
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-coordinate first-row transition metal complexes in catalysis and small molecule activation
Laurence J.
Taylor
 and
Deborah L.
Kays
*
and
Deborah L.
Kays
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: Deborah.Kays@nottingham.ac.uk
First published on 29th July 2019
Abstract
Enforcing unusually low coordination numbers on transition metals with sterically demanding ligands has long been an area of interest for chemists. Historically, the synthesis of these challenging molecules has helped to elucidate fundamental principles of bonding and reactivity. More recently, there has been a move towards exploiting these highly reactive complexes to achieve a range of transformations using cheap, earth-abundant metals. In this Perspective, we will highlight selected examples of transition metal complexes with low coordination numbers that have been used in catalysis and the activation of small molecules featuring strong bonds (N2, CO2, and CO).
 Laurence J. Taylor | Laurence graduated from the University of Cambridge with an MSci in Natural Sciences in 2013. He was then awarded the St Andrews 600th Anniversary Scholarship for his PhD studies at the University of St Andrews under Dr Petr Kilian. After completing his PhD studying air- and moisture-sensitive pnictogen compounds, he took up a postdoctoral position at the University of Nottingham under Professor Deborah Kays in late 2017. His current research focuses on the use of highly reactive complexes of the first-row transition and s-block metals as catalysts for organic transformations. |
 Deborah L. Kays | Deborah Kays received her MChem and PhD degrees in Chemistry from Cardiff University, Wales. After postdoctoral work, also at Cardiff University, she took up a Junior Research Fellowship at the University of Oxford. She has been at the University of Nottingham since 2007, as Lecturer in Inorganic Chemistry, she was promoted to Associate Professor in 2014 and to Professor in 2019. Her research interests involve the investigation of the stabilisation of highly unsaturated complexes and their reactivity under stoichiometric and catalytic regimes. For her contributions to low-coordinate transition metal chemistry, Deborah was awarded the 2018 RSC Chemistry of the Transition Metals Award. |
Introduction
The stabilisation of transition metal complexes featuring unusually low coordination numbers remains a challenging and active area of research for synthetic inorganic chemists. These species are typically stabilised by sterically demanding ligands, which shield the metal from oligomerisation or further coordination by Lewis basic ligands. To this end, a vast array of bulky ligands have been prepared and utilised for this purpose; including amides,1 alkyls,2 silyls,2 aryls,3,4 and carbenes.5,6 The development of such ligands has been instrumental in advancing the range of complexes featuring low-coordinate and highly reactive transition metal centres, and has facilitated the isolation of new classes of compounds,7 such as species featuring metal–metal multiple bonds.8As the chemistry of low-coordinate complexes has developed, there has been an increasing interest in the exploitation of their reactivity. With the 3d metals, which are cheap and earth-abundant,9–11 enforcing low-coordinate geometries can drastically alter the reactivity and properties of these elements.7 For example, two-coordinate 3d metal complexes have been shown to act as single molecule magnets,12–14 and have been employed in the synthesis of Zintl ions.15 In recent years, exciting examples of such species catalysing unusual reactions or promoting the activation of challenging substrates have started to appear in the literature. Our research group has become increasingly interested in this field, with some of our recent work looking at the use of m-terphenyl complexes in the stoichiometric activation of small molecules and the catalysis of chemical reactions.
The term “low-coordinate” can be a rather tricky one to define when applied to the transition metals. One can argue, for example, that coordination numbers of four could be considered low for high oxidation state iron or vanadium compounds. One could also discuss how to define formal coordination number, and therefore depending on your definition “low-coordinate complexes” could cover a very broad range of species indeed. Given that Perspective articles are meant to be brief, we have restricted ourselves to considering complexes with a formal coordination number of three or less where, for example, ligands such as alkyls, aryls, amides, carbenes, phosphines, alkenes, etc. are considered to occupy one coordination site, and η6-C6H6 to occupy three. This is not meant to provide a rigorous definition of “low-coordinate” but has instead been chosen to keep this discussion focused.
Even with this restriction, there are still too many examples for this article to be an exhaustive account of the field. As such, this Perspective will cover selected examples of catalysis and small molecule activation by low-coordinate complexes as defined above.
Catalysis
Given the increasing importance of sustainability in catalysis, there has been a shift away from the use of platinum group metals (such as platinum, palladium, and rhodium) towards cheaper and more abundant first-row transition metals.9–11 In particular, the use of low coordination numbers for 3d transition metal complexes is affording catalysts for a wide range of reactions. A commonly employed ligand system used to support low-coordinate first-row transition metal catalysts is the β-diketiminate; bidentate monoanionic ligands that bind through nitrogen. This ligand has been complexed to numerous metals, and such species have been shown to catalyse a wide variety of transformations including polymerisation,16,17 catalytic hydrodefluorination,18,19Z-selective alkene isomerisation,20 nitrene transfer reactions,21,22 the dehydrocoupling of phosphines,23 phosphine-boranes and amine-boranes,24 and the hydrophosphination25 and hydroboration26 of alkenes and alkynes. One example of such a catalyst is shown in Scheme 1, with the three-coordinate iron(II) catalyst (1), and the proposed mechanism by which it catalyses the dehydrocoupling of dimethylamine-borane.24 The development of β-diketiminate 3d complexes as catalysts was covered in a recent Perspective article by Ruth Webster, and we recommend consulting this article for a more in-depth look at such species.27 Thus, this section will focus on low coordinate 3d metal catalysts that do not feature β-diketiminate ligands. The discussion will be subdivided by metal, to aid clarity.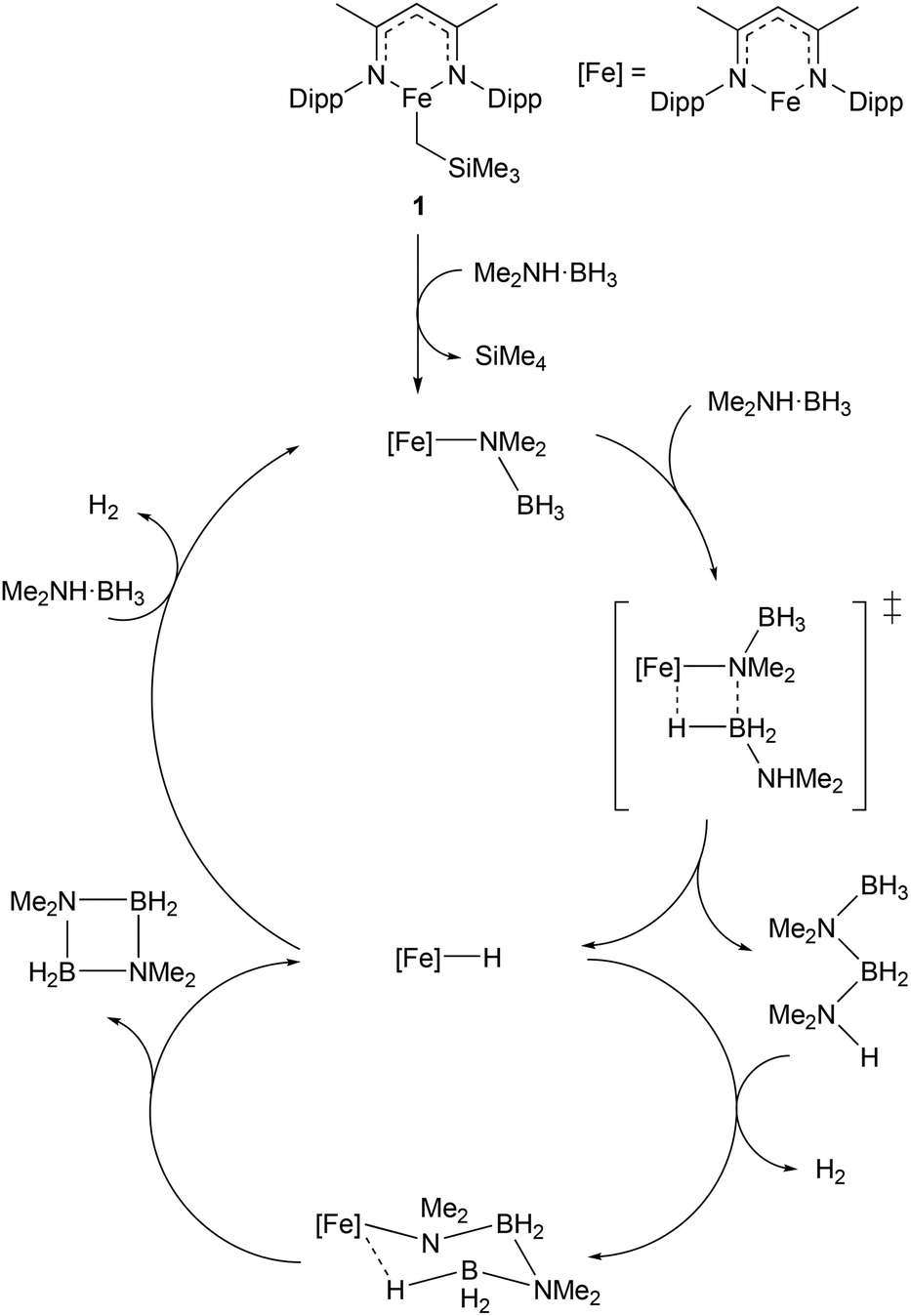 | ||
| Scheme 1 Example of a three-coordinate iron catalyst with a β-diketiminate ligand (1) and the proposed mechanism by which it catalyses the dehydrocoupling of dimethylamine-borane (Dipp = 2,6-iPr2C6H3).24 | ||
Nickel
For the most part the metal complexes covered in this review are highly reactive species which have been forced into disfavoured low-coordinate geometries by sterically demanding ligands. However, nickel is something of an exception to this. Complexes of Ni(0) are found to readily adopt two- or three-coordinate geometries when combined with neutral donor ligands such as phosphines and N-heterocyclic carbenes (NHCs), and such species have proven competent catalysts for a broad range of organic transformations. In recent years, there have been examples of C–H activation with the alkenylation,28 alkylation,29–31 and stannylation32 of aromatic systems, C–O bond cleavage,33–35 and catalytic cycloaddition chemistry.36 The catalysts are typically generated in situ from bis(cyclooctadiene)nickel(0) and the required ligand, with some examples from the groups of Hartwig and Nakao shown in Scheme 2.28,33,34 There are too many examples to cover in detail here, and we recommend that readers interested in the field consult some of the excellent in-depth reviews on this topic.37,38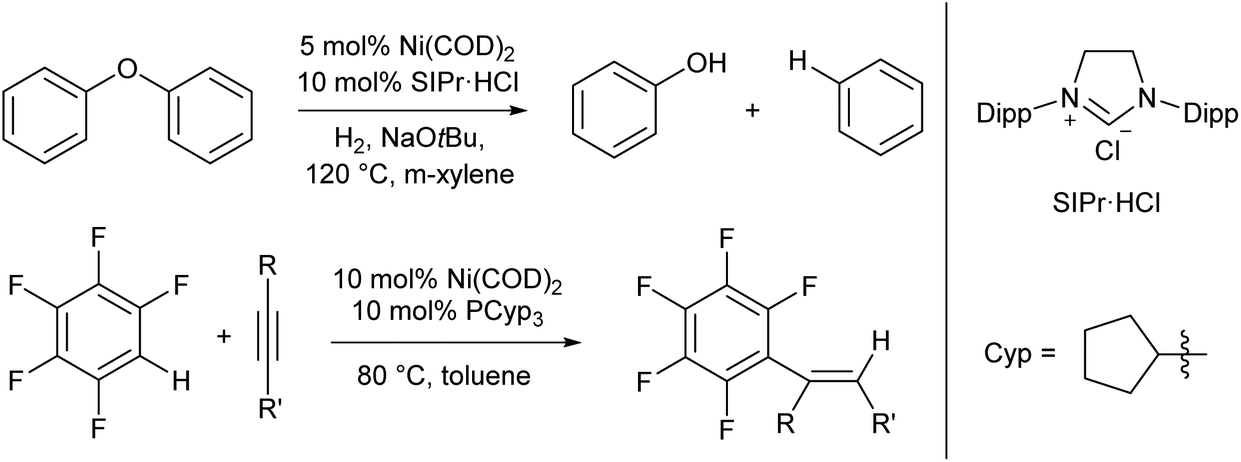 | ||
| Scheme 2 Hydrogenolysis of aryl ethers33,34 and alkenylation of arene rings28 promoted by low coordinate Ni(0) catalysts. | ||
Low-coordinate catalysts based on Ni(I) and Ni(II) are somewhat less common, although certainly not unheard of. For example, several low-coordinate Ni(I) complexes have been investigated as catalysts for Kumada cross-coupling reactions.12,39–42 The Ni(0) species 2a and 2b were reacted with chlorobenzene or bromobenzene to generate the Ni(I) NHC complexes 3a–c (with elimination of biphenyl),40,42 all of which catalyse the Kumada reaction (Scheme 3) between aryl magnesium bromides and aryl chlorides or bromides. Compounds 3b–c also catalysed the coupling of aryl boronic acids to aryl bromides (Suzuki coupling).42 These complexes are interesting as case studies for the reactivity of low coordinate metals, but more convenient and less sensitive nickel catalysts exist for such transformations.43
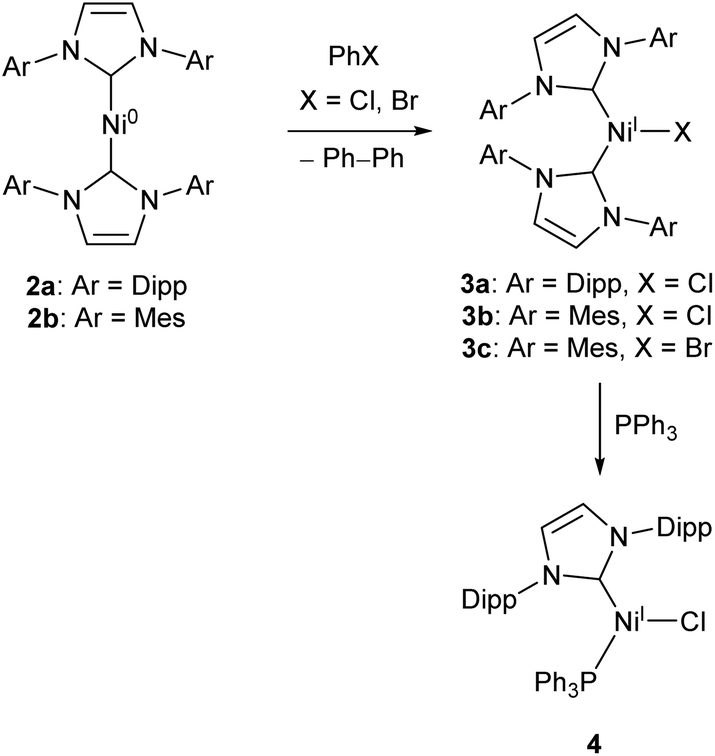 | ||
| Scheme 3 Synthesis of Ni NHC complexes for Kumada and Suzuki cross-couplings, and Buchwald–Hartwig amination (Dipp = 2,6-iPr2C6H3, Mes = 2,4,6-Me3C6H2).40–42 | ||
Matsubara and co-workers found that treatment of 3a with triphenylphosphine afforded the mixed NHC/phosphine complex 4, which catalysed the Buchwald–Hartwig amination of aryl halides (chlorides, bromides and iodides) by diphenylamine under mild conditions (40–70 °C), affording yields ranging between 41–98% dependent on substrate.41 The catalyst showed good tolerance of ketone, alkene, and nitro functional groups; providing the first example of a nickel catalyst capable of coupling diphenylamine to aryl halides to afford triphenylamine derivatives (Scheme 3).41
Following on from this work, the Matsubara group very recently published an improved Ni(I) amination catalyst (5a) which coupled a range of primary and secondary arylamines to 4-bromobenzophenone in yields ranging from 58–96%.44 The initial pre-catalyst 5a is four-coordinate, but the active species is believed to be the two-coordinate Ni(I) amide species 5b (Scheme 4). This species was also synthesised, isolated, and found to be catalytically active.44 An exchange reaction with 2,2′-biquinoline revealed that the 2,2-bipyridine ligand on 5a is labile and readily exchanged, allowing for the generation of the active two-coordinate species.44 EPR spectroscopy of a stoichiometric mixture of 5b and 4-bromoanisole provided evidence of both Ni(I) and Ni(III) species in solution, providing direct support for the involvement of a Ni(III) intermediate in the catalytic cycle. Such Ni(I)/Ni(III) redox cycles have previously been proposed for a number of nickel catalysts,45–47 but this study provides some of the best direct evidence for the existence of a Ni(III) intermediate.44 The catalytic cycle proposed by Matsubara et al. is presented in Scheme 4.
