 108.3 (Ccym), 148.0 (Ctrz–Bu), 167.1 (Ctrz–Ru), 169.2, 169.6, 170.1, 170.5 (4 × CO); FT-IR (ATR, cm−1): 2957 (w), 1747 (s, CO), 1433, 1365, 1212 (s), 1032, 922.
Ru complex 5Gal
From 3Gal (0.223 g) according to the general procedure, 5Gal was obtained (0.138 g, 60%). Anal. calcd for C31H45N3O9RuCl2 (775.683 g mol−1), C 48.00, H 5.85, N 5.42%; found C 47.37, H 5.65, N 5.00%. HRMS (m/z) (ESI+): Calculated for C31H45N3O9RuCl+m/z = 740.1882 [M − Cl]+; found m/z = 740.1886. 1H NMR (400 MHz, CDCl3): δ = 0.95 (t, 3H, 3JH,H = 7.2 Hz, butyl CH3), 1.30, 1.33 (2 × d, 3H, 3JH,H = 6.9 Hz, CHCH3), 1.40–1.55 (m, 2H, butyl CH2), 1.60–1.90 (br s, 2H, butyl CH2), 1.92 (s, 3H, C(O)CH3), 1.97 (s, 3H, cym–CH3), 1.99, 2.02, 2.23 (3 × s, 3H, C(O)CH3), 2.85–3.14 (m, 3H, butyl CH2 + CHMe2), 4.08 (s, 3H, NCH3), 4.11–4.28 (m, 3H, galactosyl C5H + C6H2), 4.97 (d, 1H, 3JH,H = 5.8 Hz, CcymH), 5.15–5.26 (m, 2H, CcymH + galactosyl CH), 5.37 (d, 1H, 3JH,H = 5.8 Hz, CcymH), 5.46–5.63 (m, 2H, CcymH + galactosyl CH), 6.17 (t, 1H, 3JH,H = 9.7 Hz, galactosyl C2H), 6.86 (br s, 1H, galactosyl C1H). 13C{1H} NMR (100 MHz, CDCl3): δ = 14.0 (butyl CH3), 18.8 (cym–CH3), 20.6, 20.90, 20.92, 21.26 (4 × C(O)CH3), 22.4 (CHCH3), 23.2 (butyl CH2), 26.3 (CHMe2), 31.1 (butyl CH2), 31.8 (butyl CH2), 37.2 (NCH3), 62.1 (galactosyl C6H2), 67.5 (galactosyl C2H), 68.4 (galactosyl C3H), 72.1 (galactosyl C4H), 73.7 (galactosyl C5H), 80.7, 81.9, 86.0, 87.0 (4 × CcymH), 87.6 (galactosyl C1H), 98.1 (Ccym), 108.3 (Ccym), 148.0 (Ctrz–Bu), 167.1 (Ctrz–Ru), 169.5, 169.9, 170.27, 170.28 (4 × CO); FT-IR (ATR, cm−1): 2956 (w), 1748 (s, CO), 1367, 1216 (s), 1051, 921.
