
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, structure and electrochemical characterization of an isopolytungstate (W4O16) held by MnII anchors within a superlacunary crown heteropolyanion {P8W48}†
Masooma
Ibrahim
 *a,
Israël M.
Mbomekallé
b,
Pedro
de Oliveira
b,
George E.
Kostakis
c and
Christopher E.
Anson
d
*a,
Israël M.
Mbomekallé
b,
Pedro
de Oliveira
b,
George E.
Kostakis
c and
Christopher E.
Anson
d
aInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann von-Helmholtz Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: masooma.ibrahim@kit.edu
bEquipe d'Electrochimie et Photo-électrochimie, Laboratoire de Chimie Physique, Université Paris-Sud, UMR 8000, CNRS-Université Paris Saclay, Orsay F-91405, France
cChemistry Department, University of Sussex, Sussex House, Falmer Brighton, BN1 9RH UK
dKIT, Institute of Inorganic Chemistry, 76131 Karlsruhe, Engesserstrasse 15, D-76131, Karlsruhe, Germany
First published on 16th July 2019
Abstract
An isopolyanion {W4O16} within an archetypal {P8W48} heteropolyanion assembly [(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]15− (Mn10W4-P8W48) has been synthesized by the reaction of the cyclic superlacunary anion [H7P8W48O184]33− and Mn(ClO4)2·6H2O in 1 M LiCl solution medium at pH 8. The isolated compound has been characterized by single-crystal X-ray crystallography, powder X-ray diffraction (PXRD), Fourier-transform infrared (FTIR) spectroscopy, elemental analysis and thermogravimetric analysis. Electrochemical studies were also performed on Mn10W4-P8W48, which confirmed the presence of Mn centres bonded to the tungstic framework. The novel polyanion [(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]15− is the first example of a macrocyclic complex, where an isopolyanion (W4O16)8− is embedded within the inner cavity of {P8W48} and is placed in position by six MnII cations as anchors, whereas the exocyclic coordination of four further MnII cations to {P8W48} yields an extended structure by linking neighbouring polyanions through {W–O–Mn–O–W} bridges. Furthermore, the polyanion Mn10W4-P8W48 is the first derivative of {P8W48} with six MnII ions (largest) coordinated to the inner side of the crown ring as anchors.
Introduction
Polyoxometalates (POMs) are inorganic compounds that are formed from oxide anions and early transition metals (M = V, Nb, Ta, Mo, and W) in their highest oxidation states and are being extensively studied because of their application in catalysis, molecular magnetism, biochemistry, analytical chemistry and separation science.1–6 Lacunary POM ligands are versatile all-inorganic molecular precursors for the construction of molecule-based materials. Meanwhile, the structural varieties and functionalities of these materials could be tuned by the incorporation of different transition metal (TM) ions,6–9 rare earth (RE) metal cations,10–16 and mixed TM–RE metal centers17–20 within the POM framework.The archetypal macrocyclic phosphotungstate [H7P8W48O184]33− {P8W48} was isolated in 1985 by Contant and Tezé.21 This polyanion has a wheel-shaped structure and is composed of four identical hexalacunary [H2P2W12O48]12− Wells–Dawson fragments which are linked to each other via oxo-bridges resulting in a cyclic arrangement. Interestingly, Contant concluded that {P8W48} is not reactive towards divalent or trivalent transition metal ions. Nevertheless, Kortz's [Cu20Cl(OH)24(H2O)12(P8W48O184)]25− in 2005 drew attention to the fact that a cyclic anionic POM {P8W48} is an excellent superlacunary “molecular container” system for “cluster within cluster” assemblies.22 Since then {P8W48} has been investigated widely, and some other 3d-, 4d-, and 4f-transition metals and main group metal derivatives of the archetypal polyanion have been synthesized such as: {Cu20-N3},23 {Cu20X (X = Br and I)},24 {VV4VIV2},25 {Fe16},26 {Mo4O4S4W},27 {Mo4O4S4},27 {Sn8},28 {As4},29 {TM4 (TM = Co, Mn, Ni, and V)},30 {Ru4},31 {Ln2 (Ln = La, Ce, Pr, and Nd)},32 and {Se4}.33 The As-analogue [H4As8W48O184]36− of the cyclic polyanion has been prepared and characterized as well.34
Furthermore, {P8W48} plays an essential role in the development of a carbon-free functional inorganic framework.35–37 An important example is K18Li6[Mn8(H2O)48P8W48O184]·108H2O, exhibiting rigid cages with an approximate volume of 7.24 nm.38 Very recently, {[Na(NO3)(H2O)]4[Al16(OH)24(H2O)8(P8W48O184)]}16− and [Ga16(OH)32(P8W48O184)]24− with the largest AlIII/GaIII-oxo clusters encapsulated within the cavity of {P8W48} have been reported, which also exhibit a high proton conductivity.39
Supramolecular assemblies through host–guest complexation have also been observed in polyoxomolybdate {Mo248}, which is formed by the addition of two {Mo36} units to the inner surface of the {Mo176} “wheel”.40
The stability of [H7P8W48O184]33− in aqueous solution over an unusually wide pH range (1–8) and its large anionic pocket (diameter of around 10 Å) are highly desirable features for host–guest chemistry, where the nucleation processes in the cavity of an archetypal POM can allow the formation of a “metal-oxo cluster within cluster” system through a self-assembly mechanism. Inspired by this, we decided to investigate the chemistry of the archetypal macrocyclic phosphotungstate {P8W48}. Herein, we report the formation of an isopolytungstate (W4O16) moiety which occurs within the cavity of {P8W48}, and is held by Mn anchors within the cavity of a cyclic molecule. The novel 4-WVI-16-oxo isopolytungstate embedded polyanion [(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]15− (Mn10W4-P8W48) has been synthesized under normal bench conditions by the reaction of Mn(ClO4)2·6H2O and K28Li5[H7P8W48O184]·92H2O {P8W48} in 1 M LiCl solution and characterized by single-crystal X-ray crystallography, powder X-ray diffraction (PXRD), Fourier-transform infrared (FTIR) spectroscopy, elemental analysis and thermogravimetric analysis. Electrochemical studies were also performed on Mn10W4-P8W48.