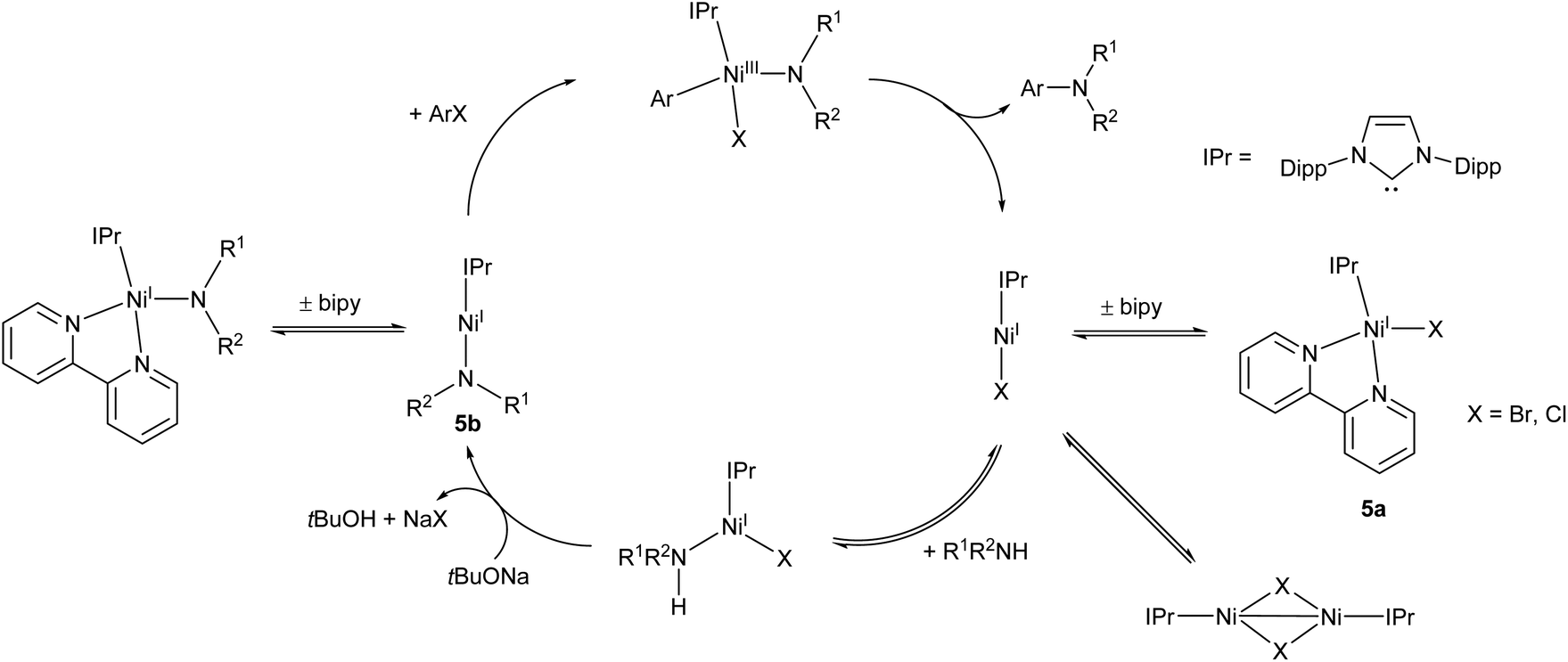 | ||
| Scheme 4 Proposed mechanism for Buchwald–Hartwig amination by pre-catalyst 5a proceeding via two-coordinate Ni(I) amide 5b.44 | ||
Nickel(I) catalysts bearing NHCs of different ring sizes (Scheme 5) were investigated for their efficacy in Kumada cross-coupling reactions, with EPR spectroscopy revealing that the magnetic properties of the complexes were strongly affected by ring size. The NHCs with the smallest ring size gave the best catalysts, although no correlation was observed with the magnetic parameters. The combination of a six-membered ring and mesityl flanking groups was found to give the best catalytic performance, with biaryl yields of 51–83% obtained at room temperature.12,39
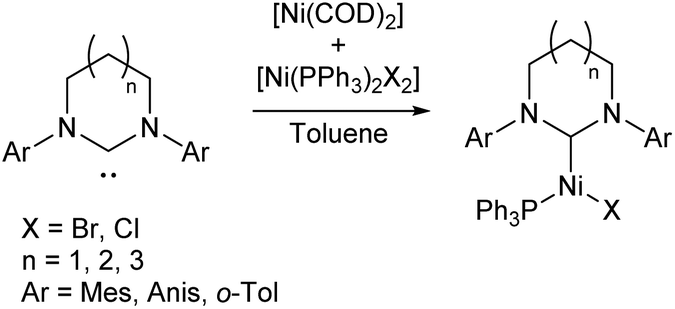 | ||
| Scheme 5 Synthesis of Ni(I) NHC complexes with varying NHC ring size for Kumada cross-coupling (Anis = 2-MeOC6H4; o-Tol = 2-MeC6H4).12,39 | ||
A two-coordinate Kumada cross-coupling catalyst is seen in the Ni(II) bis(silylamide) complex 6a reported by Lipschutz and Tilley (Scheme 6).48–50 This catalyst was more effective for electron-poor aryl halides and, notably, promoted couplings to pyridine-based heterocycles. Stoichiometric reaction of 6a with MeMgBr resulted in the rapid formation of an alkylated Ni(II) species, 6b.48 This compound was found to react over 45 minutes with 1-iodonaphthalene to afford the coupled product (1-methylnaphthalene) in low yield (13%) along with 1,1′-binaphthalene and Ni(III) methyl complex 6c. Reacting 1-iodonaphthalene with 2 equivalents of 6b, by contrast, gave clean and rapid conversion to 1-methylnaphthalene in 98% yield.48 It is believed that the formation of 1,1′-binaphthalene in the stoichiometric reaction is due to the formation of naphthyl radicals, and the second equivalent of 6b is required to trap these. The radical nature of this step is supported by a radical clock experiment coupling (iodomethyl)cyclopropane to PhMgBr, which resulted in 4-phenyl-1-butene, the expected product of a radical rearrangement.48 Finally, an anionic Ni(I) species (6e) can be obtained by chemical reduction of 6a, and the reaction of 6c and 6e results in a redox equilibrium affording 6b and 6a. Based on the results of these various stoichiometric transformations, Lipschutz and Tilley proposed the catalytic cycle shown in Scheme 6.48 Species 6a is also an effective catalyst for the hydrosilylation of 1-octene with diphenylsilane, affording (n-octyl)diphenylsilane as the sole product after 2 h at room temperature (5 mol% catalyst loading).50
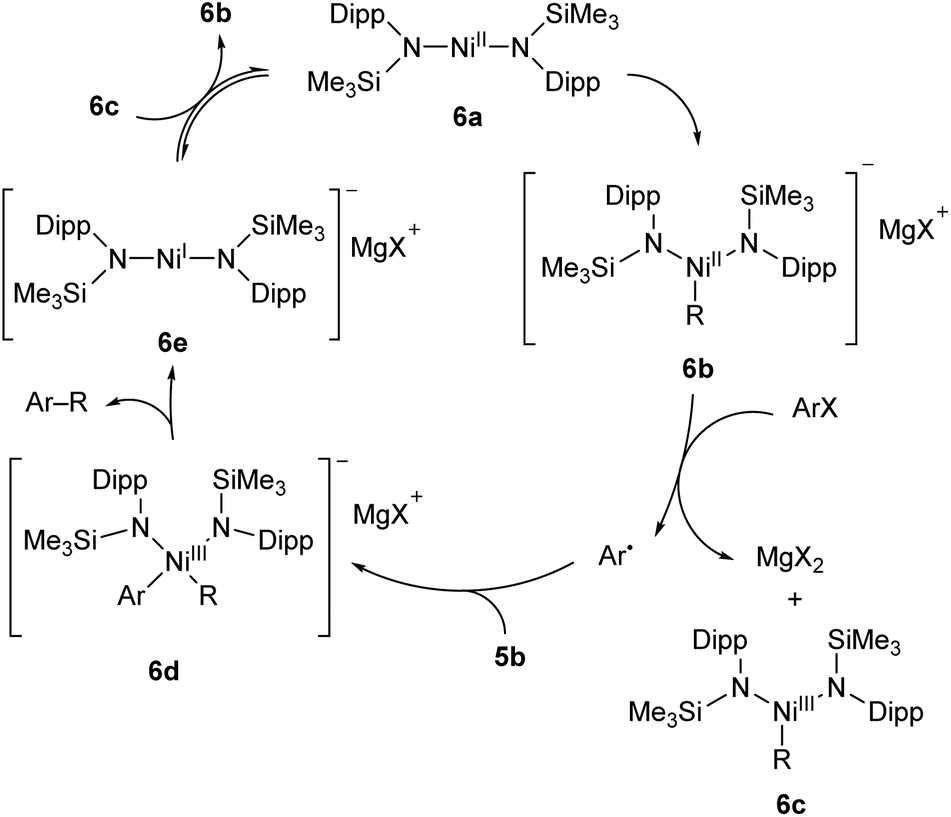 | ||
| Scheme 6 Proposed reaction mechanism for Kumada cross-coupling catalysed by low-coordinate Ni(II) species 6a.48 | ||
Treatment of the Ni(II) chloride pincer complexes 7a–b with lithium triethylborohydride affords the four-coordinate Ni(II) hydrides 8a–b.51 These complexes can reversibly lose hydrogen, and under ambient pressure exist predominantly as the three coordinate Ni(I) species 9a–b (Scheme 7a, Fig. 1). The dehydrogenation reaction is second order with respect to 8b, and likely proceeds via a biomolecular elimination reaction.51 These compounds catalyse hydrodehalogenation reactions, such as the dehalogenation of geminal dihalogenides,51 and defluorination of geminal difluorocyclopropanes to fluoroalkenes.52 Such reactions are regarded as a promising route to partially halogenated compounds from readily available perhalogenated species.53 Both reactions are stereoselective, with moderate ee (enantiomeric excess) values for the monohalides of 20–74%,51 and high Z-selectivity in the formation of alkenes from cyclopropanes.52 Reactions with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) resulted in catalyst inhibition, suggesting that the mechanism is radical in nature.51,52 This was further supported by reactions with radical clock reagents.51 The proposed catalytic cycle for the dehalogenation of geminal dihalogenides is shown in Scheme 7b.51
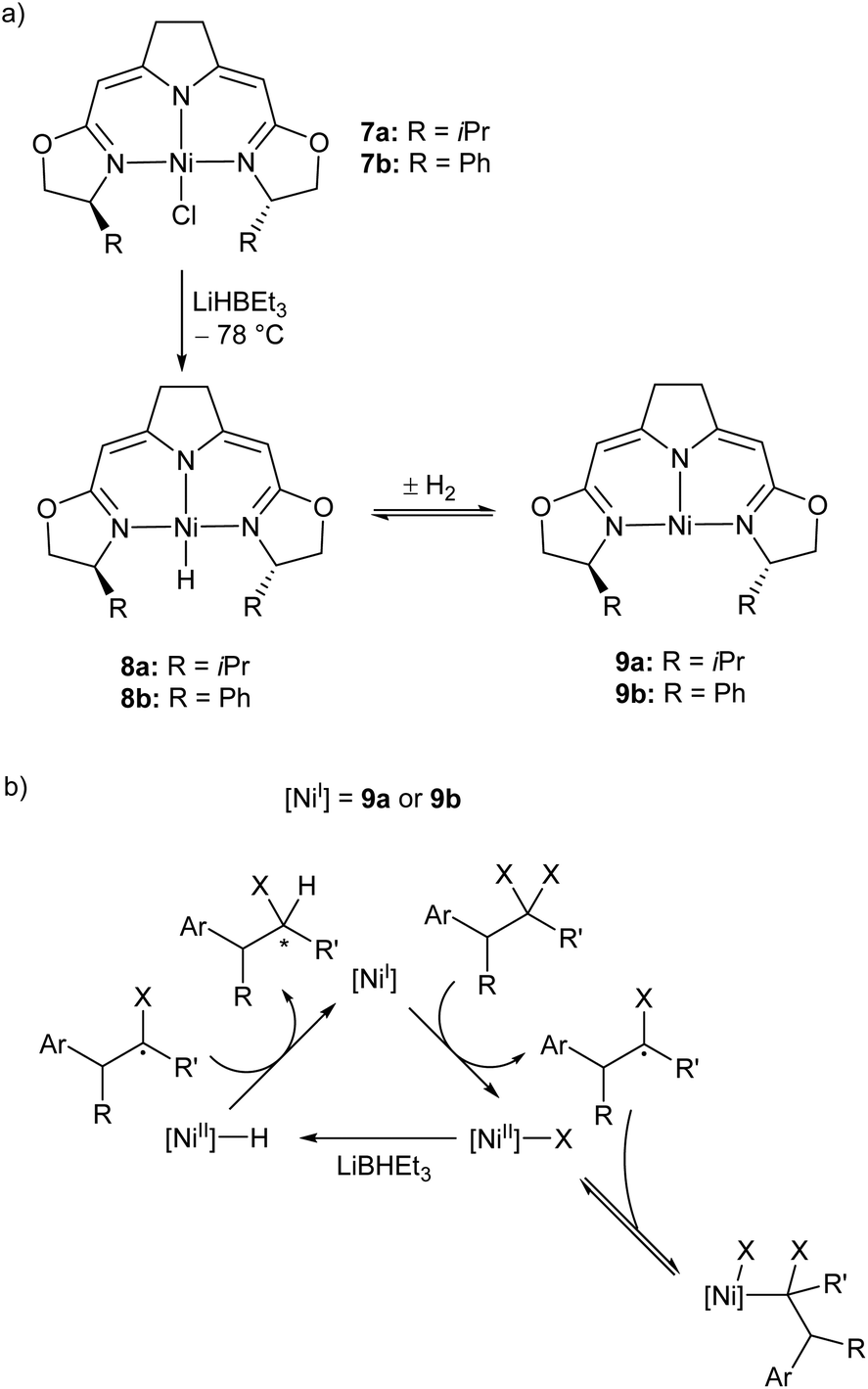 | ||
| Scheme 7 (a) Synthesis of the three-coordinate Ni(I) species 9a and 9b. (b) Proposed mechanism for catalytic, stereoselective hydrodehalogenation of geminal dihalogenides by 9a and 9b. LiBHEt3 is used in excess as a reductant and hydride source.51,52 | ||
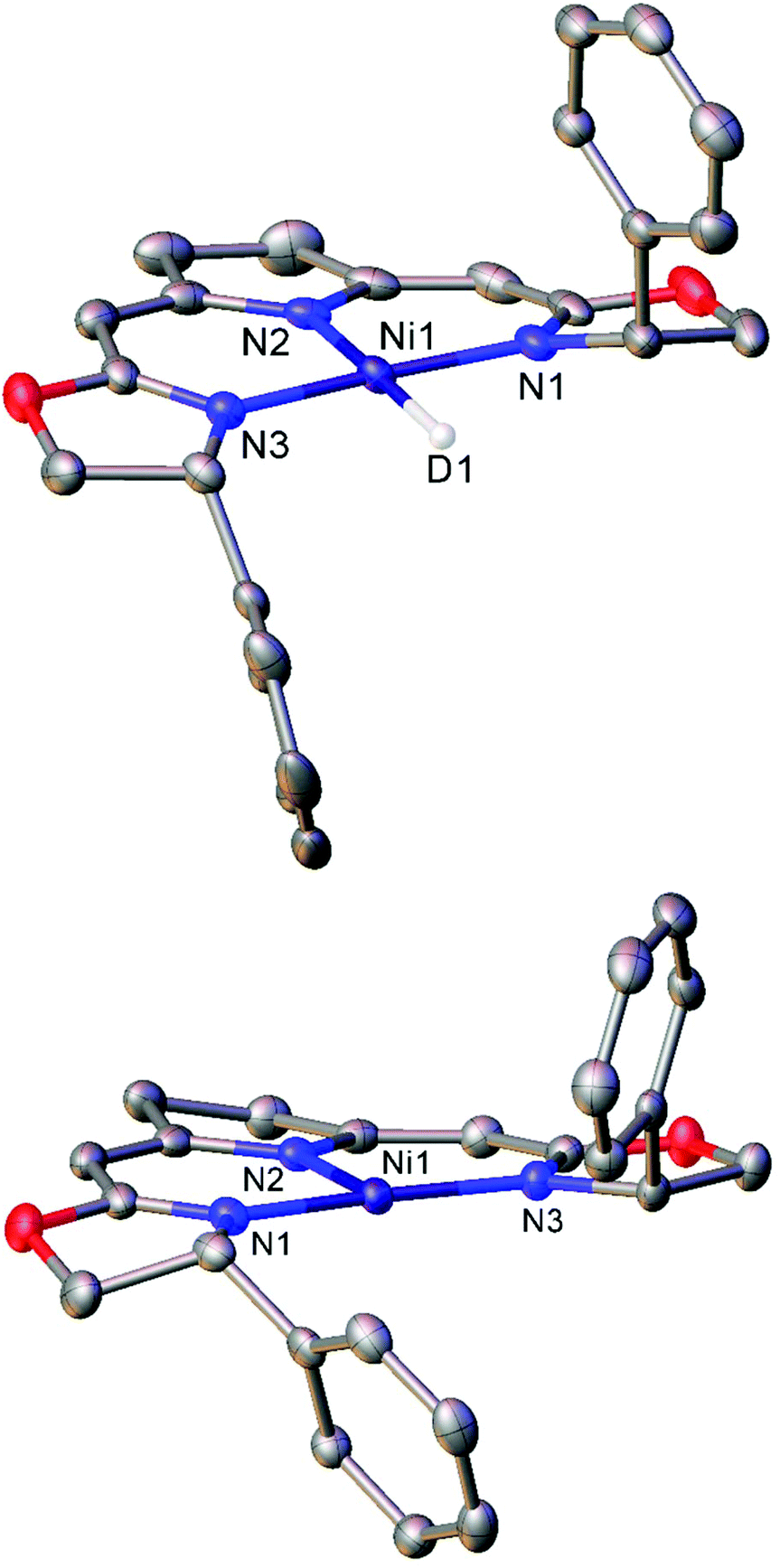 | ||
| Fig. 1 Molecular structures of the deuterium analogue of 8b (top) and three-coordinate Ni(I) complex 9b (bottom). Hydrogen atoms and second molecule in asymmetric unit (8b) omitted for clarity. Anisotropic displacement ellipsoids are set at 50% probability.51 | ||
The reaction between proligand 10, nBuLi, and trans-[Ni(PPh3)2(Ph)Cl] resulted in the formation of Ni(I) complex 11, with concomitant elimination of biphenyl (Scheme 8a).54 This spontaneous reduction of the nickel centre presumably occurs to reduce the steric demands arising from the bulky bidentate ligand. This three-coordinate species was found to be a remarkably active catalyst for the polymerisation of norbornene in the presence of a methylaluminoxane (MAO) co-catalyst (Scheme 8b), affording high molecular weight polynorbornene (Mwca. 106 g mol−1) with catalytic activities of up to 2.82 × 107 gPNP mol−1Ni h−1.54
 | ||
| Scheme 8 (a) Synthesis of three-coordinate Ni(I) complex 11 with elimination of biphenyl from a Ni(II) source. (b) Polymerisation of norbornene by 11 in the presence of MAO co-catalyst.54 | ||
Cobalt