Ru complex 6
Reaction of 4 (0.220 g) according to the general procedure afforded complex 6 (0.096 g, 30%). Anal. calcd for C33H49N3O10RuCl2·(H2O) (837.751 g mol−1), C 47.31, H 6.14, N 5.02%; found C 47.49, H 5.89, N 4.70%. HRMS (m/z) (ESI+): Calculated for C33H49N3O10RuCl+m/z = 784.2144 [M − Cl]+; found m/z = 784.2153. 1H NMR (400 MHz, CDCl3): δ = 0.96 (t, 3H, 3JH,H = 7.3 Hz, butyl CH3), 1.30 (d, 6H, 3JH,H = 6.9 Hz, CHCH3), 1.46 (m, 2H, butyl CH2), 1.55–1.70 (br s, 2H, butyl CH2), 1.98 (s, 3H, C(O)CH3), 1.99–2.04 (m, 9H, cym–CH3 + 2 × C(O)CH3), 2.07 (s, 3H, C(O)CH3), 2.86–3.09 (m, 3H, butyl CH2 + CHMe2), 3.75 (ddd, 1H, 3JH,H = 10.1, 4.6, 2.4 Hz, glucosyl C5H), 3.98 (s, 3H, NCH3), 4.08–4.19 (m, 2H, ethylene CHH + glucosyl C6HH), 4.22–4.38 (m, 2H, ethylene CHH + glucosyl C6HH), 4.65 (d, 1H, 3JH,H = 8.0 Hz, glucosyl C1H), 4.83 (dtd, 2H, 2JH,H = 14.0, 3JH,H = 8.0, 5.8 Hz, ethylene CH2), 4.95 (dd, 1H, 3JH,H = 9.5, 8.0 Hz, glucosyl C2H), 5.07–5.14 (m, 3H, glucosyl C4H + CcymH), 5.20 (t, 1H, 3JH,H = 9.5 Hz, glucosyl C3H), 5.37 (dd, 1H, 3JH,H = 5.8, 1.3 Hz, CcymH), 5.40 (dd, 1H, 3JH,H = 5.8, 1.3 Hz, CcymH). 13C{1H} NMR (100 MHz, CDCl3): δ = 13.9 (butyl CH3), 18.5 (cym–CH3), 20.59, 20.61, 20.8, 22.5 (4 × C(O)CH3), 22.8 (CHCH3), 23.1 (butyl CH2), 26.0 (CHMe2), 30.7 (butyl CH2), 31.8 (butyl CH2), 36.4 (NCH3), 53.8 (ethylene CH2), 61.9 (glucosyl C6H2), 68.3 (glucosyl CH), 68.7 (ethylene CH2), 71.4, 71.9, 72.9 (3 × glucosyl CH), 82.4, 82.9, 85.1, 85.2 (4 × CcymH), 96.7 (Ccym), 100.5 (glucosyl C1H), 107.4 (Ccym), 147.6 (Ctrz–Bu), 163.0 (Ctrz–Ru), 169.2, 169.5, 170.1, 170.6 (4 × CO).
In situ deprotection of 5Glc
Complex 5Glc (4 mg, 5 μmol) was dissolved in D2O (0.5 mL) and KOH (0.2 M in D2O, 0.05 mL, 0.05 mmol) was added, which induces an immediate color change from orange to yellow. HRMS (m/z) (ESI+): Calculated for C23H36N3O5Ru+m/z = 536.1704 [M − 2Cl − H]+; found m/z = 536.1684. 1H NMR (300 MHz, D2O): δ = 1.13 (d, 3H, 3JH,H = 6.8 Hz, CHCH3), 1.21 (t, 3H, 3JH,H = 6.9 Hz, butyl CH3), 1.29 (d, 3H, 3JH,H = 6.8 Hz, CHCH3), 1.61–2.10 (m, 4H, butyl CH2), 2.08 (s, 12H, CH3COOK) 2.24 (s, 1H, cym–CH3), 2.52–2.72 (m, 1H, CHMe2), 2.95–3.20 (m, 2H, butyl CH2), 3.47–3.60 (m, 1H, glucosyl CH), 3.60–3.76 (m, 2H, glucosyl CH), 3.76–4.03 (m, 2H, glucosyl CH), 4.15 (d, 1H, J = 10.7 Hz, glucosyl C6H), 4.35 (s, 3H, NCH3), 5.04–5.13 (m, 1H, CcymH), 5.23 (d, 1H J = 8.2 Hz, glucosyl C1H), 5.45 (d, 1H, 3JH,H = 6.1 Hz, CcymH), 5.77 (d, 1H, 3JH,H = 5.7 Hz, CcymH), 5.82 (d, 1H, 3JH,H = 5.7 Hz, CcymH).General procedure for transfer hydrogenation catalysis
The Ru complex (0.01 mmol) was dissolved in iPrOH (5 mL) and aqueous KOH was added (2 M, 0.05 mL, 0.10 mmol). Hexamethylbenzene was added as an internal standard. The solution was heated to reflux for 20 min, then the ketone (1.00 mmol) was added and stirring at reflux was continued. Aliquots of the reaction mixture were sampled at given times and analyzed by diluting into CDCl3 and measuring 1H NMR spectra to monitor reaction progress.Conflicts of interest
There are no conflicts to declare.Funding sources
We acknowledge generous support from the European Commission for a Marie Skłodowska Curie Individual Fellowship to J. P. B. (Grant 749549) and from the European Research Council (CoG 615653), as well as the Swiss-European Mobility Programme for a visiting fellowship for P. M. to Bern.References
- M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903 RSC.