Results and discussion
Synthesis
The title polyanion Mn10W4-P8W48 was prepared by the reaction of Mn(ClO4)2·6H2O with a superlacunary archetypal-type polyanion [H7P8W48O184]33− in 1 M LiCl solution medium at pH 8 and isolated as mixed cation salts, K3Li8Mn2[(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]·62H2O (KLiMn-Mn10W4-P8W48). The synthesis of KLiMn-Mn10W4-P8W48 is possible only by increasing the pH value of the reaction mixture to ca. 10 and bringing it back to pH 8. Also the use of LiCl solution as a solvent and manganese(II) perchlorate seems to play a crucial role in the formation of the title compound, which leads to a gradual increase in solubility and release of K+ cations inside the cavity of the POM. The formation of (W4O16) occurs in situ, as a result of partial decomposition of some of the superlacunary {P8W48} POM precursors in the solution. Interestingly, the addition of an equivalent amount of sodium tungstate at any point during the reaction did not result in Mn10W4-P8W48, but instead led to the formation of an unidentified yellow precipitate. The origin of the solitary Na+ in the cluster is unclear.Single-crystal X-ray structure determination
The molecular and packing structures of KLiMn-Mn10W4-P8W48 were determined by single-crystal X-ray diffraction (Fig. 1–3). The compound KLiMn-Mn10W4-P8W48 crystallizes in monoclinic symmetry in the space group C2/c with Z = 4. The polyanion Mn10W4-P8W48 is composed of crown-shaped {P8W48}, in which an isopolytungstate anion {W4O16}8− is unprecedentedly entrapped exactly at the center of the macrocycle and is anchored in position by six MnII centers attached to the inner rim of the {P8W48} POM (Fig. 1). In addition, four MnII centers were also attached to the outer surface of the ring by coordination to oxido ligands from the P8W48 crown ring (Fig. 2). The polyanion Mn10W4-P8W48 is the first example where six MnII centers have been incorporated into the inner rim of the superlacunary POM {P8W48}. The embedded isopolyanion is composed of four edge-sharing WO6 octahedra, forming a planar butterfly topology. With respect to their coordination environment, these manganese anchors are of three types: only two of the six MnII ions (Mn1) are linked to {P8W48} by two Mn1–O(W) and two Mn1–O(P) bonds each; therefore, the four phosphate groups of {P8W48} are not involved in the binding to the anionic isopolytungstate via Mn linkers. The rest of the coordination environment is filled by terminal oxygen atoms of two W centers in the (W4O16) POM, whereas two MnII ions (Mn2) each form two covalent Mn–O(Whet) and Mn–O(Wiso) bonds, and the remaining four octahedral coordination sites on each atom are occupied by H2O.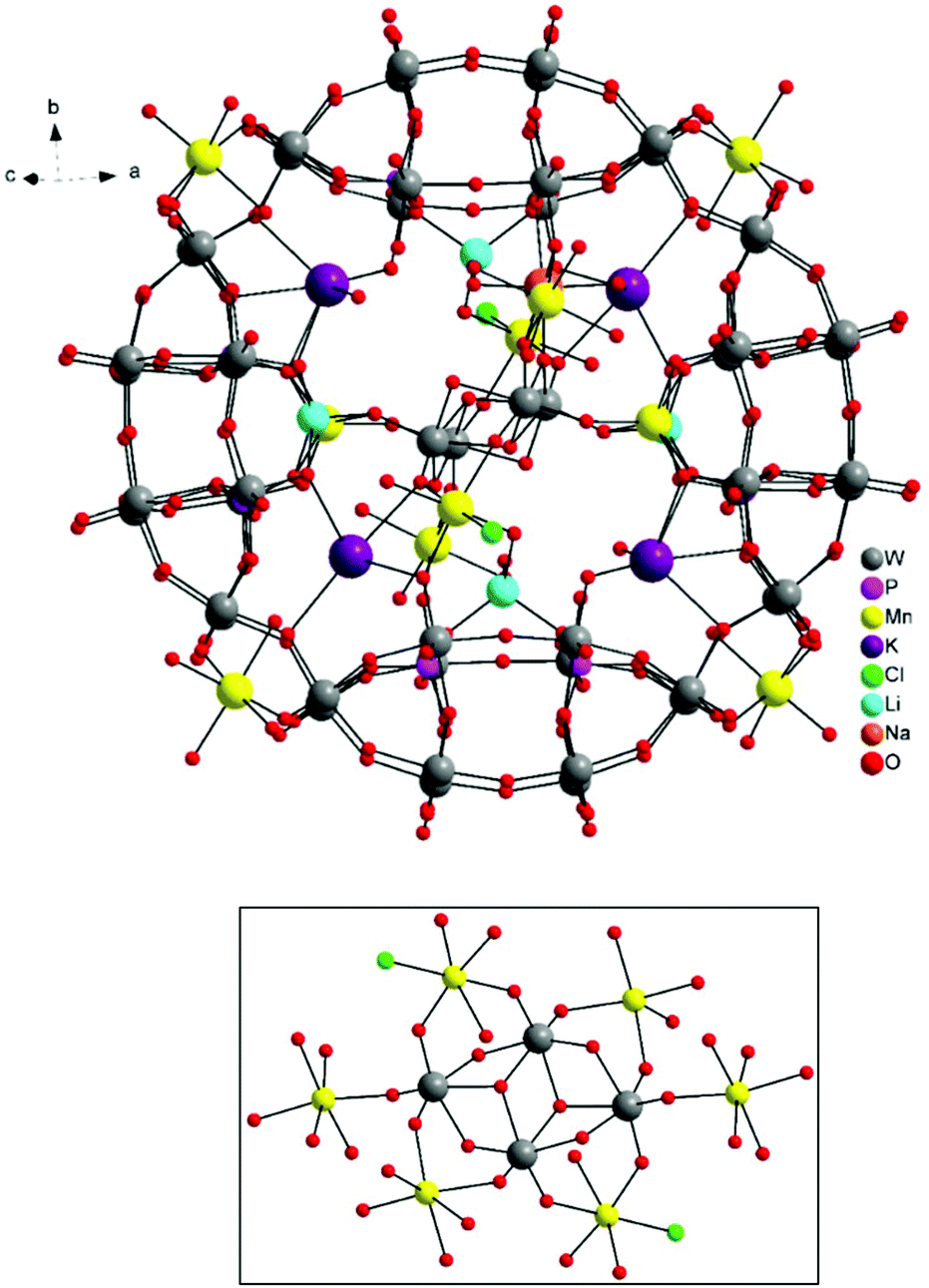 | ||
| Fig. 1 Ball-and-stick representation. Above: Polyanion [(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]15−. Below: (W4O16) isopolyanion with MnII anchors. | ||
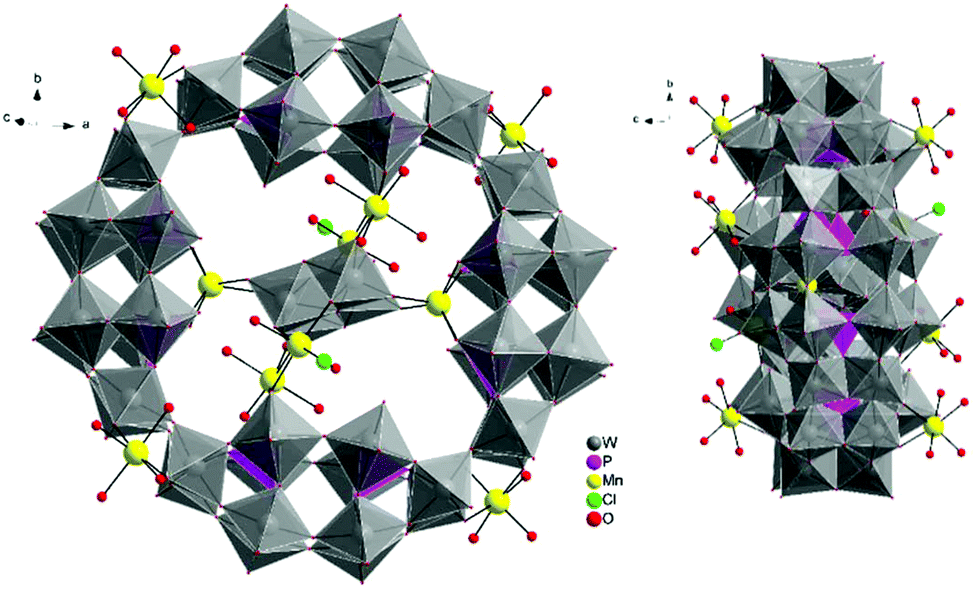 | ||
| Fig. 2 Combined polyhedral/ball-and-stick representation. Front and side views of the structure of Mn10W4-P8W48. Alkali metal cations are omitted for clarity. | ||
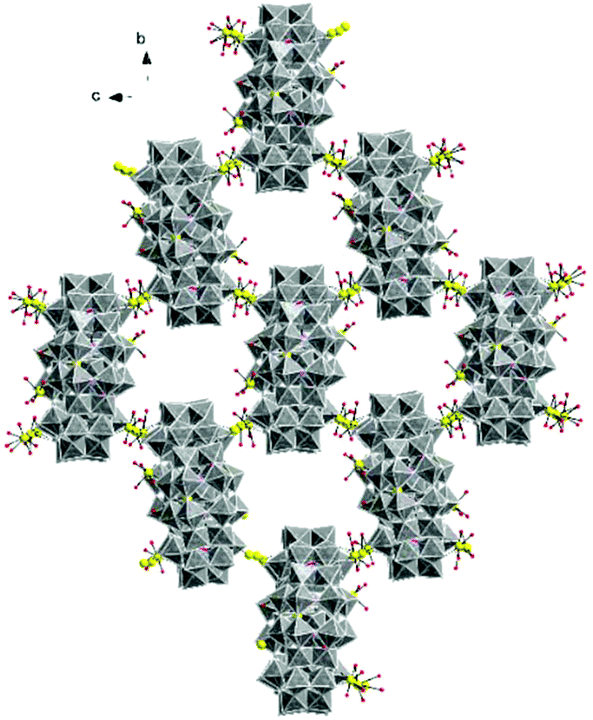 | ||
| Fig. 3 Combined polyhedral/ball-and-stick representation of the crystal packing arrangement in Mn10W4-P8W48. Alkali metal cations are omitted for clarity. WO6 octahedra (gray), O (red), P (pink), and Mn (yellow). | ||
Two MnII anchors (Mn3) each make a link between the two W atoms of the isopolyanion (W4O16) and one W atom of the crown-type heteropolyanion {P8W48} ((Wiso)O2–Mn–O(Whet)) through oxo-ligands, and the remaining two coordination spheres are filled by the two aqua ligands and one chloro ligand. The Mn–O bond lengths are all in the range of 2.087–2.315 Å, while the Mn3–Cl1 bond length is 2.365 Å. The oxidation states of the Mn cations were checked by bond valence sum analysis,46 and all were found to be MnII (calculated values: Mn1: 1.91; Mn2: 2.03; Mn3: 2.20; Mn4: 2.03; and Mn5A: 2.10).
The alkali metal cations (4K+ and 4Li+) are also coordinated to the rim of the central cavity forming a ring of alternating K+ and Li+ centers. The four K+ cations reside in the inner cavity of {P8W48} at the vacant sites between the neighboring {P2W12} subunits. The 3D framework of this assembly is achieved via intramolecular Mn–O(W) bonding and linking potassium ions (Fig. 3).
Na+ is only of half occupancy in the asymmetric unit; so there is on average one per cluster unit, though sodium ions were not added to the reaction mixture. To the best of our knowledge, Mn10W4-P8W48 is the first example of a wheel-shaped polyanion {P8W48} that incorporates an isopolyoxotungstate anion.