Several two- and three-coordinate cobalt catalysts bearing N-heterocyclic carbene (NHC) ligands have been published by Deng and co-workers.55–59 The three-coordinate Co(I) species 12 was shown to catalyse the hydrosilylation of terminal alkynes, showing broad functional group tolerance and moderate to good selectivity for formation of the E-alkene (Scheme 9a).56 Subsequently, the Co(II) amide species 13, bearing an asymmetric NHC ligand, catalysed the hydrosilylation of terminal alkenes affording predominantly anti-Markovnikov products (Scheme 9b).57 This catalyst was most effective for reactions with triethoxysilane; reactions with other silanes, including triphenylsilane and triethylsilane, gave poor conversions and low yields.57 Following this investigation, a selection of two-, three-, and four-coordinate Co(I) NHC complexes were able to achieve different selectivity (Markovnikov, anti-Markovnikov, or hydrogenation) for the reaction of diphenylsilane with terminal alkenes, dependent on the choice of catalyst (Scheme 9c).58 While cobalt hydrosilylation catalysts are widely known, those displaying Markovnikov or hydrogenation selectivity are relatively rare,58,60,61 so these cobalt NHCs provide a valuable addition to the field.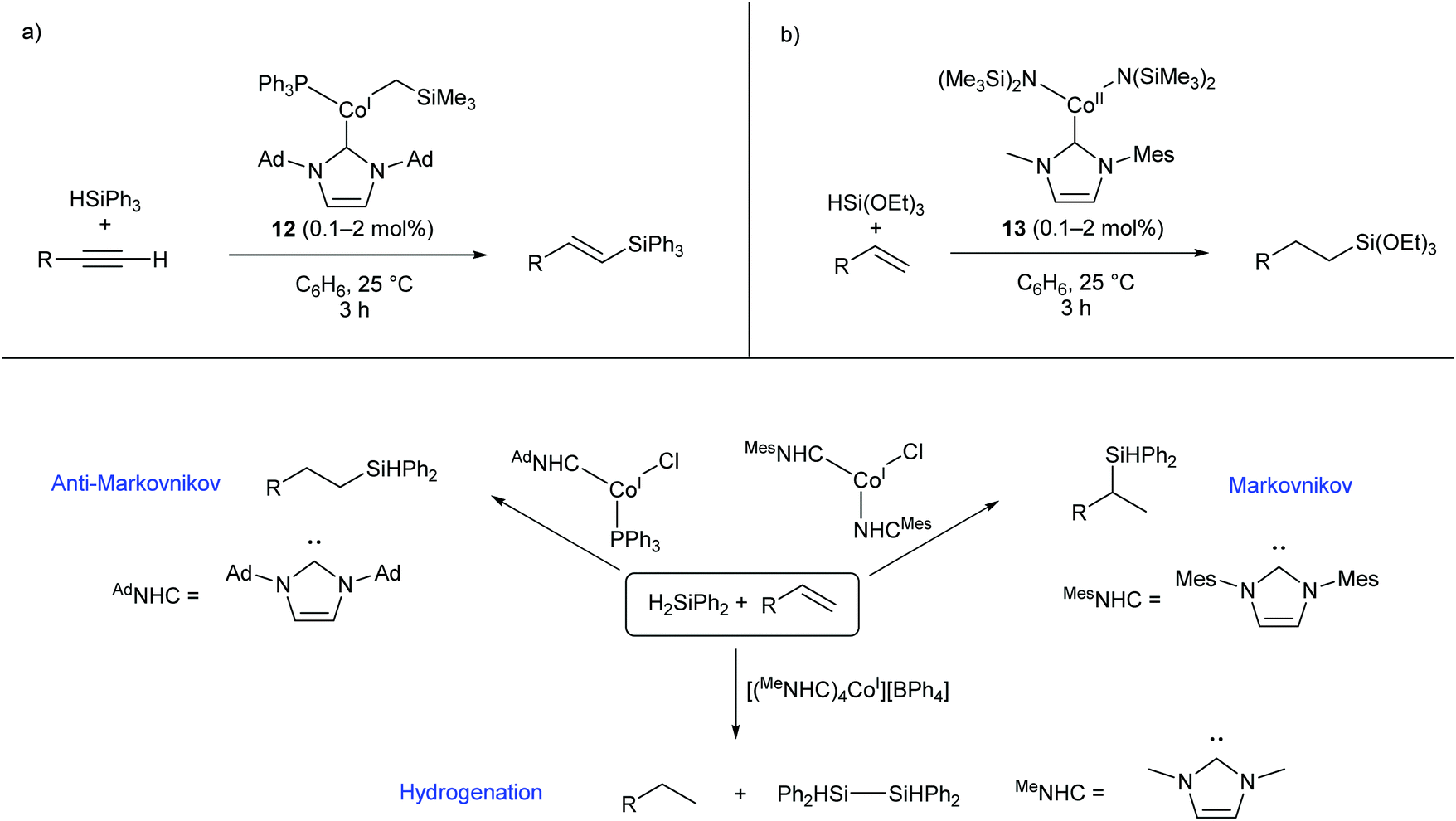 | ||
| Scheme 9 Cobalt NHC complexes in hydrosilylation reactions. (a) Complex 12, featuring an NHC and phosphine ligand, selectively gives E-alkenes from the reaction of terminal alkynes with triphenylsilane. (b) Co(II) amide NHC complex 13 gives anti-Markovnikov products in the reaction of triethoxysilane with terminal alkenes. (c) Different Co(I) NHC complexes afford different selectivities in the reaction of diphenylsilane with terminal alkenes. Ad = 1-Adamantyl.55–58 | ||
More recently, the three-coordinate Co(0) complexes 14 and 15 were shown to catalyse the dehydrocoupling of primary aryl-phosphines to the corresponding diphosphanes (Scheme 10), an unusual example of a cobalt catalyst for such a transformation.59 The catalysts afforded the coupled products in moderate to good yields (47–73%), but were ineffective with the secondary phosphine Ph2PH and primary alkyl phosphine tBuPH2 (7% and 8% yields, respectively).59
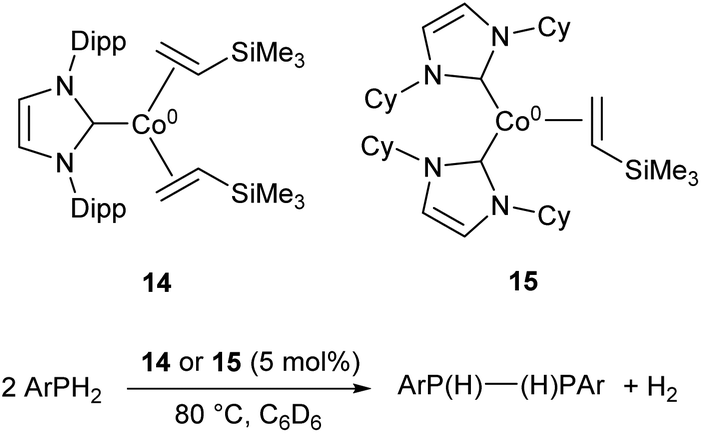 | ||
| Scheme 10 Dehydrocoupling of primary aryl phosphines by Co(0) NHC complexes 14 and 15.59 | ||
Manganese and iron
In recent work by our research group, a series of Mn(II) and Fe(II) m-terphenyl complexes were found to catalyse the cyclotrimerisation of primary aliphatic isocyanates (Scheme 11).62 The reactions proceeded cleanly under mild conditions to afford isocyanurates, which find use as polymer additives to improve their physical properties,63,64 in addition to applications in microporous materials,65,66 selective ion bonding,67 and drug delivery.68–71 Two different flanking aryl groups were employed in the m-terphenyl ligands; [2,6-Mes2C6H3]−, and the less sterically encumbering [2,6-Tmp2C6H3]− (Tmp = 2,4,5-Me3C6H2), giving catalytically active manganese and iron complexes (16–19, Scheme 11a). The Mes-substituted complexes are two-coordinate, while the less bulky Tmp-substituted ligands stabilise three-coordinate complexes, with metal-coordination by an additional THF ligand. While 17 and 19 showed no significant difference in reactivity, two-coordinate 16 was found to give a faster reaction rate than 18. Catalyst poisoning and radical trap experiments showed no evidence for the formation of catalytic nanoparticles or the involvement of radical processes, and, as a result, the mechanism is postulated to proceed via a homogeneous pathway. Kinetic experiments in the reaction of 16 with ethyl isocyanate showed a first order rate dependence in both 16 and substrate. Based on these observations, a mechanism was proposed involving Lewis acid catalysis (Scheme 9c).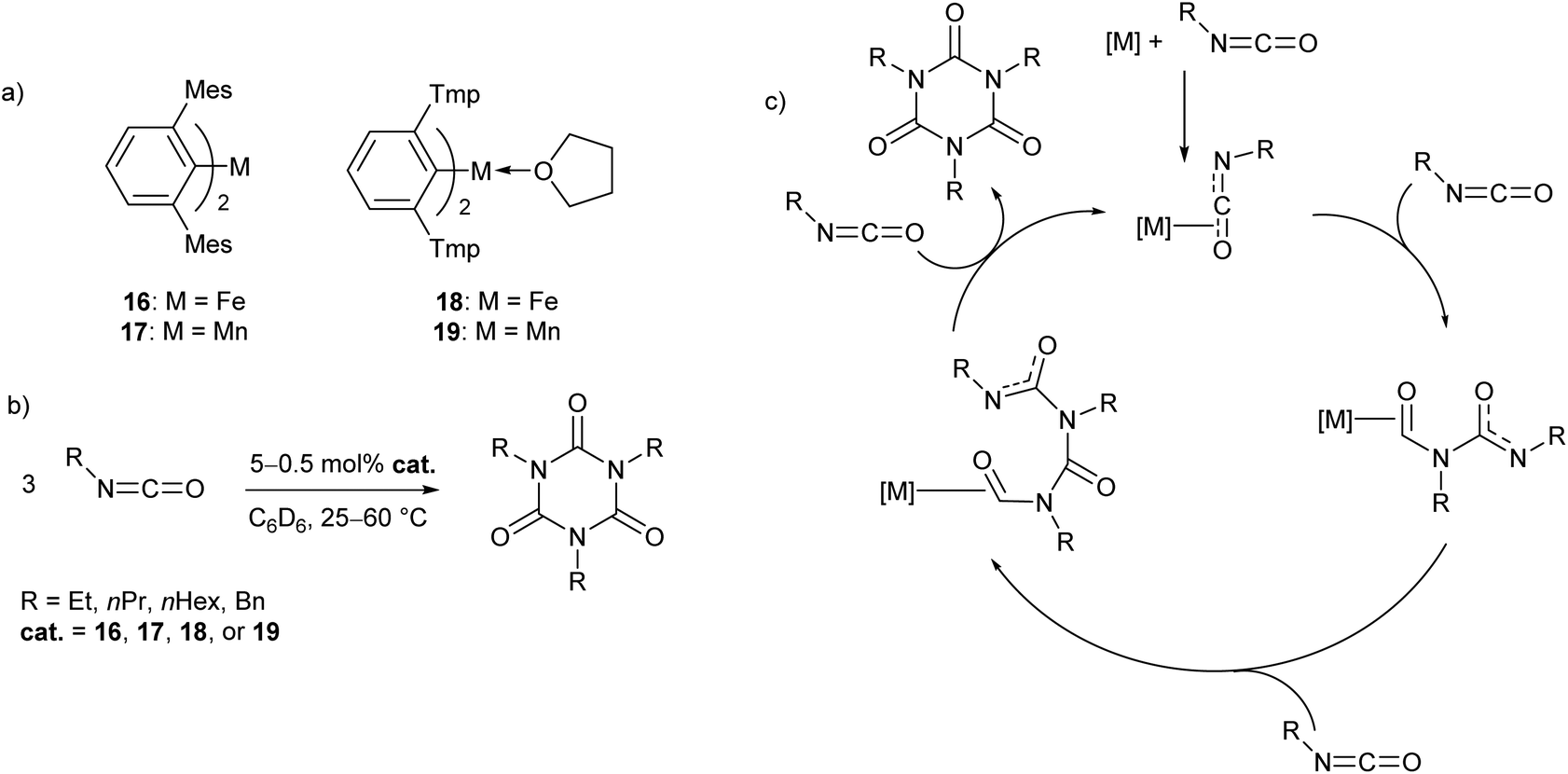 | ||
| Scheme 11 Cyclotrimerisation of isocyanates with low coordinate m-terphenyl metal complexes. (a) Metal complexes used as precatalysts in the cyclotrimerisation reaction. (b) General reaction scheme for cyclotrimerisation of primary aliphatic isocyanates. (c) Proposed Lewis acid mechanism for reaction.62 | ||
The manganese complexes 17, 19 (Scheme 11a) and the xylyl-substituted analogue catalyse the dehydrocoupling of dimethylamine-borane (Scheme 12),72 which is of interest in the chemical storage and release of hydrogen.73–76 While the reaction was slow at room temperature, increasing the reaction temperature to 60 °C provided relatively clean conversion to the cyclic dimer [Me2NBH2]2 in high yields and reasonable timeframes. The linear species Me2NH–BH2–NMe2–BH3 was also observed as a side product and intermediate in the reaction. While reactions with the two-coordinate catalyst 17 (and its xylyl analogue) showed no appreciable change upon addition of elemental mercury, reactions with the three-coordinate 19 showed a significant drop in activity. Furthermore, reaction of 19 with Me2NH·BH3 results in the rapid formation of a dark red suspension, while 17 remains a clear yellow solution. This evidence suggests that the reaction with 19 is heterogeneous in nature, involving the formation of catalytic nanoparticles, while reactions with 17 proceed via a homogeneous route. This was confirmed by isolation of the manganese nanoparticles from reactions between 19 and Me2NH·BH3, followed by characterisation by transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDX).72 This reaction serves as an example of how small changes in the steric properties of the flanking aryl groups of m-terphenyl ligands can result in significant differences in reactivity.