- V. Dragutan, I. Dragutan, L. Delaude and A. Demonceau, Coord. Chem. Rev., 2007, 251, 765–794 CrossRef CAS.
- B. Alcaide, P. Almendros and A. Luna, Chem. Rev., 2009, 109, 3817–3858 CrossRef CAS.
- L. Schwartsburd and M. K. Whittlesey, in N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley-VCH, 2014, pp. 341–370 Search PubMed.
- L. Delaude and A. Demonceau, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Diez-Gonzalez, Royal Society of Chemistry, 2nd edn, 2017, pp. 268–301 Search PubMed.
- B. K. Keitz, J. Bouffard, G. Bertrand and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8498–8501 CrossRef CAS.
- B. K. Keitz and R. H. Grubbs, Organometallics, 2010, 29, 403–408 CrossRef CAS.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- D. A. Hey, R. M. Reich, W. Baratta and F. E. Kühn, Coord. Chem. Rev., 2018, 374, 114–132 CrossRef CAS.
- M. Poyatos, J. A. Mata, E. Falomir, R. H. Crabtree and E. Peris, Organometallics, 2003, 22, 1110–1114 CrossRef CAS.
- W. Baratta, J. Schütz, E. Herdtweck, W. A. Herrmann and P. Rigo, J. Organomet. Chem., 2005, 690, 5570–5575 CrossRef CAS.
- R. Pretorius, Z. Mazloomi and M. Albrecht, J. Organomet. Chem., 2017, 845, 196–205 CrossRef CAS.
- M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Mueller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462–4473 RSC.
- S. Enthaler, R. Jackstell, B. Hagemann, K. Junge, G. Erre and M. Beller, J. Organomet. Chem., 2006, 691, 4652–4659 CrossRef CAS.
- W. W. N. O, A. J. Lough and R. H. Morris, Organometallics, 2009, 28, 6755–6761 CrossRef CAS.
- X.-Q. Guo, Y.-N. Wang, D. Wang, L.-H. Cai, Z.-X. Chen and X.-F. Hou, Dalton Trans., 2012, 41, 14557 RSC.
- M. Hollering, M. Albrecht and F. E. Kühn, Organometallics, 2016, 35, 2980–2986 CrossRef CAS.
- A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162–1167 CrossRef CAS.
- A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588–2598 CrossRef CAS.
- B. Bagh, A. M. McKinty, A. J. Lough and D. W. Stephan, Dalton Trans., 2014, 43, 12842–12850 RSC.
- R. Pretorius, J. Olguín and M. Albrecht, Inorg. Chem., 2017, 56, 12410–12420 CrossRef CAS.
- S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 2014, 3164–3171 CrossRef CAS.
- L. Bernet, R. Lalrempuia, W. Ghattas, H. Mueller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058 RSC.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052–1072 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- J. D. Crowley and P. H. Bandeen, Dalton Trans., 2010, 39, 612–623 RSC.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530 RSC.
- J. P. Byrne, S. Blasco, A. B. Aletti, G. Hessman and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2016, 55, 8938–8943 CrossRef CAS.
- J. P. Byrne, J. A. Kitchen, J. E. O'Brien, R. D. Peacock and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 1426–1439 CrossRef CAS PubMed.
- J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325 RSC.
- X. Liu, R. B. P. Elmes and K. A. Jolliffe, Aust. J. Chem., 2017, 70, 201 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS.
- C. Bouillon, A. Meyer, S. Vidal, A. Jochum, Y. Chevolot, J. P. Cloarec, J. P. Praly, J. J. Vasseur and F. Morvan, J. Org. Chem., 2006, 71, 4700–4702 CrossRef CAS.
- M. Milne, K. Chicas, A. Li, R. Bartha and R. H. E. Hudson, Org. Biomol. Chem., 2012, 10, 287–292 RSC.
