
Paddlewheel-type diruthenium(iii,iii) tetrakis(2-aminopyridinate) complexes with NIR absorption features: combined experimental and theoretical study†

Abstract
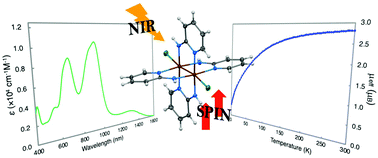
The reactions of [Ru2(O2CCH3)4Cl] with 2-aminopyridine (Hamp) and 2-amino-4-methylpyridine (Hammp) afforded two novel Ru2 complexes, [Ru2(amp)4Cl2] (1) and [Ru2(ammp)4Cl2] (2), respectively. Single crystal X-ray diffraction analyses revealed that 1 and 2 adopted typical paddlewheel-type structures, where the Ru2 units are coordinated with four aminopyridinate ligands with a cis-2:2 arrangement at the equatorial positions and two chloride ligands at the axial positions. The stabilities of 1 and 2 were supported by unrestricted density functional theory (uDFT) calculations. The zero-point energies of the three structural isomers (trans-2:2, 3:1, and 4:0 arrangements) of 1 and 2 were less stable than those of the respective cis-2:2 arrangements. Temperature-dependences of the magnetic susceptibility measurements and uDFT calculations showed that the oxidation and spin states of the Ru2 units in 1 and 2 were commonly Ru26+ and triplet states, respectively. Cyclic voltammetry showed that 1 and 2 underwent one-electron reduction processes, i.e., 1/1− and 2/2−, at redox potentials (E1/2) of −0.08 and −0.18 V vs. SCE, respectively. These results agreed well with the DFT-calculated E1/2 values of 1 (−0.08 V vs. SCE) and 2 (−0.18 V vs. SCE) and were theoretically attributed to Ru2-centred (δ*(Ru2)) redoxes. Moreover, 1 and 2 showed unique near-infrared absorption bands at approximately 1200–1500 nm, which were theoretically attributed to the ligand-to-metal charge transfer (LMCT) with π(amp or ammp) → δ*(Ru2) transition characteristics.
 Please wait while we load your content...
Please wait while we load your content...