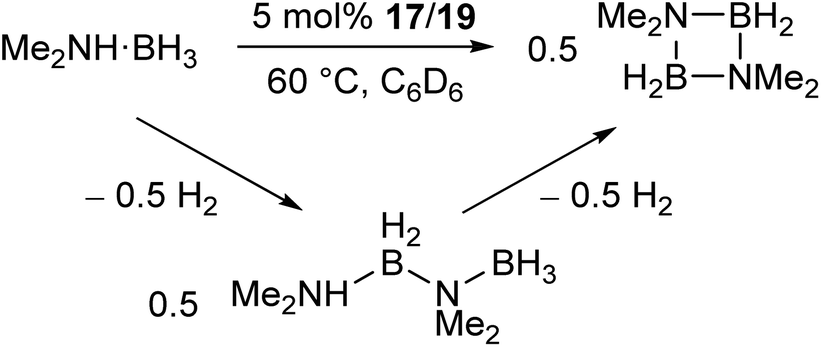 | ||
| Scheme 12 Dehydrocoupling of Me2NH·BH3 catalysed by low coordinate Mn(II) terphenyl complexes.72 | ||
The iron(II) m-terphenyl complexes 16 and 18 (Scheme 11a) are effective catalysts for the hydrophosphination of isocyanates.77 This reaction affords a mixture of phosphinocarboxamide and phosphinodicarboxamide products, corresponding to both mono- and diinsertion of the isocyanate into the P–H bond (Scheme 13a). Such diinsertion reactions are rare78 and the double insertion of an isocyanate into a P–H bond has led to a family of phosphinodicarboxamide compounds. By changing the reaction conditions, this reaction can be made selective to afford either the phosphinocarboxamide (reaction in THF solvent) or phosphinodicarboxamide (through the reaction in benzene or toluene solutions in the presence of Et3N·HCl) (Scheme 13b).
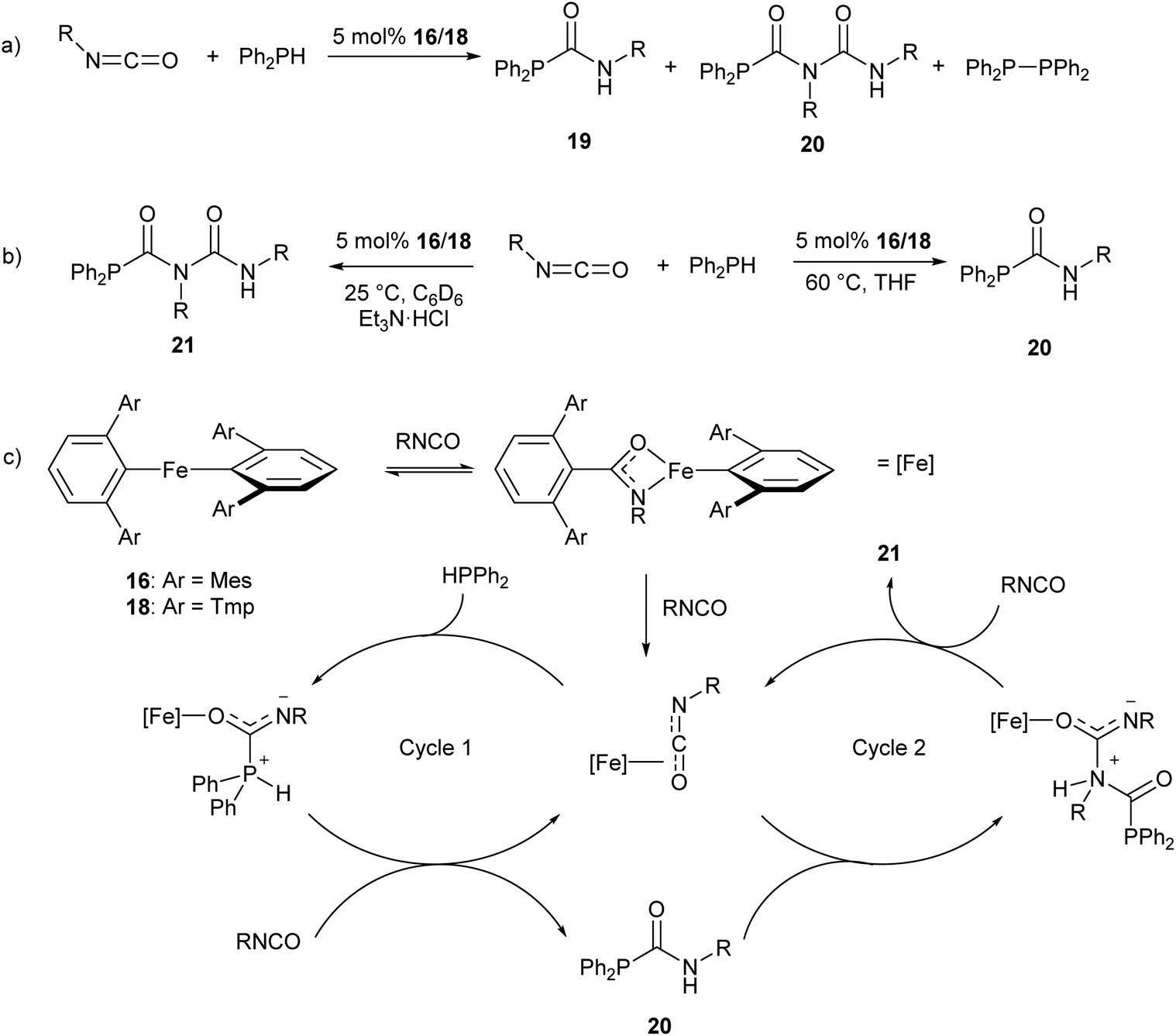 | ||
| Scheme 13 Iron(II) catalysed hydrophosphination of isocyanates. (a) Initial hydrophosphination conditions affording a mixture of products. R = Ph, p-Tol (p-Tol = 4-MeC6H4), 3,5-(OMe)2C6H3, 4-BrC6H4, nHex, Cy, iPr, tBu. (b) Optimised conditions for selective formation of monoinsertion (20) or diinsertion (21) product. (c) Proposed catalytic cycle for formation of observed products.77 | ||
Reaction monitoring through in situ1H and 31P NMR spectroscopy reveals that the three-coordinate complex 18 shows significantly higher activity than 16, which may be due to the presence of a labile THF ligand. Interestingly, reactions with 16 show an induction period of ca. 2 h, while no such induction period was observed in reactions catalysed by 18. Monitoring of stoichiometric reactions by IR spectroscopy suggest that an iron amidate complex may be the active catalytic species in this reaction (Scheme 13c). Poisoning experiments have suggested that the reaction is homogeneous and does not involve radical processes; and a catalytic cycle where the transition metal acts as a Lewis acid was proposed to account for these observations (Scheme 13c).77
The two-coordinate Fe(II) amide 22 catalyses the hydrosilylation of ketones, affording the corresponding silyl ethers in good to quantitative yields (Scheme 14b).79 The catalyst was effective for the reaction of diphenylsilane with a range of ketones, proceeding cleanly at room temperature with low catalyst loadings (0.01–2.7 mol%). However, the reaction failed with tertiary silanes or silanes with bulky substituents, presumably due to steric effects.79 Species 22 represents an early example of an iron catalyst for this industrially relevant transformation, although it has since been supplanted by more convenient systems.80
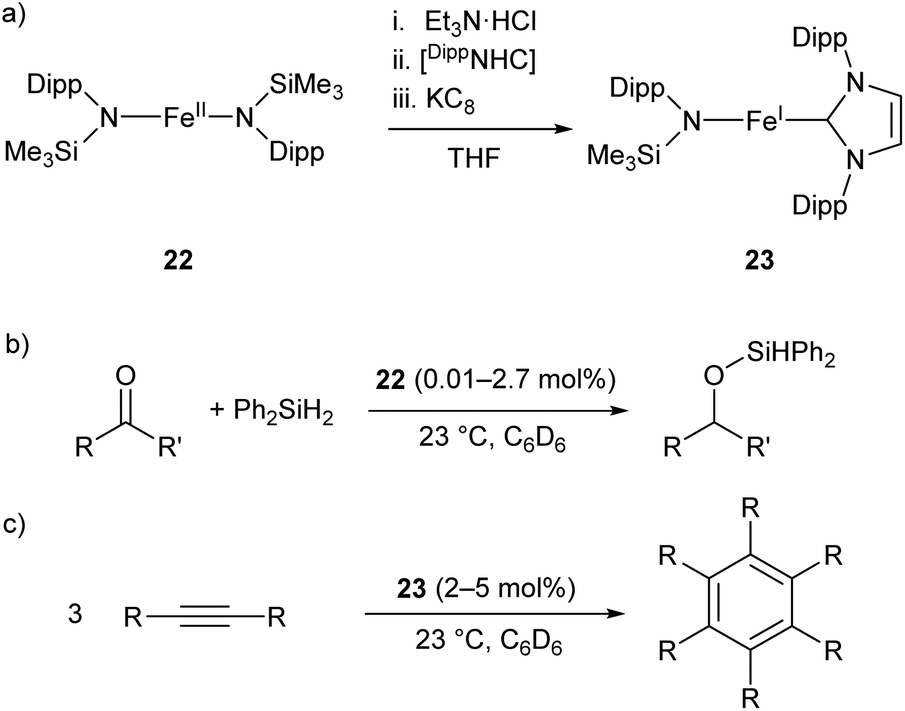 | ||
| Scheme 14 (a) One-pot synthesis of Fe(I) heteroleptic complex 23 from Fe(II) diamide 22. [DippNHC] = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. (b) Hydrosilylation of ketones catalysed by 22. (c) Cyclotrimerisation of alkynes catalysed by 23.79,81 | ||
Complex 22 was later used as the precursor for the (one-pot) synthesis of the first heteroleptic two-coordinate Fe(I) complex 23 (Scheme 14a), which was shown to catalyse the cyclotrimerisation of alkynes to arenes (Scheme 14c).81 This catalyst was effective at loadings of 2–5 mol% at room temperature, but had limited scope, showing poor reactivity towards substrates with bulky or electron-withdrawing substituents. Nonetheless, the reactivity of this complex is comparable to the handful of iron-based catalysts known for this reaction.82,83
Small molecule activation
The activation of small molecules such as N2, CO2, and CO poses significant but exciting challenges. Utilising these molecules generally involves overcoming large energy barriers owing to their high bond strengths and, in some cases, low polarity.84 However, the activation of such molecules is of significant industrial importance in reactions such as the Haber process and Fischer–Tropsch catalysis.85 The development of homogeneous systems for functionalising these relatively inert species is thus an area of considerable research interest. In this section, we will outline some examples of low-coordinate first-row transition metal complexes that are able to bind and activate these small molecules.Nitrogen activation
An early example of a low-coordinate 3d metal binding and activating dinitrogen is the nickel phosphine complex 24a, which was first synthesised in 1968,86 and crystallographically characterised in 1971 (Fig. 2).87,88 In this structure, the N2 unit is protected by a cage of cyclohexyl rings,87 and a related compound featuring PiPr3 ligands (24b) was reported in 2013.89 Another notable early example of N2 binding is seen in the mixed Fe/Mo complex 25, which was the first complex to feature a three-coordinate iron centre coordinated entirely by N2-derived ligands.90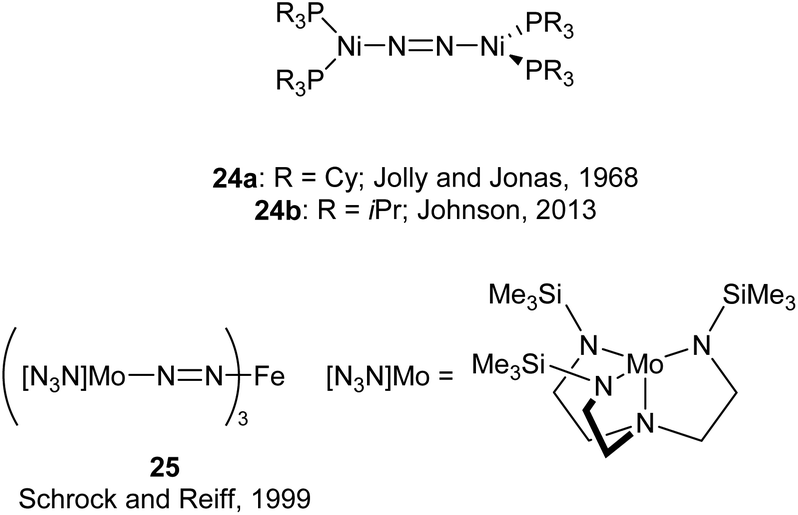 | ||
| Fig. 2 Notable early examples of 3d metal complexes which bind dinitrogen. | ||
Some elegant examples of N2 fixation and activation with low-coordinate 3d metals are seen in the series of iron and cobalt β-diketiminate complexes investigated by the Holland group. Reduction of the metal(II) chloride species (26a–c) using KC8 under a nitrogen atmosphere affords the dinuclear N2-bridged complexes 27a–c (Scheme 15).91–95 Of these N2-bridged species, complex 27b was shown to be an effective pre-catalyst for the synthesis of asymmetric carbodiimides from organoazides and isocyanates.21 The reduction of a THF solution of 26c with Reike magnesium (Mg*) under a N2 atmosphere afforded the highly unusual complex 28c, which features a magnesium bridging between two N2 ligands.96 This complex was sufficiently stable for isolation and characterisation by single crystal X-ray diffraction. The corresponding Mg* reductions with 26a and 26b afforded the metastable Fe complexes 28a and 28b, which could only be characterised in THF solution by infrared (νNN = 1808 cm−1 for 28a; 1818 cm−1 for 28b) and Mössbauer spectroscopy.