- S. Horn, C. Gandolfi and M. Albrecht, Eur. J. Inorg. Chem., 2011, 2011, 2863–2868 CrossRef CAS.
- A. Monney, G. Venkatachalam and M. Albrecht, Dalton Trans., 2011, 40, 2716 RSC.
- J. D. Crowley, A.-L. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118–1132 CrossRef CAS.
- R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS.
- T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS.
- Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS.
- W. W. N. O, A. J. Lough and R. H. Morris, Chem. Commun., 2010, 46, 8240 RSC.
- W. B. Cross, C. G. Daly, Y. Boutadla and K. Singh, Dalton Trans., 2011, 40, 9722 RSC.
- A. Bartoszewicz, R. Marcos, S. Sahoo, A. K. Inge, X. Zou and B. Martín-Matute, Chem. – Eur. J., 2012, 18, 14510–14519 CrossRef CAS.
- A. Bartoszewicz, G. González Miera, R. Marcos, P. O. Norrby and B. Martín-Matute, ACS Catal., 2015, 5, 3704–3716 CrossRef CAS.
- G. González Miera, E. Martínez-Castro and B. Martín-Matute, Organometallics, 2018, 37, 636–644 CrossRef.
- S. Woodward, M. Diéguez and O. Pàmies, Coord. Chem. Rev., 2010, 254, 2007–2030 CrossRef CAS.
- S. Castillón, C. Claver and Y. Díaz, Chem. Soc. Rev., 2005, 34, 702 RSC.
- A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2016, 14, 4008–4017 RSC.
- W. Zhao, V. Ferro and M. V. Baker, Coord. Chem. Rev., 2017, 339, 1–16 CrossRef CAS.
- K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton Trans., 2014, 43, 1443–1448 RSC.
- K. Purkait, S. Karmakar, S. Bhattacharyya, S. Chatterjee, S. K. Dey and A. Mukherjee, Dalton Trans., 2015, 44, 5969–5973 RSC.
- A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2014, 12, 9180–9183 RSC.
- H. Paulsen, Z. Györgydeák and M. Friedmann, Chem. Ber., 1974, 107, 1568–1578 CrossRef CAS.
- P. Quagliotto, G. Viscardi, C. Barolo, D. D'Angelo, E. Barni, C. Compari, E. Duce and E. Fisicaro, J. Org. Chem., 2005, 70, 9857–9866 CrossRef CAS.
- S. M. Paterson, J. Clark, K. A. Stubbs, T. V. Chirila and M. V. Baker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4312–4315 CrossRef CAS.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- D. Canseco-Gonzalez and M. Albrecht, Dalton Trans., 2013, 42, 7424 RSC.
- C. C. Yang, P. S. Lin, F. C. Liu, I. J. B. Lin, G. H. Lee and S. M. Peng, Organometallics, 2010, 29, 5959–5971 CrossRef CAS.
- Y. Imanaka, H. Hashimoto, I. Kinoshita and T. Nishioka, Chem. Lett., 2014, 43, 687–689 CrossRef CAS.
- B. Ren, M. Wang, J. Liu, J. Ge, X. Zhang and H. Dong, Green Chem., 2015, 17, 1390–1394 RSC.
- J. J. Topczewski, P. J. Cabrera, N. I. Saper and M. S. Sanford, Nature, 2016, 531, 220–224 CrossRef CAS.
- M. T. Reetz and T. Neugebauer, Angew. Chem., Int. Ed., 1999, 38, 179–181 CrossRef CAS.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J. P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213–7218 CrossRef CAS.
- J. F. Moya, C. Rosales, I. Fernández and N. Khiar, Org. Biomol. Chem., 2017, 15, 5772–5780 RSC.
- M. Coll, O. Pàmies and M. Diéguez, Adv. Synth. Catal., 2014, 356, 2293–2302 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of carbohydrate azide synthesis; 1H and 13C spectra of new compounds and HSQC spectra of deprotected complex; catalytic data; chiral gas chromatogram of acetophenone transfer hydrogenation product. See DOI: 10.1039/c9dt02614b |
| This journal is © The Royal Society of Chemistry 2019 |