Experimental section
General methods and materials
The POM ligand, K28Li5[H7P8W48O184]·92 H2O {P8W48}, was synthesized according to the literature methods and was characterized by FTIR spectroscopy.21 All reactions were carried out under aerobic conditions. All other reagents were purchased commercially and were used without further purification.Synthesis of K3Li8Mn2[(P8W48O184)(W4O16)K10Li4Mn10Na(H2O)50Cl2]·62H2O (KLiMn-Mn10W4-P8W48)
K28Li5[H7P8W48O184]·92H2O (0.37 g, 0.020 mmol) was dissolved in 15 mL of 1 M lithium chloride solution. Then, Mn(ClO4)2·6H2O (0.15 g, 0.41 mmol) was added to this solution under stirring. The pH value of the resultant turbid solution was increased to ca. 10 with 4 M LiOH. After stirring for 5 minutes, the pH value was carefully adjusted to ca. 8 with HCl. The reaction mixture was then stirred and heated at 85 °C for one hour. Later, the solution was filtered and left to slowly evaporate at room temperature, and pale yellow crystals were obtained after approximately three weeks. Yield 0.047 g. IR (2% KBr pellet, ν/cm−1): 1133 (wk), 1105 (wk), 1076 (wk), 1041 (wk), 1015 (wk), 921 (shp), 786 (brd), and 668 (brd). Elemental analysis (%) calc. (found): K 3.11 (3.07), Li 0.51 (0.52), Na 0.15 (0.05), W 58.53 (58.30), P 1.52 (1.81), and Mn 4.04 (4.05).X-ray crystallography
Data on a single crystal of KLiMn-Mn10W4-P8W48 were collected at 180 K using a Stoe IPDS II area detector diffractometer with graphite-monochromated Mo-Kα radiation. Semi-empirical absorption corrections were applied using XPREP in SHELXTL.41 Structure solution was achieved by dual-space direct methods (SHELXT),42 followed by full-matrix least-squares refinement (SHELXL-2018)43 within the Olex2 platform (Table 1).44 Anisotropic temperature factors were used for all ordered non-H atoms, except for Li+ cations and some oxygen atoms of lattice water molecules. The counter cations within or on the periphery of the cluster were ordered and could be refined anisotropically with their coordinated water molecules. Furthermore, in the cavities between the clusters, other counter cations and water molecules were badly disordered and could not be modeled; their contribution to the structure factors was calculated using SQUEEZE.45 The overall formulation was based on analytical data, and this formula is given in the ESI.† The oxidation numbers of the Mn cations were checked using bond valence sum analysis.46| Compound | KLiMn-Mn10W4-P8W48 |
|---|---|
| Formula | Cl2H224K13Li12Mn12NaO312P8W52 |
| Formula weight/g mol−1 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370.49 370.49 |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a/Å | 39.2947(14) |
| b/Å | 30.2573(6) |
| c/Å | 27.7045(10) |
| β/° | 122.850(2) |
| U/Å3 | 27 672.1(16) |
| Z | 4 |
| T/K | 180(2) |
| F(000) | 29264 |
| D c/g cm−3 | 3.929 |
| μ(Mo-Kα)/mm−1 | 22.436 |
| Data measured | 95098 |
| Unique data | 25538 |
| R int | 0.0748 |
| Data with I ≥ 2σ(I) | 15912 |
| wR2 (all data) | 0.1081 |
| S (all data) | 0.885 |
| R 1 [I ≥ 2σ(I)] | 0.0452 |
| Parameters/restraints | 1616/20 |
| Biggest diff. peak/hole/e Å−3 | +2.32/−2.66 |
| FIZ/CSD number | 1922312 |
Further details of the crystal structure investigation may be obtained from FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany, https://www.ccdc.cam.ac.uk/structures/, on quoting the deposition number CSD 1922312.
Powder X-ray diffraction (PXRD) analysis
Powder X-ray diffraction (PXRD) analysis was carried out on KLiMn-Mn10W4-P8W48 to confirm the identity and phase purity of the crystalline material. As shown in Fig. 4, the experimental pattern is consistent with the simulated one (on the basis of the single-crystal structure), which indicated the phase purity of the sample. The minor shift in the peaks reflects the changes in the unit cell due to the fact that single-crystal measurement was carried out at 100 K, whereas the PXRD data were collected at room temperature. There might be some loss of lattice water molecules during PXRD measurement.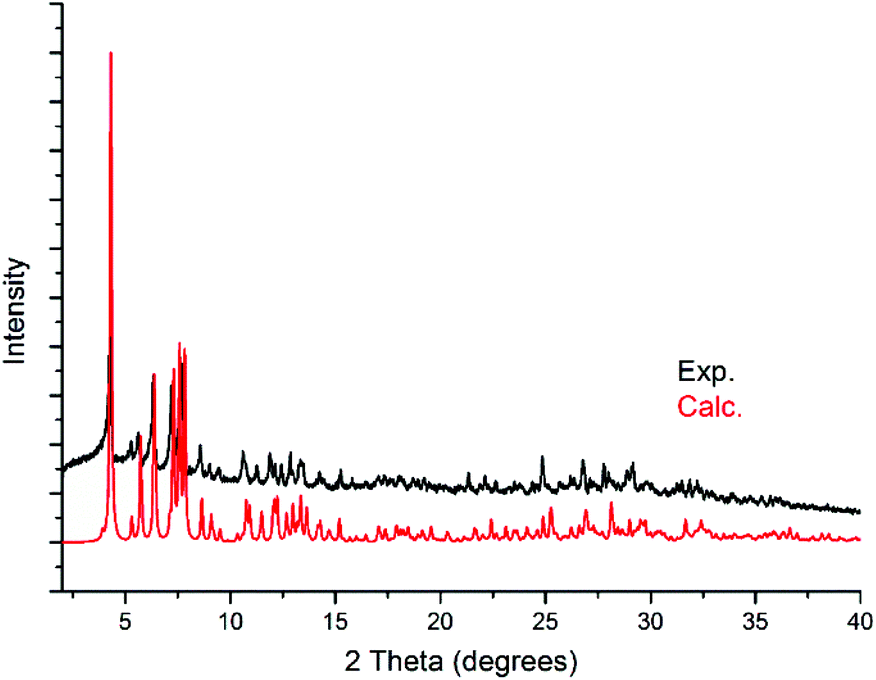 | ||
| Fig. 4 Experimental powder diffraction pattern of KLiMn-Mn10W4-P8W48 and the calculated powder diffraction pattern from the single-crystal X-ray diffraction structure of KLiMn-Mn10W4-P8W48. | ||
Fourier-transform infrared (FTIR) spectroscopy
Infrared spectroscopy is another frequently employed analytical tool for the characterization of polyoxometalates due to its characteristic peaks in the 1200–450 cm−1 region, which is called the fingerprint region for the POM skeleton and results from the stretch vibration frequency of the metal–oxygen bonds. FTIR spectra were recorded on the precursor {P8W48} and the isolated product KLiMn-Mn10W4-P8W48 in order to further confirm the structural information and bulk purity. In the IR spectrum of {P8W48}, a set of bands in the range of 1135–400 cm−1 are the characteristic bands of the {P8W48} skeleton. The characteristic peaks at 1138, 1083, and 1015 cm−1 are attributed to the P–O vibrations of a polyoxoanion precursor. The strong peak at 925 cm−1 belongs to the stretching vibrations of the terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The characteristic peaks at 820–400 cm−1 can be assigned to the W–O–W and W–O–P vibrations. The changes and splits observed in the stretching frequencies of the IR spectrum of KLiMn-Mn10W4-P8W48 in comparison with the superlacunary precursor {P8W48} indicated the formation of a new compound (Fig. S1†).
O bonds. The characteristic peaks at 820–400 cm−1 can be assigned to the W–O–W and W–O–P vibrations. The changes and splits observed in the stretching frequencies of the IR spectrum of KLiMn-Mn10W4-P8W48 in comparison with the superlacunary precursor {P8W48} indicated the formation of a new compound (Fig. S1†).