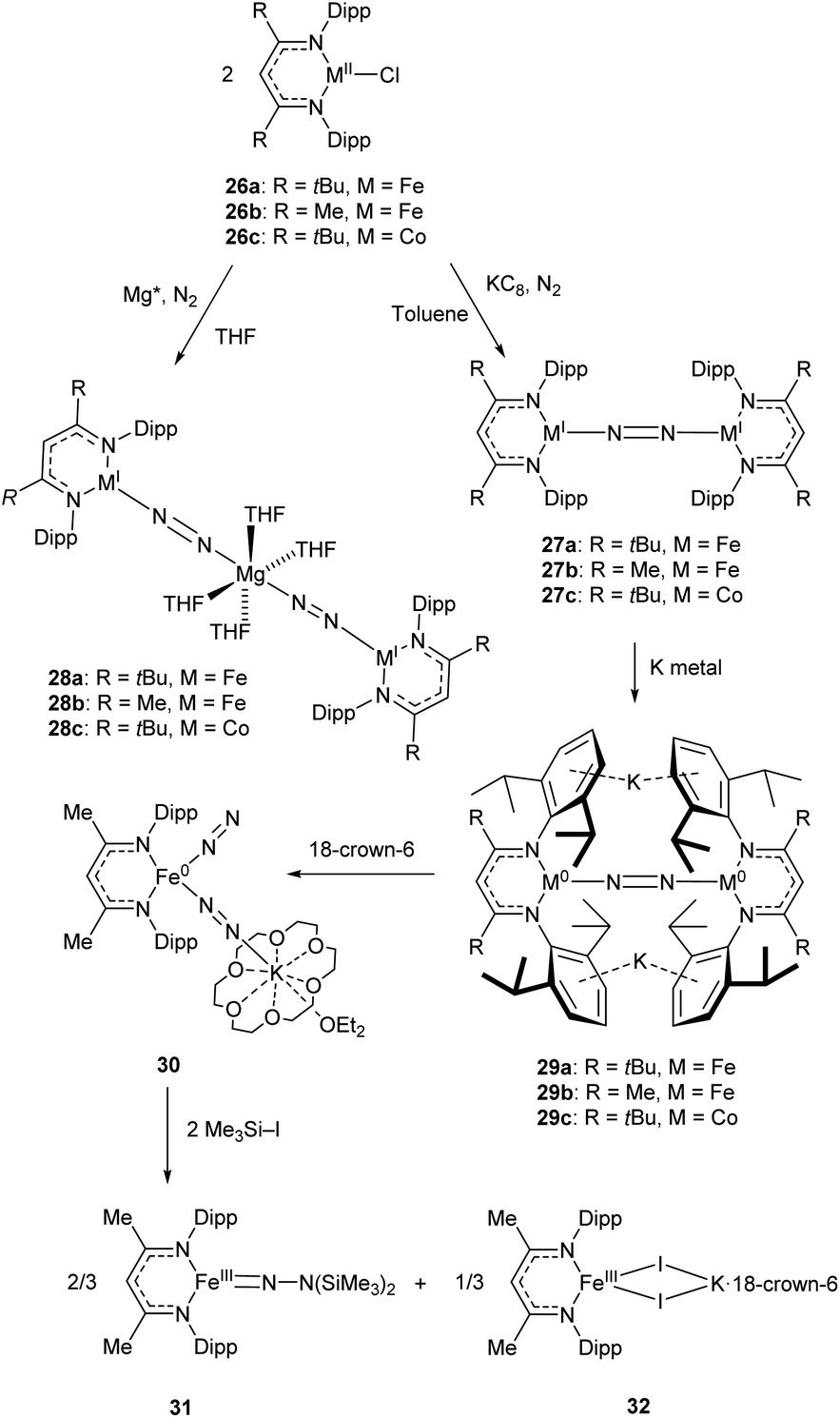 | ||
| Scheme 15 Synthesis and subsequent reactivity of three-coordinate Fe and Co dinitrogen bridged complexes.91–97 | ||
Complexes 27a–c can also be reduced by potassium to afford the metal(0) species 29a–c, which features two K+ ions coordinated to the aromatic Dipp rings.91–95 The side-on coordination of K+ results in elongated N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds [29a 1.233(6) Å; 29b 1.215(6) Å; 29c 1.220(2) Å],91–95 compared to those observed in 27a–c [27a 1.189(4) Å; 27b 1.186(7) Å; 27c 1.139(2) Å].91–95 Treatment of the reduced iron species 29b with 18-crown-6 affords a new four-coordinate complex 30, in which potassium is coordinated by an N2 ligand.97 This species is active towards silylation, reacting with Me3SiI to afford the three-coordinate Fe(III) hydrazido species 31 along with Fe(II) iodide 32.97 It should be noted that reacting 29b with Me3SiI directly affords 31 in low yields (ca. 5%) with long reaction times.
N bonds [29a 1.233(6) Å; 29b 1.215(6) Å; 29c 1.220(2) Å],91–95 compared to those observed in 27a–c [27a 1.189(4) Å; 27b 1.186(7) Å; 27c 1.139(2) Å].91–95 Treatment of the reduced iron species 29b with 18-crown-6 affords a new four-coordinate complex 30, in which potassium is coordinated by an N2 ligand.97 This species is active towards silylation, reacting with Me3SiI to afford the three-coordinate Fe(III) hydrazido species 31 along with Fe(II) iodide 32.97 It should be noted that reacting 29b with Me3SiI directly affords 31 in low yields (ca. 5%) with long reaction times.
Complexes 27a–c can be compared with the closely related chromium complex 34, prepared by Theopold and co-workers.98 Despite having a very similar ligand framework, in 34 the two nickel atoms bind dinitrogen side-on rather than end on as in 27a–c. Complex 34 was prepared by magnesium reduction of the Cr(II) iodide 33 in THF solution under a nitrogen atmosphere (Scheme 16). This side-on coordination results in greater lengthening of the N–N bond (1.249(5) Å in 34)98 compared with the end-on coordination of 27a–c.
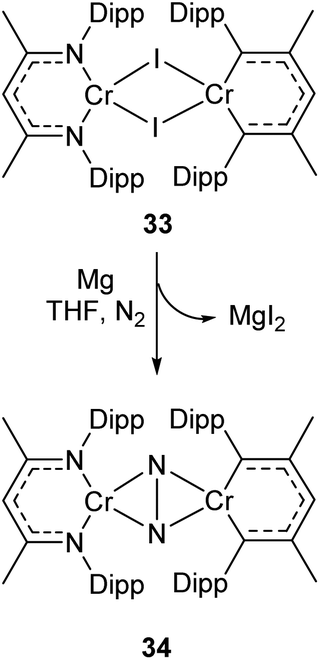 | ||
| Scheme 16 Synthesis of chromium complex 34, featuring side-on N2 coordination.98 | ||
While the binding and partial reduction of dinitrogen in this manner presents exciting possibilities, the full reduction of nitrogen to ammonia by a homogeneous catalyst remains a challenging prospect.99,100 One example of a low-coordinate complex which is capable of reducing N2 in this manner is the Fe cyclic alkyl amino carbene (CAAC) complex 35, recently published by Peters, Ung and co-workers.101,102 This two-coordinate complex was capable of binding N2 reversibly at low temperatures (ca. −78 °C) which resulted in significant changes in the UV/Vis spectra of a solution in pentane.102 The reaction between this complex and KC8 at −95 °C in the presence of 18-crown-6 facilitated the isolation of the Fe(−I) complex 36 (Scheme 17), the structure of which was confirmed by X-ray crystallography (Fig. 3). Attempting this reaction at temperatures above −78 °C resulted in decomposition to a complex mixture of products. The treatment of 35 with an excess of both KC8 and HBArF4·OEt2 ([BArF4]− = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) at −95 °C in diethyl ether under a dinitrogen atmosphere allowed for the catalytic generation of ammonia, albeit with modest turnover numbers (3.3 ± 1.1 equivalents of NH3 per Fe). Reactions at temperatures above −95 °C were ineffective, which is attributed to the poor binding of N2 to 35 at elevated temperatures.102
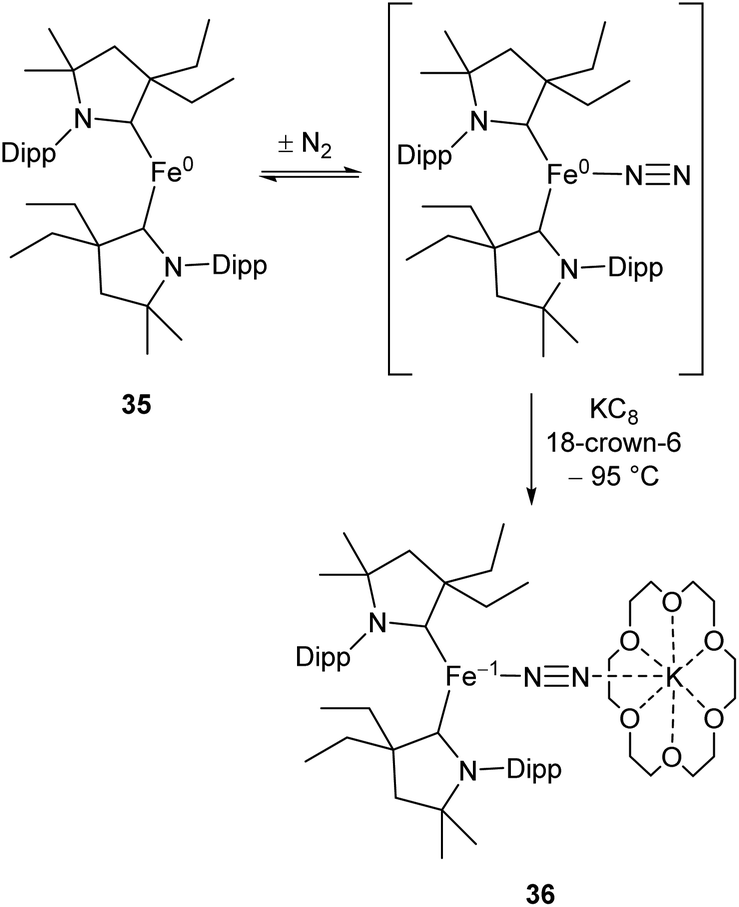 | ||
| Scheme 17 Reversible binding of N2 by Fe(CAAC) species 35 and subsequent reduction by KC8.102 | ||
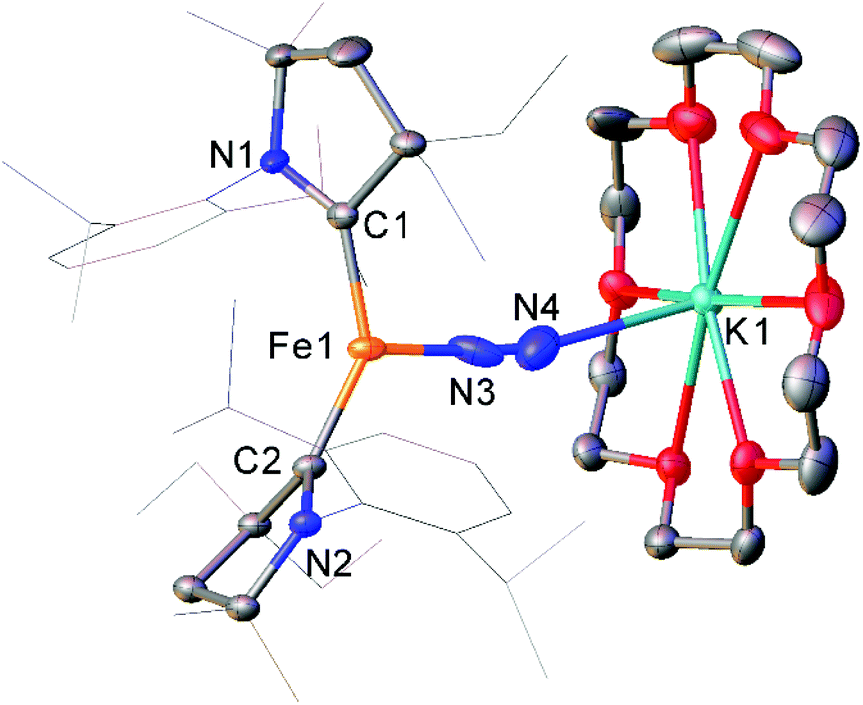 | ||
| Fig. 3 Molecular structure of 36. Hydrogen atoms omitted and flanking groups of CAAC ligands are depicted as wireframe for clarity. Anisotropic displacement ellipsoids are set at 50% probability.102 | ||
Carbon dioxide activation
One common example of carbon dioxide activation by low-coordinate 3d metals is the co-polymerisation of CO2 and epoxides by Zn β-diketiminate complexes, with a significant number of reports on this subject.103–108 The reaction is postulated to be an example of Lewis acid catalysis, and recent work has shown that introducing electron withdrawing CF3 groups into the ligand framework can drastically increase turnover frequency for these catalysts.106–108A Ni complex (37), closely related to the Fe and Co complexes 27a–c shown in Scheme 15, was synthesised by Limberg and co-workers by reduction of the corresponding nickel(II) bromide with potassium triethylborohydride under a nitrogen atmosphere.109 This complex can be further reduced to the Ni(0) species 38 by reaction with KC8. Both complexes were able to activate carbon dioxide, undergoing reductive coupling and cleavage to generate Ni(I)CO (39), Ni(II)CO3 (40), and Ni(II)C2O4Ni(II) (41) species (Scheme 18).110,111 Complex 40 forms a macrocyclic structure in the solid state, consisting of six nickel and six potassium cations, which was characterised by X-ray diffraction (Fig. 4). All of the Ni(II) centres in this structure are square planar and possess low spin configurations. Complex 40 was also synthesised by reacting a Ni(0)CO complex with either N2O or O2, a highly unusual example of CO oxidation at nickel.112 The iron dinitrogen complex 27a (Scheme 15) reacted with CO2 in a similar manner, affording the first four-coordinate iron dicarbonyl complex, and a carbonate-bridged diiron complex.113
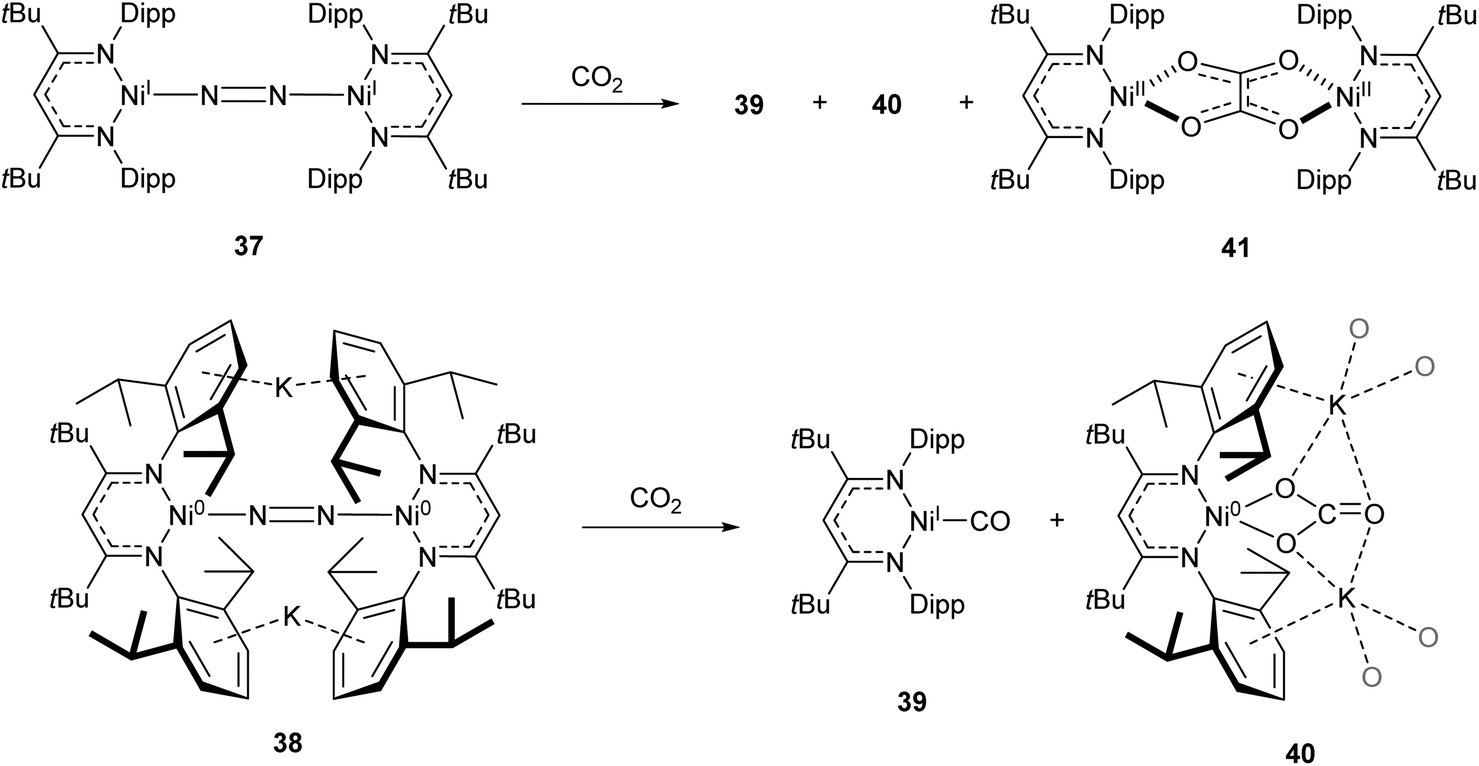 | ||
| Scheme 18 CO2 fixation and activation by dinitrogen-bridged nickel complexes 37 and 38.109–111 | ||
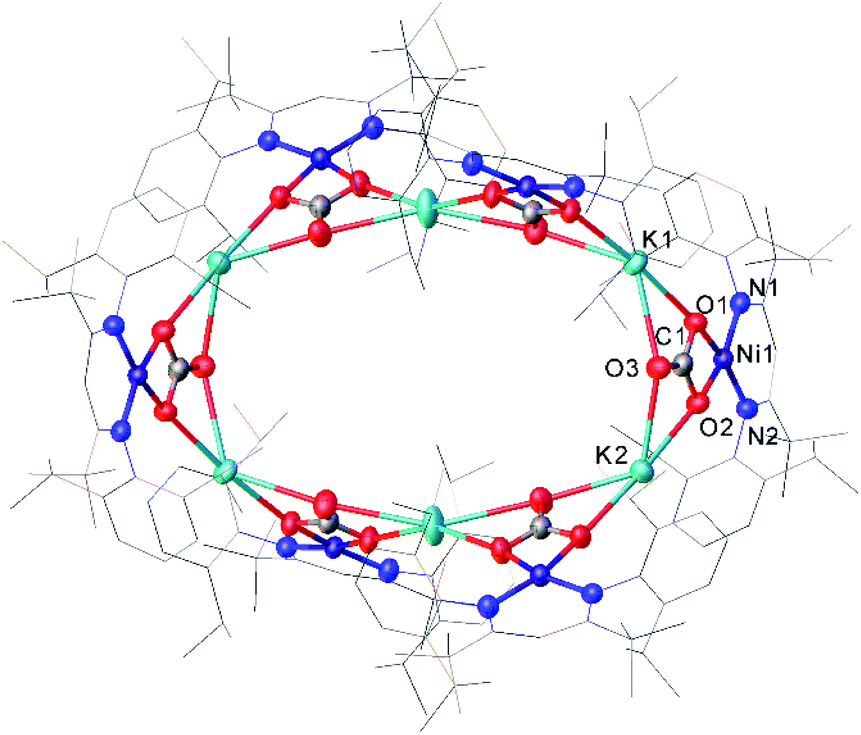 | ||
| Fig. 4 Molecular structure of 40. Hydrogen atoms and co-crystallised hexane omitted and carbon atoms of β-diketiminate ligands are depicted as wireframe for clarity. Anisotropic displacement ellipsoids are set at 50% probability.112 | ||
Similar reactivity towards CO2 was demonstrated by the Co(I) β-diketiminate complex 42, which features a highly unusual slipped κN,η6-arene coordination mode.114,115 The reaction between this compound and CO2 affords the monocarbonyl complex 43 and dicobalt carbonate complex 44 (Scheme 19). The mechanism has been probed by DFT calculations, and is considered to proceed via the oxo-bridged dimer 45, which was synthesised independently by reaction of 43 with N2O. The reaction between 45 and CO2 afforded 44 as the sole product (Scheme 19).115
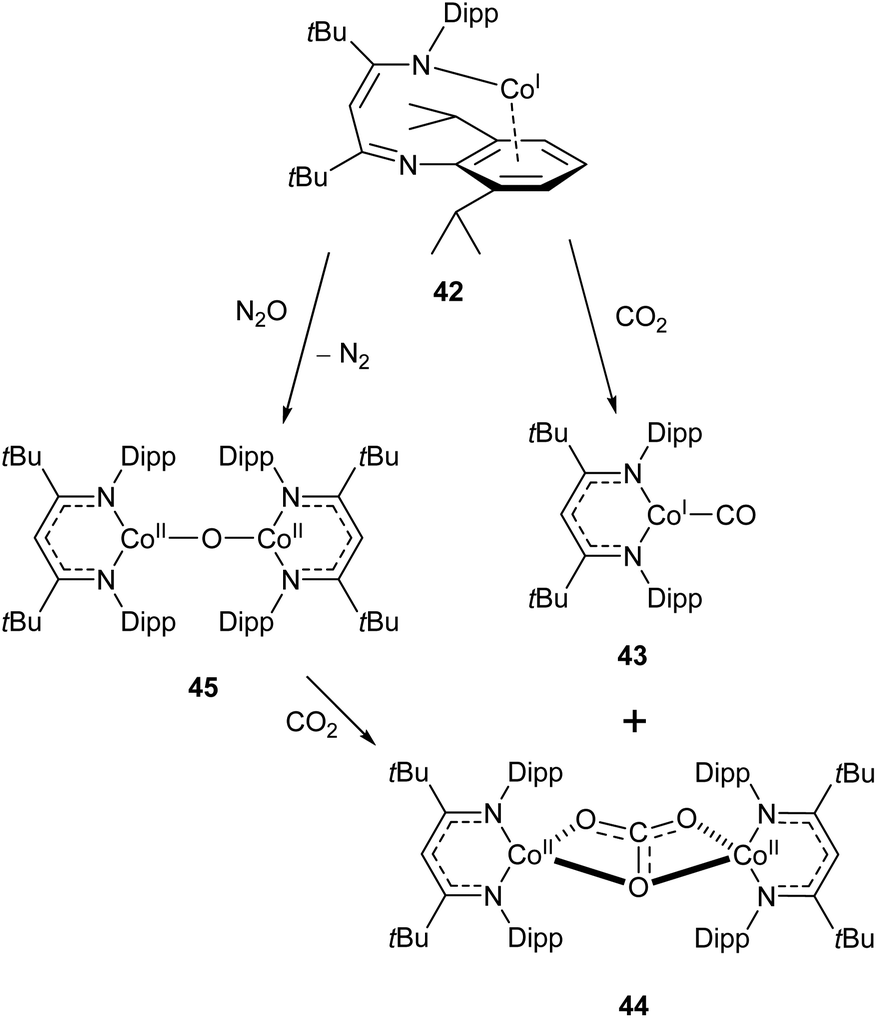 | ||
| Scheme 19 Reactivity of “slipped” Co(I) β-diketiminate complex 42 with CO2 and N2O.114,115 | ||
Reactions between low-coordinate metal amides and carbon dioxide can afford isocyanates, carbodiimides, or (often) a mixture of the two. While the reaction has been performed with metals from the s-,116,117 p-,118–120 and f-block,121,122 zinc- and iron-based systems have shown some of the greatest selectivities,123,124 with a recent iron silylamide (46) affording the corresponding isocyanate with >95% selectivity at CO2 pressures as low as 0.01 atm (Scheme 20a).124 A related reaction is seen with the PNP-pincer Ni(I) complex 47,125 which undergoes a transposition of the ligand N atom on reaction with CO2 to afford a POP-pincer and Ni(I) isocyanate (Scheme 18b).126
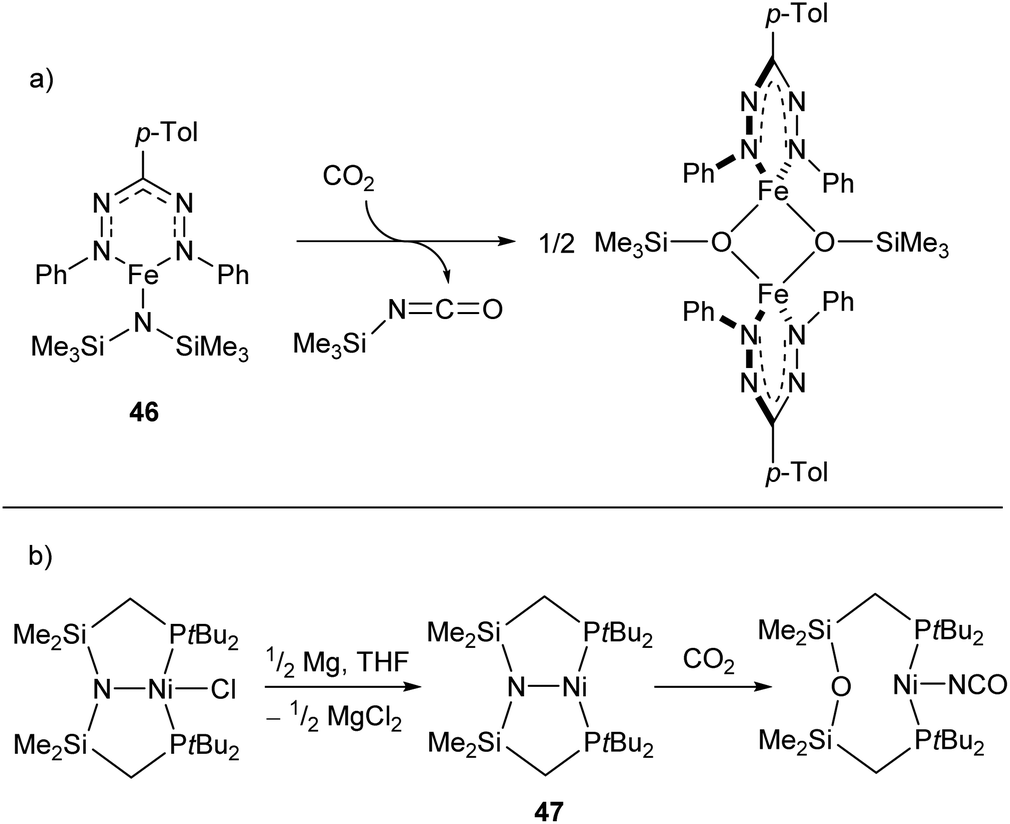 | ||
| Scheme 20 (a) Formation of isocyanate from reaction of CO2 with iron-silylamide complex 46. (b) Transposition reaction of Ni complex 47 with CO2 to generate Ni(I) isocyanate.124,125 | ||
The T-shaped Ni(I) complex 48, which features a rigid acridane-based ligand, was recently investigated by Yoo and Lee.127 This species was obtained by reduction of the corresponding Ni(II) chloride by sodium naphthalenide, and proved capable of activating a diverse range of small molecules under mild conditions. Notably, 48 reduced carbon dioxide under ambient conditions, affording the carboxylate-bridged species 49 (Fig. 5). The complex can also reduce ethene to an ethane bridge, and causes homolytic bond cleavage of a range of molecules; including dihydrogen, methanol, phenol, diphenyl disulfide, methyl iodide, hydrazine, and acetonitrile.127
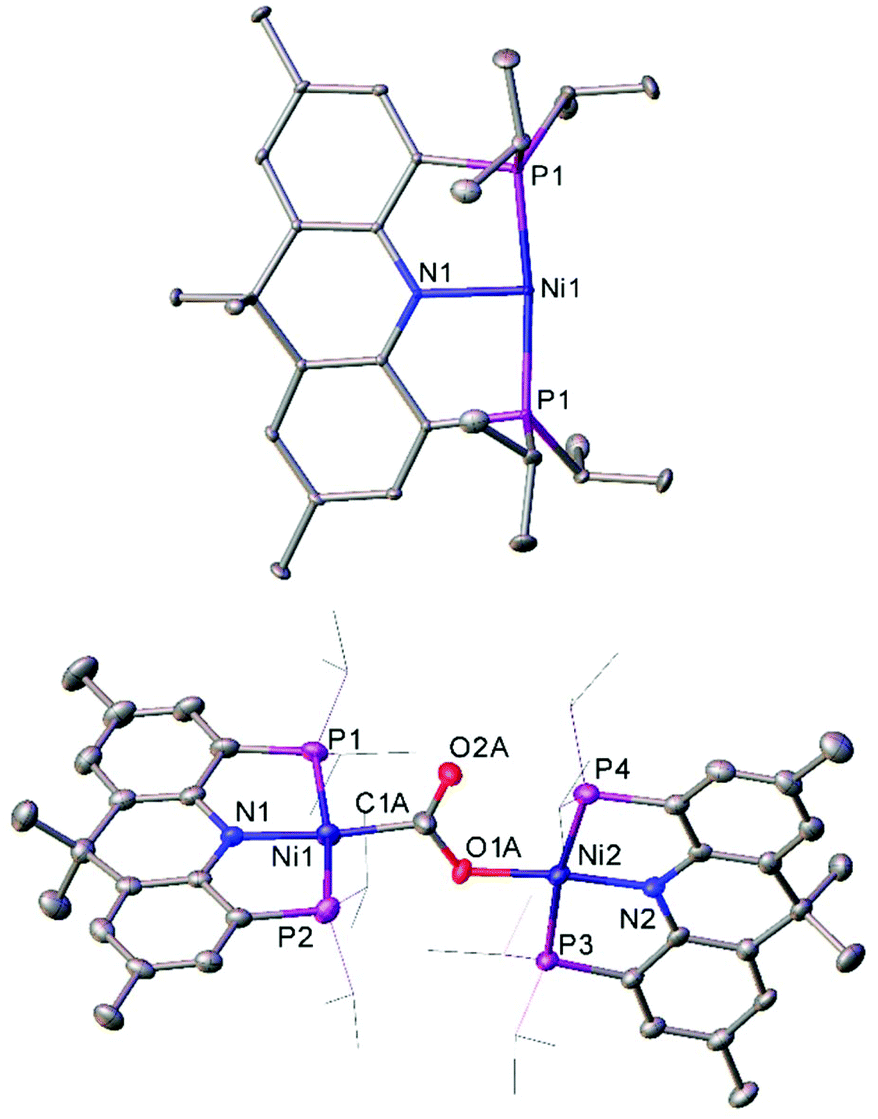 | ||
| Fig. 5 Molecular structure of T-shaped Ni(I) complex 48 (top), and the species obtained after treating with CO2 (49, bottom). Hydrogen atoms, co-crystallised naphthalene (48) and disorder in carboxylate group (49) omitted, and iPr groups (49) are depicted as wireframe for clarity. Anisotropic displacement ellipsoids are set at 50% probability.127 | ||
Reactivity towards carbon monoxide
The activation of carbon monoxide by low-coordinate 3d species is less explored than that of CO2 or N2, with the majority of low-coordinate complexes simply binding CO to give the corresponding metal carbonyls.89,92,125,127 Examples of more interesting reactivity include the chromium dinitrogen complex 34, which reacts with CO to afford the bridged complex 50,98 a relatively rare example of an isocarbonyl complex.128 Compound 50 features activated CO molecules bridging through both the carbon and oxygen atoms with a diamagnetic mixed valent Cr(II)/Cr(0) core (Scheme 21a).98 The three-coordinate Ni(0) carbonyl complex 51 (Scheme 21b) is capable of oxidising CO in the presence of N2O or O2 to give the macrocyclic carbonate complex 40,112 previously seen in Scheme 18 and Fig. 4 as the result of CO2 activation.110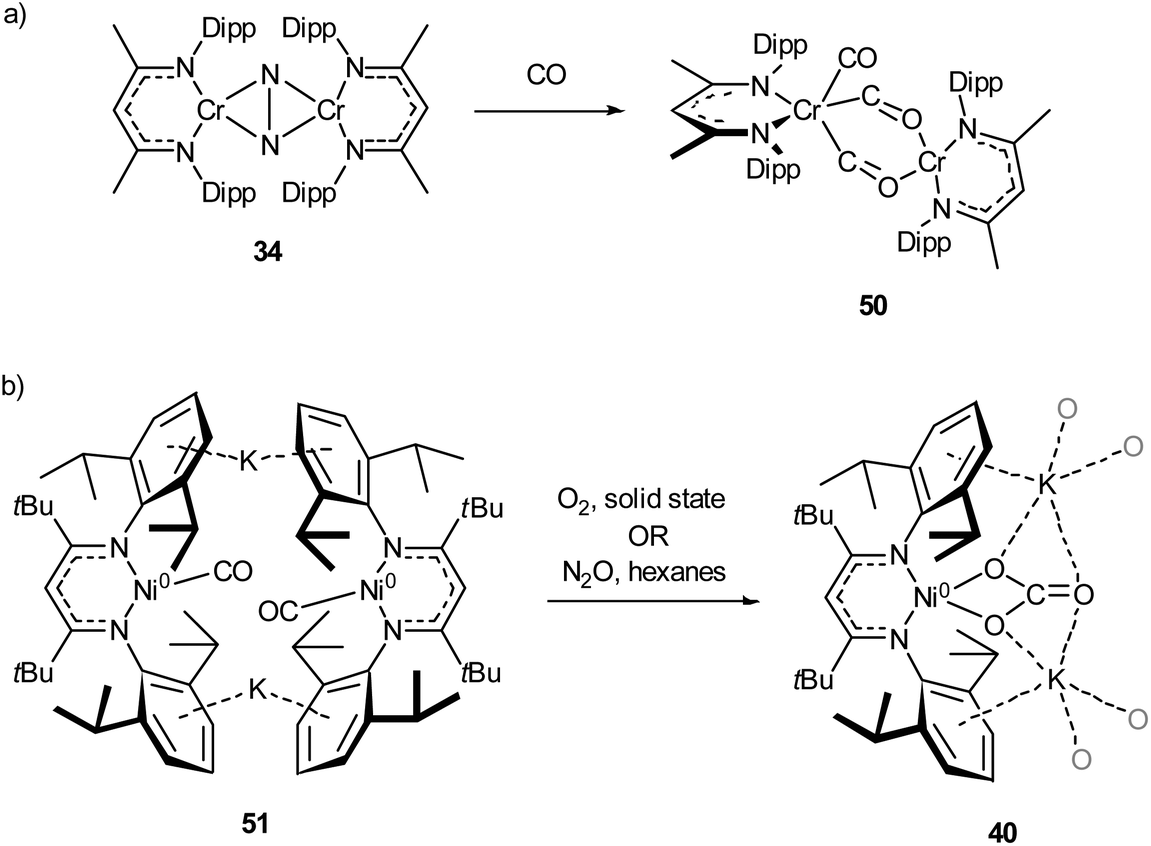 | ||
| Scheme 21 (a) Reaction of chromium dinitrogen complex 34 with CO to give bridged isocarbonyl complex 50.98 (b) Oxidation of CO to carbonate by reaction with O2 or N2O with 51.112 | ||
Of the low-coordinate 3d complexes investigated for CO activation, m-terphenyl complexes have arguably shown the most promise. There are examples of such complexes undergoing CO insertion reactions to afford carbonyl complexes, such as metal acyl species.129,130 The cobalt(II) complexes 52a and 52b reacted with CO to afford sterically encumbered ketones.131 These reactions proceeded cleanly at room temperature affording either a benzophenone (53) or a keto-fluorenone (54) depending on the flanking aryl group (Scheme 22).131
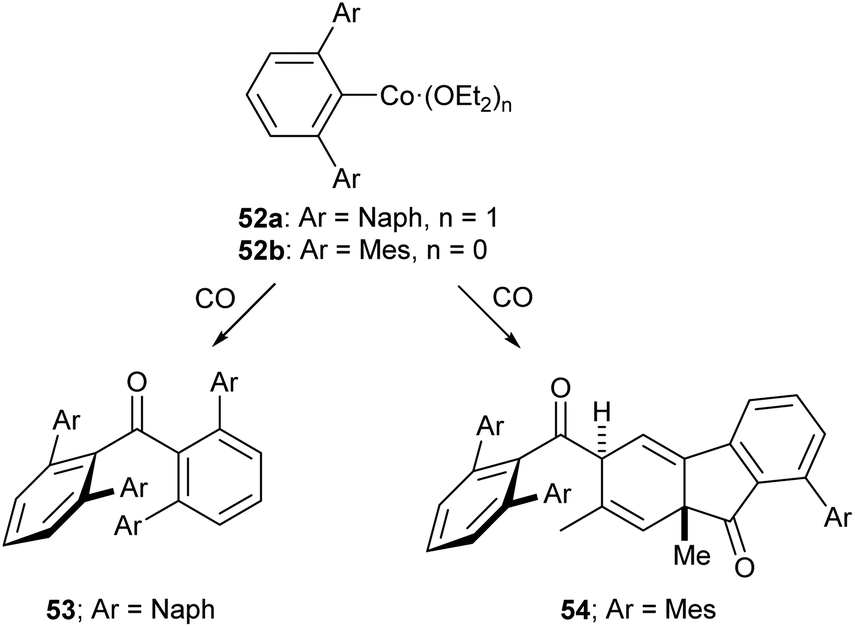 | ||
| Scheme 22 Reactions of Co m-terphenyl complexes with CO to afford sterically encumbered ketones. Naph = 1-Naphthyl, 1-C10H7.131 | ||
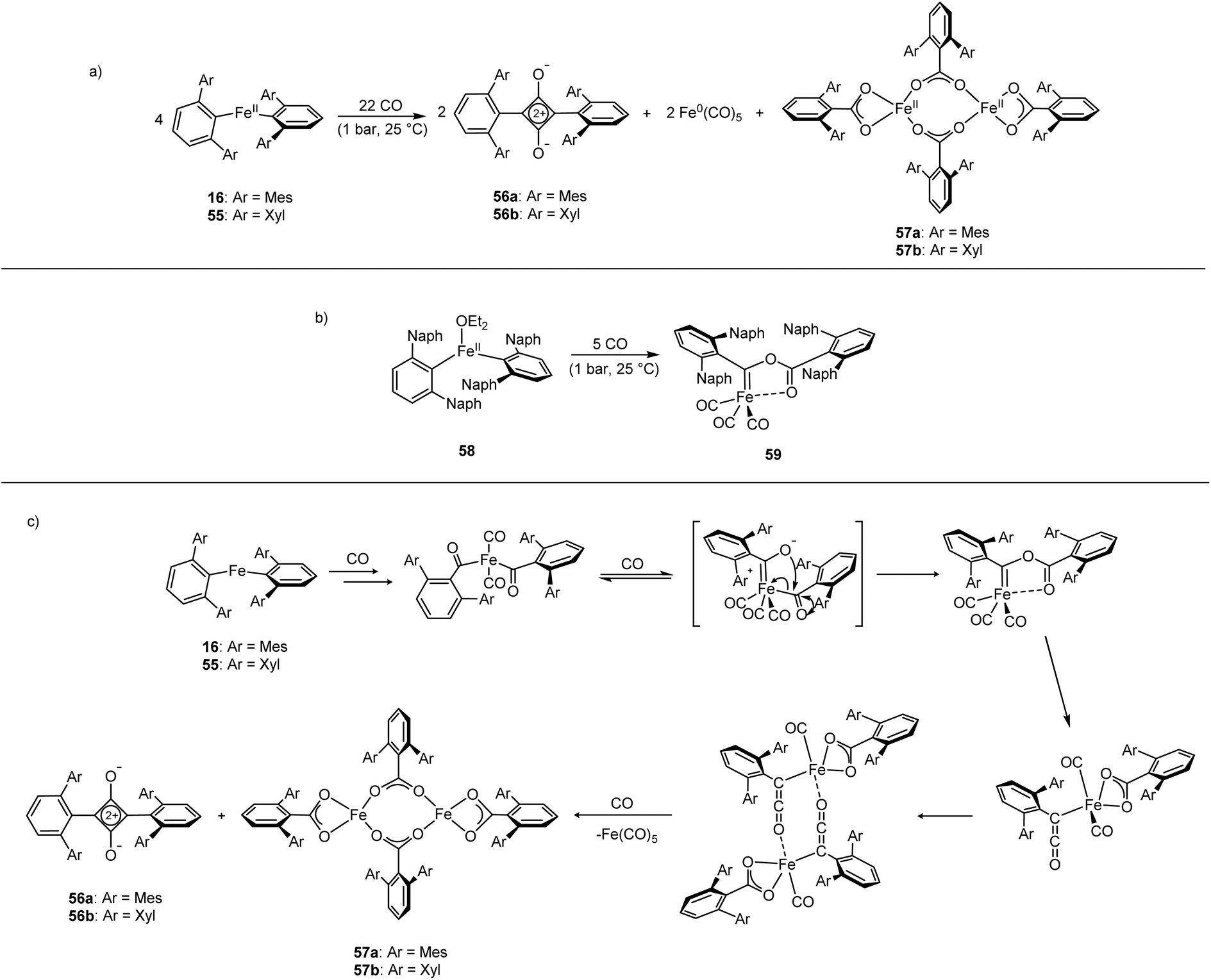
The Fe(II) complexes 16 and 55 reduce CO with complete scission of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond, affording the highly unusual squaraines 56a–b, with concomitant formation of the Fe(II) carboxylate complexes 57a–b and Fe(CO)5 (Scheme 23a, Fig. 6).132 This reaction proceeds cleanly at room temperature and 1 bar pressure, and is both the first example of reductive cleavage of CO by a low-coordinate iron complex133,134 and C4 ring formation from CO with complete CO bond cleavage.135 The squaraines 56a–b feature broken conjugation due to the steric bulk of the aryl substituents, which forces them out-of-plane with the C4O2 ring. This results in less delocalisation into the aromatic rings, giving an unusually high CO IR stretching frequency (56a = 1673 cm−1) and carbonyl chemical shift (56aδc = 269.7 ppm) in comparison to other squaraines.136–139 Reactions with 13CO proved that all four carbon atoms in the central squaraine ring are derived from CO, and monitoring by IR spectroscopy has suggested that ketene or ketenyl (CCO) intermediates may be formed during the reaction. The analogous reaction using the related m-terphenyl complex 58, which features flanking 1-naphthyl substituents, has facilitated the isolation of Fe(II) carbene 59 (Scheme 23b), which is postulated to be an intermediate in the formation of squaraines from 16 and 55. It is proposed that the reaction halts at 59 due to the increased steric demands of the flanking naphthyl groups, which prevent further reaction. Indeed, naphthyl-substituted m-terphenyl complexes are known to display conformational isomerism due to restricted rotation,140 and the results of DFT calculations support the notion that these flanking groups halt the reaction at species 59. Based upon these observations, a mechanism was proposed that accounts for the formation of these products, and fits all the available mechanistic data (Scheme 23c).132