Thermogravimetric analysis
In order to estimate the total content of the water molecules in the bulk sample, thermogravimetric analysis (TGA) was conducted. The TG curve of the compound KLiMn-Mn10W4-P8W48 in the range of room temperature−950 °C exhibits two weight loss steps up to ca. 550 °C. The total weight loss is ca. 12%, which corresponds to the removal of the crystal water molecules (62) and the coordinated water molecules (50) (Fig. S2†). The overall elemental composition of the bulk material was also determined by complete elemental analysis.Electrochemical experiments
Pure water was obtained using a Milli-Q Intregral 5 purification set. All reagents were of high-purity grade and were used as purchased without further purification: CH3CO2H (Glacial, Prolabo Normapur), H2SO4 (Sigma-Aldrich), Li2SO4·H2O (Acros Organics) and LiCH3CO2·2H2O (Acros Organics). The composition of the various media is as follows: for pH 3.0, 0.5 M Li2SO4 + H2SO4 and for pH 5.0, 1.0 M LiCH3CO2 + CH3CO2H. The stability of the different compounds in solution was assessed by cyclic voltammetry.Electrochemical data were obtained using an EG & G 273 A potentiostat driven by a PC with the M270 software. A one-compartment cell with a standard three-electrode configuration was used for cyclic voltammetry experiments. The reference electrode was a saturated calomel electrode (SCE) and the counter electrode was platinum gauze with a large surface area; both electrodes were separated from the bulk electrolyte solution via fritted compartments filled with the same electrolyte. The working electrodes were of 3 mm outer diameter, EPG from Mersen, France. The pre-treatment of the electrode before each experiment is adapted from a method described elsewhere.47 Prior to each experiment, solutions were thoroughly de-aerated for at least 30 min with pure argon. A positive pressure of this gas was maintained during subsequent work. All cyclic voltammograms (CVs) were recorded at a scan rate of 10 mV s−1 and potentials were quoted against SCE, unless otherwise stated. The polyanion concentration was 0.07 mM. All experiments were performed at room temperature, which is controlled and fixed for the laboratory at 20 °C. Results were very reproducible from one experiment to the other and slight variations observed over successive runs are rather attributed to the uncertainty associated with the detection limit of our equipment (potentiostat, hardware and software) and not to the working electrode pre-treatment or the possible fluctuations in temperature.
As is often the case, the medium used for the synthesis is not the sole one in which the POM is stable, fortunately. The CVs recorded at pH 8 show poorly defined waves, and therefore present no real interest for the study. The media selected for the study (at pH 3 and 5) are usually more appropriate to obtain unambiguous electrochemical responses with POMs, making the comparison of the redox properties of species belonging to the same family easier.
The stability of Mn10W4-P8W48 was assessed by cyclic voltammetry both at pH 3 and 5 upon recording several CVs at regular times for 6 hours. No major evolution of the curves, which could be indicative of the decay of the POM in these media, was observed during this time span.
Therefore, in order to make the electrochemical features of the species Mn10W4-P8W48 more obvious, we carried out a comparative study with its parent compound {P8W48}.21 The purpose was to reveal the influence of the MnII centres present in the new species on its electrochemical behaviour. The comparison of the CVs of the two compounds recorded under the same experimental conditions, both at pH 3 and 5, showed no marked differences as far as their shapes are concerned on the negative side of the potential scale (Fig. S3†). The reduction waves peak at almost the same potential values, namely, at pH 3 (Table 2). On the positive side of the potential scale, the CV of Mn10W4-P8W48 exhibits an oxidation wave attributed to the MnII centres, which is also the case for the majority of POMs containing Mn centres.30,48–53 The shape of this oxidation wave, as well as that of its associated reduction wave, clearly indicates the involvement of film adsorption and desorption phenomena taking place on the surface of the working electrode, probably made of manganese oxides.
| W | Mn | |||||
|---|---|---|---|---|---|---|
| E c1 | E c2 | E c1 | E c2 | E a1 | ||
| pH 3 | Mn10W4-P8W48 | −0.51 | −0.64 | −0.05 | 0.82 | 1.07 |
| P8W48 | −0.52 | −0.65 | ||||
| E c | E c | E a1 | E a2 | ||
|---|---|---|---|---|---|
| pH 5 | Mn10-P8W48 | −0.91 | 0.57 | 0.82 | 1.36 |
| P8W48 | −0.97 | ||||
At pH 3, and upon successive cycling, there is another reduction wave peaking at −0.05 V vs. SCE from the second cycle onwards (Fig. 5A, red curve), which is assigned to the reduction and concomitant desorption of some remnants of the layer of manganese oxides that may not have been totally eliminated at the reduction step at +0.82 V vs. SCE. The thickness of the film increases upon successive cycling (Fig. 5B), but the application of sufficiently negative potentials (<−0.3 V vs. SCE) allows an almost total regeneration of the surface of the working electrode, and therefore of its previous electrochemical response. This behaviour has been analysed in more detail in a recent study using a quartz crystal microbalance and XPS spectroscopy.50
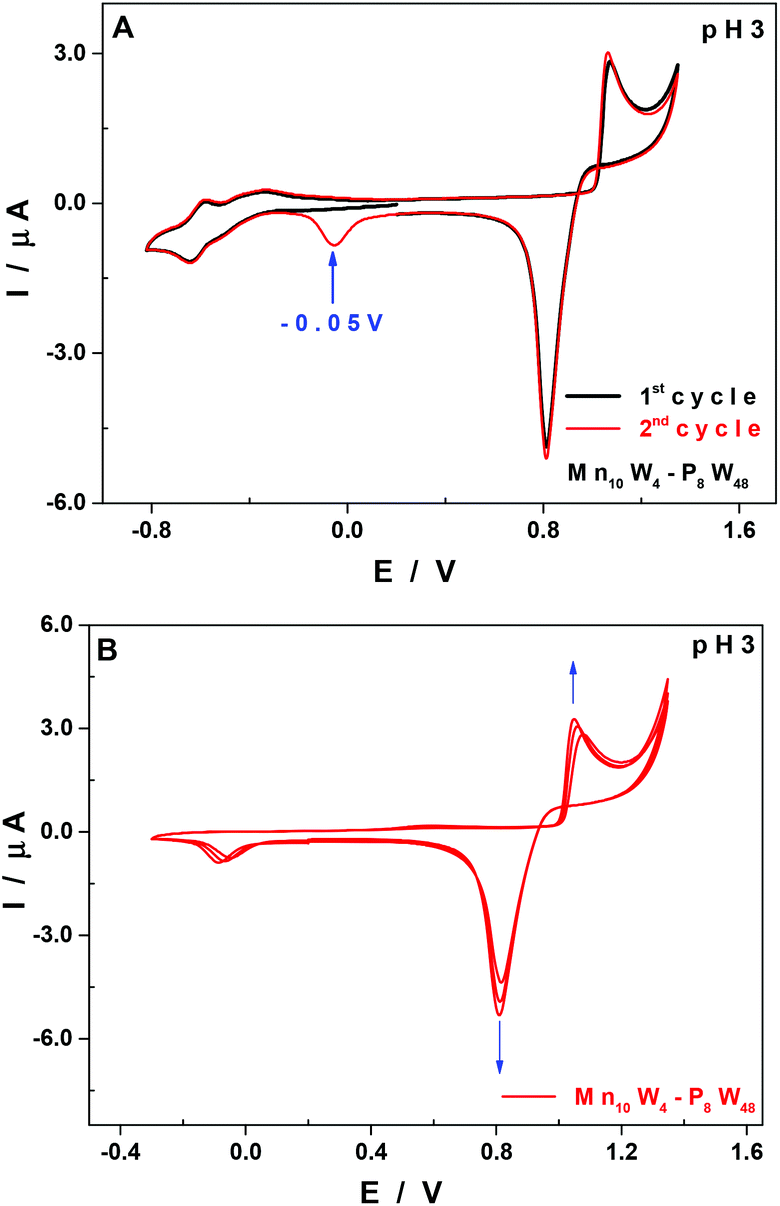 | ||
| Fig. 5 CVs of Mn10W4-P8W48 in 0.5 M Li2SO4 + H2SO4 at pH 3.0. POM concentration: 0.07 mM. (A) Potential range was between 1.35 V and −0.82 V. The scan was started at +0.2 V towards the negative potentials. (B) Potential range was between 1.35 V and −0.30 V. The scan was started at +0.2 V towards the positive potentials. Working electrode: EPG; counter electrode: Pt wire; and reference electrode: SCE. Scan rate: 10 mV s−1. | ||
The formation of manganese oxides has already been observed and described for POMs having MnII centres,48,50,51,53 namely, a species having the same structure as Mn10W4-P8W48 but containing just 4 MnII centres.30 If all the MnII centres were removed from Mn10W4-P8W48, possibly as manganese oxides which would be deposited on the surface of the working electrode, the species remaining in solution would be {P8W48}, which is stable from pH 0 to 8.21,54 The reduction and re-dissolution of these oxides may regenerate the parent compound, but the quantities of Mn10W4-P8W48 involved under our experimental conditions are so small that no difference would be noticed in the CVs if the concentration happens to decrease slightly.
At pH 5, other than the expected shift of the pH-dependent waves towards more negative potentials when compared to pH 3 (Table 2 and Fig. S4†), there are two points worth mentioning. First, the second re-dissolution step of the film of manganese oxides is no longer observed, with the whole process taking place in a single reduction wave peaking at 0.57 V vs. SCE (Fig. 6A).
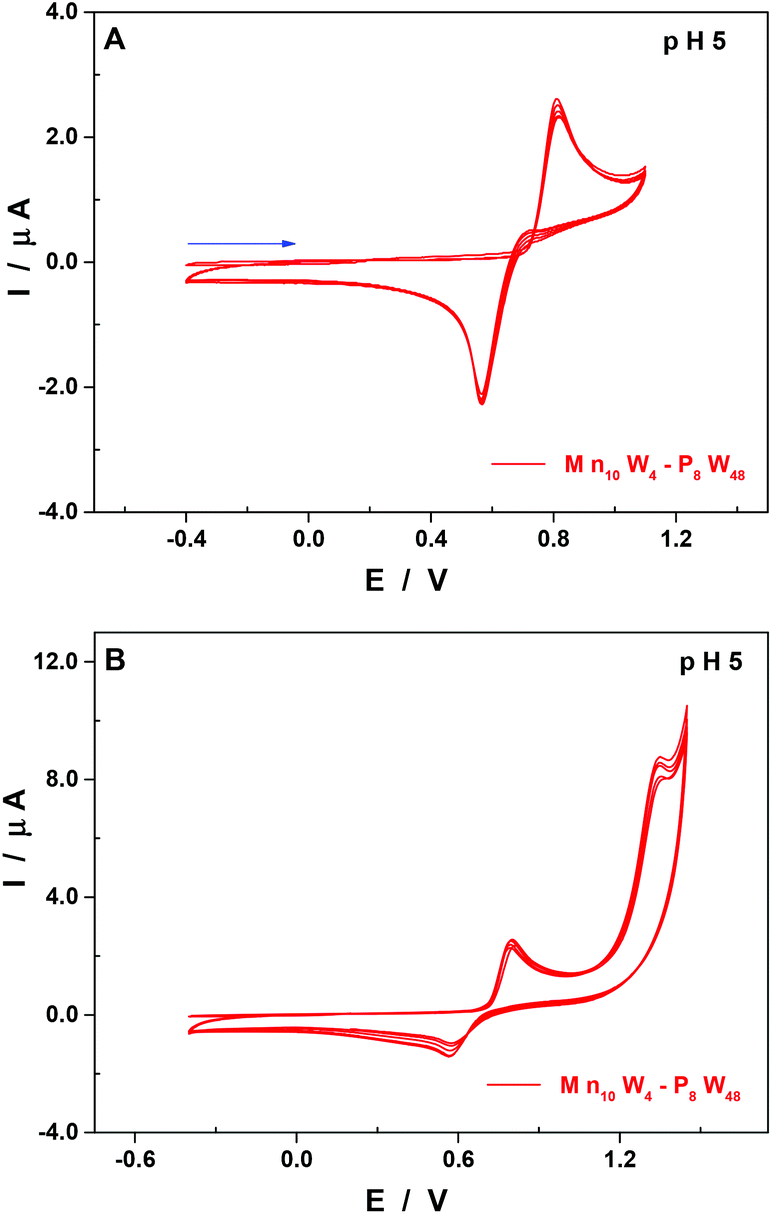 | ||
| Fig. 6 CVs of Mn10W4-P8W48 in 1.0 M LiCH3CO2 + CH3CO2H at pH 5.0. POM concentration: 0.07 mM. (A) Potential range was between 1.10 V and −0.40 V. The scan was started at +0.2 V towards the negative potentials. (B) Potential range was between 1.45 V and −0.40 V. The scan was started at +0.2 V towards the positive potentials. Working electrode: EPG; counter electrode: Pt wire; and reference electrode: SCE. Scan rate: 10 mV s−1. | ||
Second, the general cathodic shift allows the exploration of another oxidation wave peaking at 1.36 V vs. SCE and being irreversible. This observation is in good agreement with other studies which have demonstrated that the MnII centres could be oxidised beyond the oxidation state +IV and be involved in the electro-catalytic oxidation of water.50,53 In this respect, the behaviour of the compound Mn10W4-P8W48 is distinct from that of the homologous species having just four Mn centres, Mn4-P8W48.54 Indeed, the oxidation peak potentials of the MnII centre waves of the two compounds are very close, at pH 5: 0.82 V for Mn10W4-P8W48 and 0.83 V for Mn4-P8W48. No second oxidation wave has been described for the latter. The presence of this second wave in the case of Mn10W4-P8W48 may be explained by a higher number of Mn centres in the POM and by their location in the molecular scaffold. As is often the case for Mn-containing POMs, the formation of a film made up of manganese oxides on the surface of the working electrode makes the determination by electrolysis of the number of electrons involved in the oxidation steps difficult. As a consequence, it is not possible to confirm the number of Mn centres present in the POM using electrochemical methods.
Conclusions
In summary, we have demonstrated here that an unprecedented isopolyanion (W4O16) can be embedded within the cavity of the archetypal superlacunary POM {P8W48} by the direct reaction of MnII ions with [H7P8W48O184]33− in basic medium using a simple, one-pot procedure. The obtained POM has been characterized by single-crystal X-ray crystallography, powder X-ray diffraction (PXRD), Fourier-transform infrared (FTIR) spectroscopy, elemental analysis and thermogravimetric analysis. Electrochemical studies performed on Mn10W4-P8W48 in aqueous media confirmed the presence of Mn centers and showed their influence on its redox behavior. Single X-ray diffraction data revealed that polyanion Mn10W4-P8W48 was isolated in the solid state as the hydrated mixed potassium/lithium/manganese salt KLiMn-Mn10W4-P8W48. The (W4O16) unit is held in the central cavity of {P8W48} by six MnII ions as linkers/anchors. The polyanion Mn10W4-P8W48 is the first example where an isopolyanion has been inserted within the {P8W48} cavity; furthermore, the largest number of MnII centers have been incorporated into the inner cavity of the macrocyclic ligand. Currently, we are investigating the possibility of encapsulation of other transition metal assemblies within the cavity of a cyclic {P8W48} POM. The host–guest architectures based on the macrocyclic ring {P8W48} allow for the construction of various structures which might display an interesting set of properties in various fields such as ion exchange, gas storage, catalysis and medicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. I. thanks Prof. Annie. K. Powell for her continuous support and guidance. M. I. acknowledges support from the Helmholtz Society through the program Science and Technology of Nanosystems (STN). M. I. also thanks the University of Balochistan, Quetta, Pakistan, for allowing her to pursue her Ph.D. and postdoctoral work at Jacobs University and KIT, respectively. We also thank Sven Stahl and Dr Thomas Bergfeldt for performing TGA measurements and elemental analysis, respectively. This work was partially carried out with the support from the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz research infrastructure at the Karlsruhe Institute of Technology. We also thank the Université Paris-Sud and the CNRS for financial support.Notes and references
- D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 Search PubMed.
- C. Vonci and M. Boskovic, Aust. J. Chem., 2014, 67, 1542–1552 CrossRef.
- M. Ibrahim, Y. Lan, B. S. Bassil, Y. Xiang, A. Suchopar, A. K. Powell and U. Kortz, Angew. Chem., Int. Ed., 2011, 50, 4708–4711 CrossRef CAS PubMed.
- X. Han, Y. Li, Z. Zhang, H. Tan, Y. Lu and E. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS PubMed.
- O. Oms, A. Dolbecq and P. Mialane, Chem. Soc. Rev., 2012, 41, 7497–7536 RSC.
- A. Haider, M. Ibrahim, B. S. Bassil, A. M. Carey, A. N. Viet, X. Xing, W. W. Ayass, J. F. Miñambres, R. Liu, G. Zhang, B. Keita, V. Mereacre, A. K. Powell, K. Balinski, A. T. N'Diaye, K. Küpper, H.-Y. Chen, U. Stimming and U. Kortz, Inorg. Chem., 2016, 55, 2755–2764 CrossRef CAS PubMed.
- M. Ibrahim, A. Haider, Y. Xiang, B. S. Bassil, A. M. Carey, L. Rullik, G. B. Jameson, F. Doungmene, I. M. Mbomekallé, P. De Oliveira, V. Mereacre, G. E. Kostakis, A. K. Powell and U. Kortz, Inorg. Chem., 2015, 54, 6136–6146 CrossRef CAS PubMed.
- B. S. Bassil, M. Ibrahim, R. Al-Oweini, M. Asano, Z. Wang, J. Van Tol, N. S. Dalal, K. Y. Choi, R. Ngo Biboum, B. Keita, L. Nadjo and U. Kortz, Angew. Chem., Int. Ed., 2011, 50, 5961–5964 CrossRef CAS PubMed.
- M. Ibrahim, S. S. Mal, B. S. Bassil, A. Banerjee and U. Kortz, Inorg. Chem., 2011, 50, 956–960 CrossRef CAS PubMed.
- M. Ibrahim, B. Bassil and U. Kortz, Inorganics, 2015, 3, 267–278 CrossRef CAS.
- C. M. Granadeiro, B. De Castro, S. S. Balula and L. Cunha-Silva, Polyhedron, 2013, 52, 10–24 CrossRef CAS.
- B. S. Bassil, M. H. Dickman, I. Römer, B. Von Der Kammer and U. Kortz, Angew. Chem., Int. Ed., 2007, 46, 6192–6195 CrossRef CAS PubMed.
- U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS.
- J. Zhao, Y. Li, L. Chen and G. Yang, Chem. Commun., 2016, 52, 4418–4445 RSC.
- B. S. Bassil and U. Kortz, Z. Anorg. Allg. Chem., 2010, 636, 2222–2231 CrossRef.
- S. Reinoso, Dalton Trans., 2011, 40, 6610–6615 RSC.
- S. Reinoso, J. R. Galán-Mascarós and L. Lezama, Inorg. Chem., 2011, 50, 9587–9593 CrossRef CAS PubMed.
- M. Ibrahim, V. Mereacre, N. Leblanc, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2015, 54, 15574–15578 CrossRef CAS PubMed.
- T. Yu, H. Ma, C. Zhang, H. Pang, S. Li and H. Liu, Dalton Trans., 2013, 42, 16328–16333 RSC.
- R. Contant and A. Teze, Inorg. Chem., 1985, 24, 4610–4614 CrossRef CAS.
- S. S. Mal and U. Kortz, Angew. Chem., Int. Ed., 2005, 3, 3777–3780 CrossRef PubMed.
- C. Pichon, P. Mialane, A. Dolbecq, J. Marrot, E. Rivière, B. Keita, L. Nadjo and F. Sécheresse, Inorg. Chem., 2007, 46, 5292–5301 CrossRef CAS PubMed.
- S. S. Mal, B. S. Bassil, M. Ibrahim, S. Nellutla, J. Van Tol, N. S. Dalal, J. A. Fernández, X. López, J. M. Poblet, R. N. Biboum, B. Keita and U. Kortz, Inorg. Chem., 2009, 48, 11636–11645 CrossRef CAS PubMed.
- A. Müller, M. T. Pope, A. M. Todea, H. Bögge, J. Van Slageren, M. Dressel, P. Gouzerh, R. Thouvenot, B. Tsukerblat and A. Bell, Angew. Chem., Int. Ed., 2007, 46, 4477–4480 CrossRef PubMed.
- S. S. Mal, M. H. Dickman, U. Kortz, A. M. Todea, A. Merca, H. Bögge, T. Glaser, A. Müller, S. Nellutla, N. Kaur, J. Van Tol, N. S. Dalal, B. Keita and L. Nadjo, Chem. – Eur. J., 2008, 14, 1186–1195 CrossRef CAS PubMed.
- V. S. Korenev, S. Floquet, J. Marrot, M. Haouas, I. M. Mbomekallé, F. Taulelle, M. N. Sokolov, V. P. Fedin and E. Cadot, Inorg. Chem., 2012, 51, 2349–2358 CrossRef CAS PubMed.
- N. V. Izarova, L. Klaß, P. De Oliveira, I. M. Mbomekalle, V. Peters, F. Haarmann and P. Kögerler, Dalton Trans., 2015, 44, 19200–19206 RSC.
- X. Yi, N. V. Izarova and P. Kögerler, Inorg. Chem., 2017, 56, 13822–13828 CrossRef CAS PubMed.
- B. S. Bassil, M. Ibrahim, S. S. Mal, A. Suchopar, R. N. Biboum, B. Keita, L. Nadjo, S. Nellutla, J. Van Tol, N. S. Dalal and U. Kortz, Inorg. Chem., 2010, 49, 4949–4959 CrossRef CAS PubMed.
- S. S. Mal, N. H. Nsouli, M. H. Dickman and U. Kortz, Dalton Trans., 2007, 186, 2627–2630 RSC.
- M. Zimmermann, N. Belai, R. J. Butcher, M. T. Pope, E. V. Chubarova, M. H. Dickman and U. Kortz, Inorg. Chem., 2007, 46, 1737–1740 CrossRef CAS PubMed.
- K. Y. Wang, S. Zhang, D. Ding, T. Ma, U. Kortz and C. Wang, Eur. J. Inorg. Chem., 2019, 2019, 512–516 CrossRef CAS.
- I. M. Mbomekallé, B. S. Bassil, A. Suchopar, B. Keita, L. Nadjo, M. Ammam, M. Haouas, F. Taulelle and U. Kortz, J. Cluster Sci., 2014, 25, 277–285 CrossRef.
- H. N. Miras, L. Vilà-Nadal and L. Cronin, Chem. Soc. Rev., 2014, 43, 5679–5699 RSC.
- S. G. Mitchell, D. Gabb, C. Ritchie, N. Hazel, D. L. Long and L. Cronin, CrystEngComm, 2009, 11, 36–39 RSC.
- T. Boyd, S. G. Mitchell, D. Gabb, D. Long, Y. Song and L. Cronin, J. Am. Chem. Soc., 2017, 139, 5930–5938 CrossRef CAS PubMed.
- S. G. Mitchell, C. Streb, H. N. Miras, T. Boyd, D. L. Long and L. Cronin, Nat. Chem., 2010, 2, 308–312 CrossRef CAS PubMed.
- P. Yang, M. Alsufyani, A. H. Emwas, C. Chen and N. M. Khashab, Angew. Chem., Int. Ed., 2018, 57, 13046–13051 CrossRef CAS PubMed.
- A. Müller, S. Q. N. Shah, H. Bögge and M. Schmidtmann, Nature, 1999, 397, 48–50 CrossRef.
- G. M. Sheldrick, SHELXTL 6.14, Bruker AXS Inc., 6300 Enterprise Lane, Madison, WI 53719-1173, USA, 2003.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- W. Liu and H. H. Thorp, Inorg. Chem., 1993, 32, 4102–4105 CrossRef CAS.
- N. Vilà, P. A. Aparicio, F. Sécheresse, J. M. Poblet, X. López and I. M. Mbomekallé, Inorg. Chem., 2012, 51, 6129–6138 CrossRef PubMed.
- I. M. Mbomekalle, B. Keita, L. Nadjo, P. Berthet, W. A. Neiwert, C. L. Hill, M. D. Ritorto and T. M. Anderson, J. Chem. Soc., Dalton Trans., 2003, 3, 2646–2650 RSC.
- M. Lebrini, I. M. Mbomekallé, A. Dolbecq, J. Marrot, P. Berthet, J. Ntienoue, F. Sécheresse, J. Vigneron and A. Etcheberry, Inorg. Chem., 2011, 50, 6437–6448 CrossRef CAS PubMed.
- A.-L. Teillout, P. de Oliveira, J. Marrot, R. Howell, N. Vilà, A. Walcarius and I. Mbomekallé, Inorganics, 2019, 7, 15 CrossRef CAS.
- J. Friedl, R. Al-Oweini, M. Herpich, B. Keita, U. Kortz and U. Stimming, Electrochim. Acta, 2014, 141, 357–366 CrossRef CAS.
- R. Al-Oweini, B. S. Bassil, J. Friedl, V. Kottisch, M. Ibrahim, M. Asano, B. Keita, G. Novitchi, Y. Lan, A. Powell, U. Stimming and U. Kortz, Inorg. Chem., 2014, 53, 5663–5673 CrossRef CAS PubMed.
- B. Keita, P. Mialane, F. Sécheresse, P. de Oliveira and L. Nadjo, Electrochem. Commun., 2007, 9, 164–172 CrossRef CAS.
- B. Keita, Y. W. Lu, L. Nadjo and R. Contant, Electrochem. Commun., 2000, 2, 720–726 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1922312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02478f |
| This journal is © The Royal Society of Chemistry 2019 |