O bond, affording the highly unusual squaraines 56a–b, with concomitant formation of the Fe(II) carboxylate complexes 57a–b and Fe(CO)5 (Scheme 23a, Fig. 6).132 This reaction proceeds cleanly at room temperature and 1 bar pressure, and is both the first example of reductive cleavage of CO by a low-coordinate iron complex133,134 and C4 ring formation from CO with complete CO bond cleavage.135 The squaraines 56a–b feature broken conjugation due to the steric bulk of the aryl substituents, which forces them out-of-plane with the C4O2 ring. This results in less delocalisation into the aromatic rings, giving an unusually high CO IR stretching frequency (56a = 1673 cm−1) and carbonyl chemical shift (56aδc = 269.7 ppm) in comparison to other squaraines.136–139 Reactions with 13CO proved that all four carbon atoms in the central squaraine ring are derived from CO, and monitoring by IR spectroscopy has suggested that ketene or ketenyl (CCO) intermediates may be formed during the reaction. The analogous reaction using the related m-terphenyl complex 58, which features flanking 1-naphthyl substituents, has facilitated the isolation of Fe(II) carbene 59 (Scheme 23b), which is postulated to be an intermediate in the formation of squaraines from 16 and 55. It is proposed that the reaction halts at 59 due to the increased steric demands of the flanking naphthyl groups, which prevent further reaction. Indeed, naphthyl-substituted m-terphenyl complexes are known to display conformational isomerism due to restricted rotation,140 and the results of DFT calculations support the notion that these flanking groups halt the reaction at species 59. Based upon these observations, a mechanism was proposed that accounts for the formation of these products, and fits all the available mechanistic data (Scheme 23c).132
 | ||
| Scheme 23 Reactions of Fe(II) m-terphenyl complexes with CO. (a) Mes and Xyl substituted compounds (16 and 55) react to give squaraines (56a–b), iron carboxylates (57a–b), and Fe(CO)5. (b) Naphthyl substituted 58 reacts with CO to afford iron-carbene 59. (c) Proposed mechanism for the reaction between 16 or 55 with CO.132 | ||
 | ||
| Fig. 6 Molecular structure of sterically encumbered squaraine 56b (top), and iron carboxylate 57b (bottom). Hydrogen atoms omitted and flanking xylyl groups depicted as wireframe for clarity. Anisotropic displacement ellipsoids are set at 50% probability.132 | ||
Conclusions and outlook
Although the use of low-coordinate, first-row metal complexes in catalysis and small molecule activation is a relatively young field, this strategy shows considerable promise for achieving reactivity that would be challenging by other means. Already, a vast array of diverse and exciting transformations has been discovered, and we feel confident that this research area will continue to grow and develop in the future. One thing we noted while composing this perspective is the relatively narrow range of 3d metals that dominate this area. Cobalt, nickel and iron are by far the most thoroughly investigated elements, with manganese, chromium and copper141 appearing less frequently, for example. It is possible that the less-explored elements could yield as-yet unseen reactivity, and we hope to see more work in this area in the future.It is highly likely that the preparation of new bulky ligands with different electronic properties will be key to the development of future low-coordinate catalysts and reagents. Of particular interest are the relatively new class of cyclic (alkyl)(amino)carbenes (CAACs), which are more σ-donating and π-accepting than N-heterocyclic carbenes;142 and the recently developed [2,6-(2,4,6-tBu3C6H2)2C6H3]−, an incredibly sterically encumbering m-terphenyl ligand,143 which was recently exploited in the synthesis of several Sn–Sn bonded compounds.144
Finally, we note the increasing interest in mechanistic investigations of these catalysts, with more researchers undertaking kinetic measurements of these systems rather than simply viewing the reaction as a “black box”. Hopefully this will lead to a greater understanding of the factors that underpin the reactivity of low-coordinate metal species and allow for the future rational design of improved catalytic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Engineering and Physical Sciences Research Council [grant number EP/R004064/1] and the University of Nottingham for their support.References
- D. L. Kays, Chem. Soc. Rev., 2016, 45, 1004–1018 RSC.
- P. P. Power, J. Organomet. Chem., 2004, 689, 3904–3919 CrossRef CAS.
- D. L. Kays, Dalton Trans., 2011, 40, 769–778 RSC.
- D. L. Kays, in Organometallic Chemistry, 2010, vol. 36, pp. 56–76 Search PubMed.
- L. Falivene, A. Poater and L. Cavallo, in N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley-VCH, 1st edn., 2014, pp. 25–38 Search PubMed.
- A. Schmidt, S. Wiechmann and C. F. Otto, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Elsevier Ltd, 2016, vol. 119, pp. 143–172 Search PubMed.
- P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef CAS PubMed.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed.
- L. L. Schafer, P. Mountford and W. E. Piers, Dalton Trans., 2015, 44, 12027–12028 RSC.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- M. J. Page, W. Y. Lu, R. C. Poulten, E. Carter, A. G. Algarra, B. M. Kariuki, S. A. MacGregor, M. F. Mahon, K. J. Cavell, D. M. Murphy and M. K. Whittlesey, Chem. – Eur. J., 2013, 19, 2158–2167 CrossRef CAS PubMed.
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577–581 CrossRef CAS PubMed.
- Y. S. Meng, Z. Mo, B.-W. Wang, Y.-Q. Zhang, L. Deng and S. Gao, Chem. Sci., 2015, 6, 7156–7162 RSC.
- B. Zhou, M. S. Denning, D. L. Kays and J. M. Goicoechea, J. Am. Chem. Soc., 2009, 131, 2802–2803 CrossRef CAS PubMed.
- V. C. Gibson, E. L. Marshall, D. Navarro-Llobet, A. J. P. White and D. J. Williams, J. Chem. Soc., Dalton Trans., 2002, 4321–4322 RSC.
- M.-S. Zhou, S.-P. Huang, L.-H. Weng, W.-H. Sun and D.-S. Liu, J. Organomet. Chem., 2003, 665, 237–245 CrossRef CAS.
- J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857–7870 CrossRef CAS PubMed.
- N. M. Hein, F. S. Pick and M. D. Fryzuk, Inorg. Chem., 2017, 56, 14513–14523 CrossRef CAS PubMed.
- C. Chen, T. R. Dugan, W. W. Brennessel, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 945–955 CrossRef CAS PubMed.
- R. E. Cowley, M. R. Golder, N. A. Eckert, M. H. Al-Afyouni and P. L. Holland, Organometallics, 2013, 32, 5289–5298 CrossRef CAS.
- S. Wiese, M. J. B. Aguila, E. Kogut and T. H. Warren, Organometallics, 2013, 32, 2300–2308 CrossRef CAS.
- A. K. King, A. Buchard, M. F. Mahon and R. L. Webster, Chem. – Eur. J., 2015, 21, 15960–15963 CrossRef CAS PubMed.
- N. T. Coles, M. F. Mahon and R. L. Webster, Organometallics, 2017, 36, 2262–2268 CrossRef CAS.
- M. Espinal-Viguri, A. K. King, J. P. Lowe, M. F. Mahon and R. L. Webster, ACS Catal., 2016, 6, 7892–7897 CrossRef CAS.
- M. Espinal-Viguri, C. R. Woof and R. L. Webster, Chem. – Eur. J., 2016, 22, 11605–11608 CrossRef CAS PubMed.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 16170–16171 CrossRef CAS PubMed.
- J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098–13101 CrossRef CAS PubMed.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215–12218 CrossRef CAS PubMed.
- S. A. Johnson, Dalton Trans., 2015, 44, 10905–10913 RSC.
- N. I. Saper and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 17667–17676 CrossRef CAS PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- A. Thakur and J. Louie, Acc. Chem. Res., 2015, 48, 2354–2365 CrossRef CAS PubMed.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed.
- N. A. Harry, S. Saranya, S. M. Ujwaldev and G. Anilkumar, Catal. Sci. Technol., 2019, 9, 1726–1743 RSC.
- E. Carter and D. M. Murphy, Top. Catal., 2015, 58, 759–768 CrossRef CAS.
- S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932–1934 RSC.
- S. Nagao, T. Matsumoto, Y. Koga and K. Matsubara, Chem. Lett., 2011, 40, 1036–1038 CrossRef CAS.
- K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC.
- L. Iffland, A. Petuker, M. van Gastel and U.-P. Apfel, Inorganics, 2017, 5, 78 CrossRef.
- I. Kalvet, Q. Guo, G. J. Tizzard and F. Schoenebeck, ACS Catal., 2017, 7, 2126–2132 CrossRef CAS PubMed.
- X. Lin and D. L. Phillips, J. Org. Chem., 2008, 73, 3680–3688 CrossRef CAS PubMed.
- M. I. Lipschutz and T. D. Tilley, Angew. Chem., Int. Ed., 2014, 53, 7290–7294 CrossRef CAS PubMed.
- A. N. Vedernikov, ChemCatChem, 2014, 6, 2490–2492 CrossRef CAS.
- M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC.
- C. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. – Eur. J., 2014, 20, 9657–9665 CrossRef CAS PubMed.
- J. Wenz, C. A. Rettenmeier, H. Wadepohl and L. H. Gade, Chem. Commun., 2016, 52, 202–205 RSC.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef PubMed.
- H.-Y. Wang, X. Meng and G. Jin, Dalton Trans., 2006, 2579–2585 RSC.
- J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS.
- Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414–17417 CrossRef CAS PubMed.
- Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798–1801 CrossRef CAS PubMed.
- Y. Gao, L. Wang and L. Deng, ACS Catal., 2018, 8, 9637–9646 CrossRef CAS.
- D. Wang, Q. Chen, X. Leng and L. Deng, Inorg. Chem., 2018, 57, 15600–15609 CrossRef CAS PubMed.
- B. Raya, S. Jing, V. Balasanthiran and T. V. RajanBabu, ACS Catal., 2017, 7, 2275–2283 CrossRef CAS PubMed.
- B. Raya, S. Biswas and T. V. Rajanbabu, ACS Catal., 2016, 6, 6318–6323 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, H. E. L. Williams, T. J. Blundell, W. Lewis, A. J. Blake and D. L. Kays, Chem. Commun., 2017, 53, 937–940 RSC.
- J. M. Kenny, L. Torre and L. M. Chiacchiarelli, J. Appl. Polym. Sci., 2015, 132, 42750 CrossRef.
- G. Wegener, M. Brandt, L. Duda, J. Hofmann, B. Klesczewski, D. Koch, R. Kumpf, H. Orzesek, H.-G. Pirkl, C. Six, C. Steinlein and M. Weisbeck, Appl. Catal., A, 2001, 221, 303–335 CrossRef CAS.
- Y. Zhang, S. N. Riduan and J. Y. Ying, Chem. – Eur. J., 2009, 15, 1077–1081 CrossRef CAS PubMed.
- E. Preis, N. Schindler, S. Adrian and U. Scherf, ACS Macro Lett., 2015, 4, 1268–1272 CrossRef CAS.
- M. Mascal, I. Yakovlev, E. B. Nikitin and J. C. Fettinger, Angew. Chem., Int. Ed., 2007, 46, 8782–8784 CrossRef CAS PubMed.
- P. Gibbons, D. Love, T. Craig and C. Budke, Vet. Parasitol., 2016, 218, 1–4 CrossRef CAS PubMed.
- A. Bosco, L. Rinaldi, G. Cappelli, A. Saratsis, L. Nisoli and G. Cringoli, Vet. Parasitol., 2015, 212, 408–410 CrossRef CAS PubMed.
- A. P. Murray and M. J. Miller, J. Org. Chem., 2003, 68, 191–194 CrossRef CAS PubMed.
- M. Ghosh and M. J. Miller, J. Org. Chem., 1994, 59, 1020–1026 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, T. J. Blundell, F. R. Hastings, M. W. Fay, G. A. Rance, W. Lewis, A. J. Blake and D. L. Kays, Catal. Sci. Technol., 2018, 8, 229–235 RSC.
- S. Bhunya, T. Malakar, G. Ganguly and A. Paul, ACS Catal., 2016, 6, 7907–7934 CrossRef CAS.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, W. Lewis, A. J. Blake and D. L. Kays, Angew. Chem., Int. Ed., 2017, 56, 4845–4848 CrossRef CAS PubMed.
- Y. Sun, Z. Zhang, X. Wang, X. Li, L. Weng and X. Zhou, Dalton Trans., 2010, 39, 221–226 RSC.
- J. Yang and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 10186–10188 CrossRef CAS PubMed.
- B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435–1445 RSC.
- M. I. Lipschutz, T. Chantarojsiri, Y. Dong and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 6366–6372 CrossRef CAS PubMed.
- A. Casitas, H. Krause, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 1521–1526 CrossRef CAS PubMed.
- B. A. Frazier, V. A. Williams, P. T. Wolczanski, S. C. Bart, K. Meyer, T. R. Cundari and E. B. Lobkovsky, Inorg. Chem., 2013, 52, 3295–3312 CrossRef CAS PubMed.
- W. B. Tolman, Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, Wiley, 1st edn, 2006 Search PubMed.
- J. Hagen, Industrial Catalysis: A Practical Approach, Wiley, 3rd edn, 2015 Search PubMed.
- P. W. Jolly and K. Jonas, Angew. Chem., 1968, 80, 705 CrossRef.
- P. W. Jolly, K. Jonas, C. Krüger and Y.-H. Tsay, J. Organomet. Chem., 1971, 33, 109–122 CrossRef CAS.
- This initial complex was followed by some other interesting nickel N2 complexes by the same authors; see: (a) K. Jonas, Angew. Chem., Int. Ed. Engl., 1973, 12, 997–998 CrossRef; (b) C. Kruger and Y.-H. Tsay, Angew. Chem., Int. Ed. Engl., 1973, 12, 998–999 CrossRef; (c) K. Jonas, D. J. Brauer, C. Kruger, P. J. Roberts and Y.-H. Tsay, J. Am. Chem. Soc., 1976, 98, 74–81 CrossRef CAS.
- R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Dalton Trans., 2013, 42, 1461–1475 RSC.
- M. B. O'Donoghue, W. M. Davis, R. R. Schrock and W. M. Reiff, Inorg. Chem., 1999, 38, 243–252 CrossRef.
- J. M. Smith, R. J. Lachicotte, K. A. Pittard, T. R. Cundari, G. Lukat-Rodgers, K. R. Rodgers and P. L. Holland, J. Am. Chem. Soc., 2001, 123, 9222–9223 CrossRef CAS PubMed.
- J. M. Smith, A. R. Sadique, T. R. Cundari, K. R. Rodgers, G. Lukat-Rodgers, R. J. Lachicotte, C. J. Flaschenriem, J. Vela and P. L. Holland, J. Am. Chem. Soc., 2006, 128, 756–769 CrossRef CAS PubMed.
- S. F. McWilliams and P. L. Holland, Acc. Chem. Res., 2015, 48, 2059–2065 CrossRef CAS PubMed.
- P. L. Holland, Acc. Chem. Res., 2008, 41, 905–914 CrossRef CAS PubMed.
- K. Ding, A. W. Pierpont, W. W. Brennessel, G. Lukat-Rodgers, K. R. Rodgers, T. R. Cundari, E. Bill and P. L. Holland, J. Am. Chem. Soc., 2009, 131, 9471–9472 CrossRef CAS PubMed.
- T. R. Dugan, K. C. MacLeod, W. W. Brennessel and P. L. Holland, Eur. J. Inorg. Chem., 2013, 3891–3897 CrossRef CAS PubMed.
- S. F. McWilliams, E. Bill, G. Lukat-Rodgers, K. R. Rodgers, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2018, 140, 8586–8598 CrossRef CAS PubMed.
- W. H. Monillas, G. P. A. Yap, L. A. Macadams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090–8091 CrossRef CAS PubMed.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
- G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed.
- G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532–535 CAS.
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- M. Reiter, A. Kronast, S. Kissling and B. Rieger, ACS Macro Lett., 2016, 5, 419–423 CrossRef CAS.
- M. Reiter, S. Vagin, A. Kronast, C. Jandl and B. Rieger, Chem. Sci., 2017, 8, 1876–1882 RSC.
- S. Pfirrmann, C. Limberg, C. Herwig, R. Stößer and B. Ziemer, Angew. Chem., Int. Ed., 2009, 48, 3357–3361 CrossRef CAS PubMed.
- B. Horn, C. Limberg, C. Herwig and B. Braun, Chem. Commun., 2013, 49, 10923–10925 RSC.
- P. Zimmermann and C. Limberg, J. Am. Chem. Soc., 2017, 139, 4233–4242 CrossRef CAS PubMed.
- B. Horn, C. Limberg, C. Herwig, M. Feist and S. Mebs, Chem. Commun., 2012, 48, 8243–8245 RSC.
- A. R. Sadique, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2008, 47, 784–786 CrossRef CAS PubMed.
- T. R. Dugan, X. Sun, E. V. Rybak-Akimova, O. Olatunji-Ojo, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 12418–12421 CrossRef CAS PubMed.
- L. Roy, M. H. Al-Afyouni, D. E. DeRosha, B. Mondal, I. M. DiMucci, K. M. Lancaster, J. Shearer, E. Bill, W. W. Brennessel, F. Neese, S. Ye and P. L. Holland, Chem. Sci., 2019, 10, 918–929 RSC.
- V. U. Wannagat, H. Kuckertz, C. Krüger and J. Pump, Z. Anorg. Allg. Chem., 1964, 333, 54–61 CrossRef.
- D. A. Dickie, K. B. Gislason and R. A. Kemp, Inorg. Chem., 2012, 51, 1162–1169 CrossRef CAS PubMed.
- L. R. Sita, J. R. Babcock and R. Xi, J. Am. Chem. Soc., 1996, 118, 10912–10913 CrossRef CAS.
- J. R. Babcock and L. R. Sita, J. Am. Chem. Soc., 1998, 120, 5585–5586 CrossRef CAS.
- C. A. Stewart, D. A. Dickie, B. Moasser and R. A. Kemp, Polyhedron, 2012, 32, 14–23 CrossRef CAS.
- H. Yin, P. J. Carroll and E. J. Schelter, Chem. Commun., 2016, 52, 9813–9816 RSC.
- C. Camp, L. Chatelain, C. E. Kefalidis, J. Pécaut, L. Maron and M. Mazzanti, Chem. Commun., 2015, 51, 15454–15457 RSC.
- A. M. Felix, B. J. Boro, D. A. Dickie, Y. Tang, J. A. Saria, B. Moasser, C. A. Stewart, B. J. Frost and R. A. Kemp, Main Group Chem., 2012, 11, 13–29 CAS.
- D. L. J. Broere, B. Q. Mercado and P. L. Holland, Angew. Chem., Int. Ed., 2018, 57, 6507–6511 CrossRef CAS PubMed.
- M. J. Ingleson, B. C. Fullmer, D. T. Buschhorn, H. Fan, M. Pink, J. C. Huffman and K. G. Caulton, Inorg. Chem., 2008, 47, 407–409 CrossRef CAS PubMed.
- B. C. Fullmer, H. Fan, M. Pink and K. G. Caulton, Inorg. Chem., 2008, 47, 1865–1867 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Angew. Chem., Int. Ed., 2017, 56, 9502–9506 CrossRef CAS PubMed.
- Isocarbonyl Complexes in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Atwood and R. A. Scott, 2006 Search PubMed.
- H. Lei, B. D. Ellis, C. Ni, F. Grandjean, G. J. Long and P. P. Power, Inorg. Chem., 2008, 47, 10205–10207 CrossRef CAS PubMed.
- C. Ni and P. P. Power, Chem. Commun., 2009, 5543–5545 RSC.
- B. M. Gridley, A. J. Blake, A. L. Davis, W. Lewis, G. J. Moxey and D. L. Kays, Chem. Commun., 2012, 48, 8910–8912 RSC.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- Z. Mo and L. Deng, Coord. Chem. Rev., 2017, 350, 285–299 CrossRef CAS.
- D. Benito-Garagorri, I. Lagoja, L. F. Veiros and K. A. Kirchner, Dalton Trans., 2011, 40, 4778–4792 RSC.
- It should be noted that the formation of squarates (C4O4)2− from CO is known, but this does not require the cleavage of CO bonds. See: N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS.
- G. Xia and H. Wang, J. Photochem. Photobiol., C, 2017, 31, 84–113 CrossRef CAS.
- V. Maltese, S. Cospito, A. Beneduci, B. C. De Simone, N. Russo, G. Chidichimo and R. A. J. Janssen, Chem. – Eur. J., 2016, 22, 10179–10186 CrossRef CAS PubMed.
- G. Chen, H. Sasabe, Y. Sasaki, H. Katagiri, X.-F. Wang, T. Sano, Z. Hong, Y. Yang and J. Kido, Chem. Mater., 2014, 26, 1356–1364 CrossRef CAS.
- T. Maeda, S. Mineta, H. Fujiwara, H. Nakao, S. Yagi and H. Nakazumi, J. Mater. Chem. A, 2013, 1, 1303–1309 RSC.
- B. M. Gridley, G. J. Moxey, W. Lewis, A. J. Blake and D. L. Kays, Chem. – Eur. J., 2013, 19, 11446–11453 CrossRef CAS PubMed.
- It should be noted that, while not mentioned in this review, there are some interesting examples of low-coordinate copper β-diketiminate complexes in catalysis. Please refer to Webster's recent Dalton Perspective (ref. 27).
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- K. V. Bukhryakov, R. R. Schrock, A. H. Hoveyda, P. Müller and J. Becker, Org. Lett., 2017, 19, 2607–2609 CrossRef CAS PubMed.
- L. G. Perla, J. M. Kulenkampff, J. C. Fettinger and P. P. Power, Organometallics, 2018, 37, 4048–4054 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |