
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-supported and -assisted stereoselective cooperative photoredox catalysis†
Jasmin
Busch
 a,
Daniel M.
Knoll
a,
Christoph
Zippel
a,
Stefan
Bräse
*ab and
Claudia
Bizzarri
*a
a,
Daniel M.
Knoll
a,
Christoph
Zippel
a,
Stefan
Bräse
*ab and
Claudia
Bizzarri
*a
aInstitute of Organic Chemistry, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: Stefan.braese@kit.edu; claudia.bizzarri@kit.edu
bInstitute of Toxicology and Genetics, Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 11th September 2019
Abstract
In this perspective, we review those stereoselective photocatalytic reactions that use synergy between photoredox catalysts and transition metal catalysts. In particular, we highlight the orchestrated interaction between two and more metals which not only enhance the turnover numbers, but also lead to increased selectivities. Aspects of green chemistry and sustainable developments are included. In this review, C–C, C–O, C–N and C–S forming reactions are discussed and a perspective on future developments is given.
Introduction
In the light of the current public discussion and the one in academia on the economical use of resources and also the use of sustainable energies such as sunlight, photochemical processes have emerged as one of the focal points in contemporary research. With the advent of novel and selective photochemical transformations in the last decade, the field of photochemistry witnessed a dramatic and dynamic change. For a long time, enantioselective photochemical reactions have been an intriguing target for synthetic chemists.1,2 Different approaches have been pursued—from the use of a chiral environment (e.g., chiral solvent and chiral cavities in heterogeneous supports) to chiral auxiliary. Asymmetric photochemical reactions have been tried, even by employing circularly polarized light.Photocatalysis is a very broad term, in general referring to any reaction that requires the presence of a catalyst able to activate a reagent upon the absorption of photons. To elaborate, the catalytic species is active in its excited state, which is able to transfer an electron (e.g., single electron transfer, SET) or an atom (e.g., atom transfer radical addition, ATRA) or simply energy (photoinduced energy transfer) to the reagents.
In particular, photoredox catalysis has emerged as a very powerful tool in organic synthesis – more than 2000 publications witness its growing interest. The application ranges from natural product chemistry,3–7 drug development and crop protection agents8 to materials sciences (e.g., for polymers).9–13
A particular feature of the last few years has been the development of visible light photoredox catalysis.14–19
In the last 100 years of preparative photochemistry (from the key discoveries of Paternò20 and Ciamician21), novel approaches have been used to address various issues. The combination of photoredox catalysis with transition metal catalysis offers access to a vast range of organic synthesis. There is synergy between the photoredox catalytic cycle and the transition metal catalyst. Therefore they depend on each other for the formation of new bonds. Recently, some reviews covering the aspects of metals in photocatalysis22–28 and another review on enantioselective photoinduced organocatalysis29 were published. We ask the reader to refer to these reviews for a comprehensive survey regarding dual catalysis via photoirradiation. In this perspective, we would like to focus only on those cooperative photoredox and transition metal catalysts that offer an alternative pathway for the synthesis of organic molecules inducing stereoselectivity. We will give an overview of the key aspects, including: yields (quantum yields, stereochemical yields), wavelength (visible light), turnover numbers (associated with photostability), solvents (stability, sustainability) and reaction setup (flow and micro reactors,30–33 scalability). When it comes to enantiocontrol, chiral ligands and additives play a crucial role in asymmetric synthesis. In most cases the conditions and efficiencies are comparable from one transformation to another, however some reaction conditions are given for optimized transformation (Scheme 1). By merging two powerful techniques – photoredox catalysis and transition metal catalysis – one can get the best of both worlds. In this review, we will bridge these techniques with a focus on the use of transition metal complexes (and by purpose excluding lanthanides), which can act as both photosensitizers and/or catalysts.
 | ||
| Scheme 1 General scheme for a photochemical reaction. | ||
A typical photochemical reaction is shown in Scheme 2. Two metals, e.g. ruthenium and scandium, are used as catalysts. Ruthenium as a photoredox catalyst (bulb) and a chiral ligand (hand) are used in catalytic amounts.
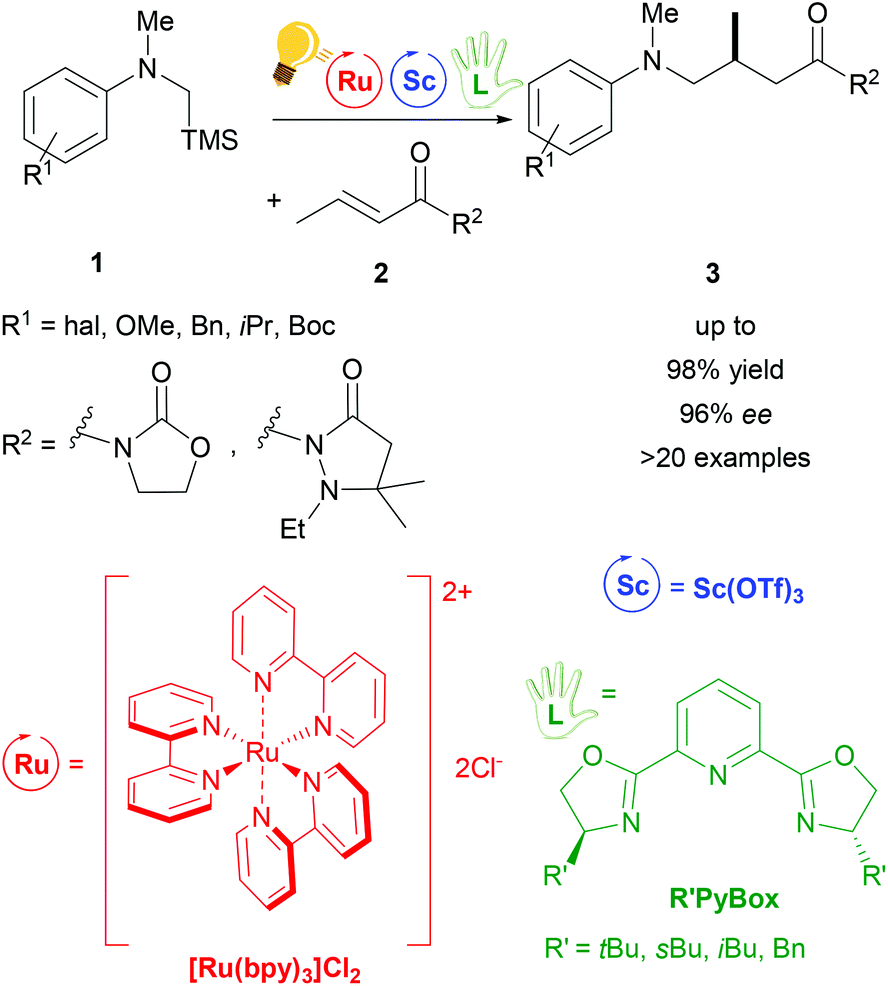 | ||
| Scheme 2 Typical asymmetric photocatalysis with two metals as catalysts. Two metals – one as a photocatalyst (bulb) and a chiral (hand) ligand giving the perfect match. Reagents and conditions are given here for every reaction. In this particular example: cooperative photoredox/Sc catalysis for asymmetric α-amino radical addition. Reagents and conditions: 1 (1.00 equiv.), 2 (1.50 equiv.), Ru-photocat. (2.0 mol%), Sc-cat. (15.0 mol%), chiral ligand (20.0 mol%), TBACl (30.0 mol%) MeCN, fluorescent lamp (23 W) irradiation, 18 h. | ||
In principle, various bond forming reactions can be envisaged. However, only a few have been addressed so far (Fig. 1).
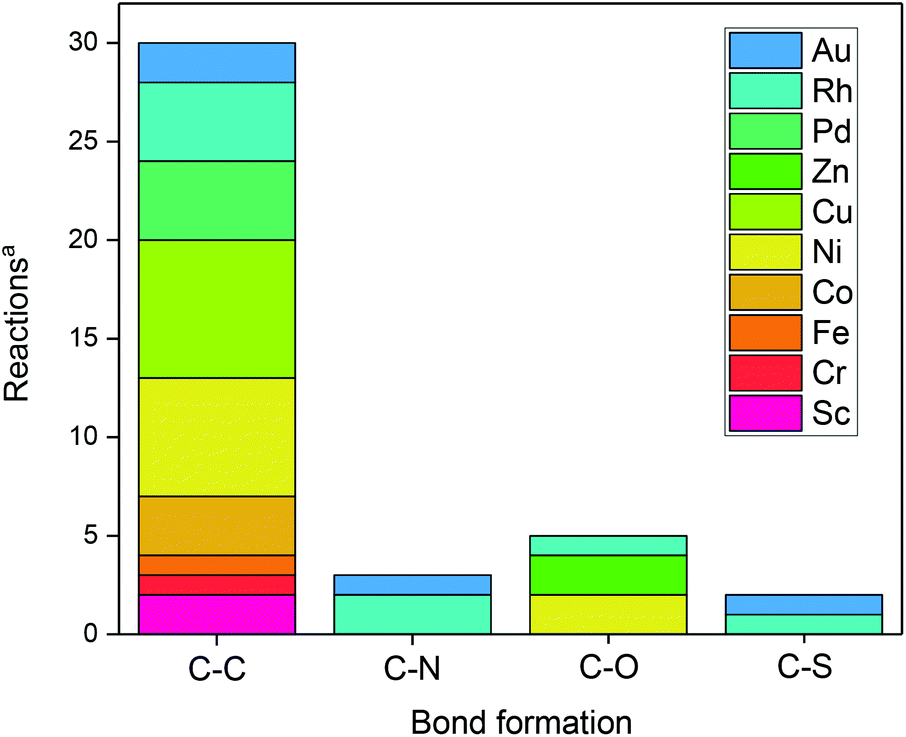 | ||
| Fig. 1 Bond-forming reactions using metal-assisted photoredox catalysis. aAccording to/covered in this review. | ||
In this review, we focus on recent developments (for some recent accounts, see ref. 27). We sort them out by increasing the atom number of the metal. Metal-free catalytic reactions – although also very important – are excluded.
Scandium(III) complexes
The use of Lewis Acids (LAs) in chemical reactions has played an important role in organic synthesis. Stereochemical control can easily be achieved by the introduction of a chiral ligand, which directs the interaction between the substrate and the LA in a favourable way. Scandium(III) is one of the most successfully employed LAs in enantioselective catalysis, via a cooperative photo-redox/LA approach.24,34 In particular, an asymmetric addition of α-amino radicals to α,β-unsaturated carbonyl compounds 2 was obtained by the cooperative approach of the [Ru(bpy)3]Cl2 photo-catalyst and Sc(OTf)3.35 Scalemic 2,6-bis(oxazol-2′-yl)pyridine derivatives (R′Pybox) were used in order to optimize stereoselectivity in the reaction of α-silylmethyl amino radicals (Scheme 2). The photoredox activation by ruthenium generates the α-amino radical, without taking part in the enantioselective addition step.The group of Yoon reported on a highly enantioselective [2+2] photocycloaddition of 2′-hydroxychalcones 4 and a diene 5, which resulted from the use of a dual-catalytic system made of the chiral Sc(III) complex and [Ru(bpy)3]2+ as photosensitizers (Scheme 3).36 A high enantiomeric excess (up to 98%) was provided by the addition of the chiral ligand tBu-PyBOX to Sc(OTf)3. In contrast to what was reported from the same group some years before, where the LA cocatalysts in the asymmetric [2 + 2] photocycloaddition were Gd(OTf)3 and Eu(OTf)3,37 it was observed that sensitization occurring from the excited [Ru(bpy)3]* happens by triplet energy transfer rather than photoinduced electron transfer. In fact, the organic substrate 2′-hydroxychalcone, upon association with Sc, possesses a lower triplet state energy (33 kcal mol−1), which lies reasonably accessible from the triplet state of [Ru(bpy)3]* (46 kcal mol−1). The evidence of this triplet sensitization was shown by the successful photoreaction also using an electron-deficient photocatalyst such as [Ru(deeb)3](PF6)2 (deeb: 2,2′-bipyridinyl-4,4′-dicarboxylic acid diethyl ester), or using benzil as an organic triplet sensitizer. The photoredox process was also ruled out because the addition of reductants or oxidants did not lead to any cycloaddition products. Very recently, Yoon and coworkers expanded the scope of the [2 + 2] photocycloaddition by triplet sensitization to other α,β-unsaturated carbonyl compounds such as cinnamate esters, using oxazaborolidine as a LA catalyst,38 successfully used in previous reports for cyclobutane synthesis by Bach39,40 and Brown.41
 | ||
Scheme 3 Cooperative photo-/Sc catalysis for the enantioselective (2 + 2) photocycloaddition of 2′-hydroxychalcones. Reagents and conditions: 4 (1.00 equiv.), 5 (10.0 equiv.), Ru-photocat. (2.5 mol%), Sc-cat. (10.0 mol%), chiral ligand (15.0 mol%), i-PrOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeCN (3:1), fluorescent lamp (23 W) irradiation, 20 h.36 :MeCN (3:1), fluorescent lamp (23 W) irradiation, 20 h.36 | ||
Chromium(II) complexes
Chromium salts are usually not considered appealing reagents, as they are famous for generating the toxic hexavalent chromium, Cr(VI). Although chromium is an essential trace element in the human body, chronic exposure may be harmful.42 The use of Cr in catalytic amounts is therefore attractive. The Nozaki–Hiyama–Kishi (NHK) reaction is a well-established chromium mediated allylation of aldehyde 8. Further studies in order to render the use of Cr catalytic are therefore highly motivated. Glorius and coworkers have recently published the combination of photoredox with Cr catalysis for the diastereoselective NHK reaction (Scheme 4).43 Blue LED irradiation generates the excited state of the Ir(III) photosensitizer, which is then reductively quenched by an allyl arene. Thus, after the deprotonation of 7+˙, an aryl radical cation is formed that is trapped by the CrCl2 salt. The following step is the insertion of aldehyde 8 to generate the chromium alkoxide that is the diastereoselective step (anti > 19:1 dr), because of the formation of a Zimmerman–Traxler transition state. Regeneration of the photocatalyst and the Cr(II) catalyst occurs by redox reaction between the Ir(II) and the Cr(III) species. Control experiments showed that in the absence of the photocatalyst or light irradiation no reaction occurs, although a different quenching mechanism (oxidative) of the photocatalyst could not be ruled out by the authors. The addition of a chiral bisoxazoline ligand in a test reaction (Scheme 4 – centre) gave a moderate enantiomeric excess (20% ee) of 9a.
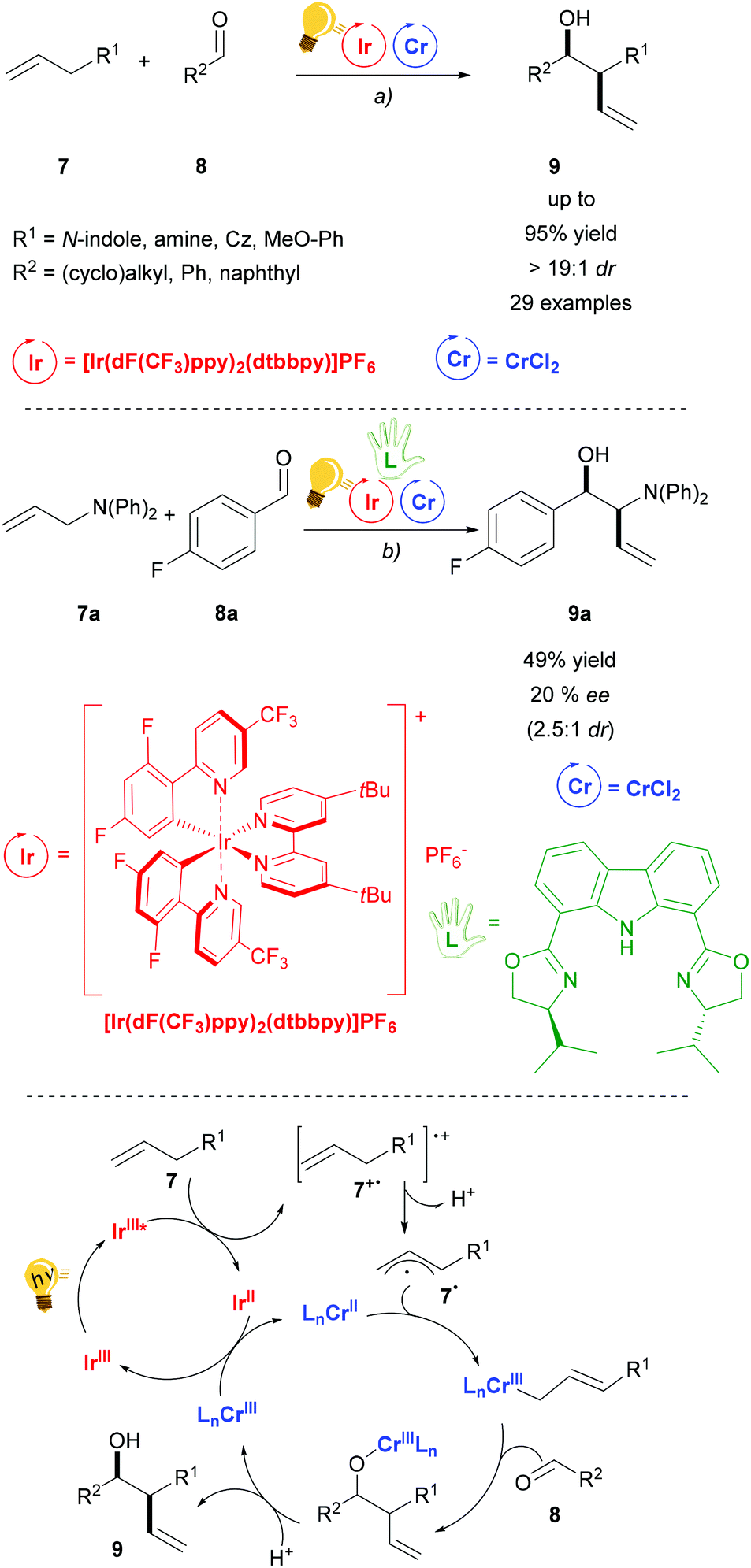 | ||
| Scheme 4 Diastereoselective allylation of aldehydes and mechanism (bottom). Reagents and conditions: (a) Ir-photocat. (2.0 mol%), Cr salt (10.0 mol%), Li2CO3 (20.0 mol%), DMF, 25 °C, Ar, blue LED (5 W), 24 h; (b) same conditions as (a) plus chiral bisoxazoline (10.0 mol%).43 | ||
Cobalt(II) complexes
The catalytic activity of cobalt salts and complexes is very rich.44–46 Nevertheless, the merger of Co catalysis and photoactivation remains mainly confined to artificial photosynthesis.47 The synergy between an organic photoredox and cobalt catalysis was employed in order to achieve regioselective carboxylation of substituted styrenes with moderate yield.48Cooperative photoredox and cobalt catalysis was employed by Rovis and coworkers in the regioselective hydroaminoalkylation of unactivated dienes.49 The photosensitizer used is the commercial [Ir(dF-CF3ppy)2(dtbbpy)]PF6, which upon excitation by blue light, undergoes reductive quenching by the tertiary amine, generating after deprotonation by a base (e.g. pivalate) an α-amino alkyl radical. The photosensitizer can be then regenerated by two possible redox reactions: (a) reducing the Co(II) salt to the Co(I) species (In Scheme 5, I); (b) reducing the Co(III)-allyl species (III in Scheme 5) to the Co(II)-allyl species (IV in Scheme 5). The Co(I) species is protonated by the in situ formed carboxylic acid, followed by a migratory insertion of the diene into the Co–H bond of the transient hydride species, forming the species III. The α-amino alkyl radical previously generated either coordinates the Co(II) metal core and the product is yielded upon reductive elimination or it may form a bond directly with the allyl ligand in IV. The reaction conditions are tolerant of many functionalities and the stereoselectivity is up to 20:1 (trans:cis).
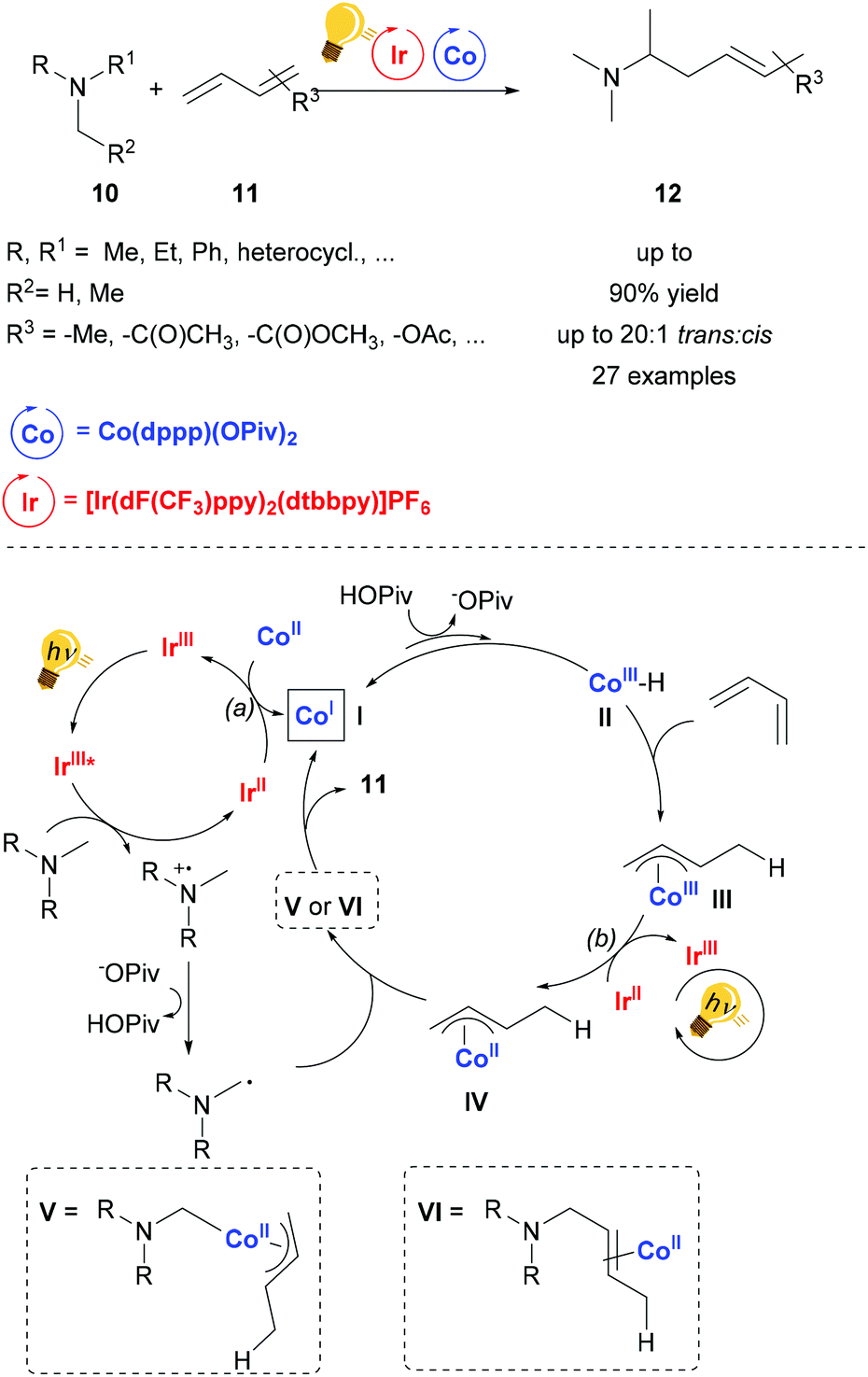 | ||
| Scheme 5 Synergetic photoredox and Co catalysis for hydroaminoalkylation of conjugated dienes. Reagents and conditions: diene 11 (1.00 equiv.), tertiary amine 10 (2.00 equiv.), Ir-photocat. (0.5 mol%), Co salt (10 mol% equiv.), base (0.4 equiv.), blue LED, 12 h. (dppp = diphenylphosphino phenyl).49 | ||
Very recently, a dual system Ir/Co driven by visible-light (440 nm) has been employed for the alkene–alkyne coupling (Scheme 6).50 Similar to the previous hydroaminoalkylation reaction, the excited photosensitizer, [Ir(dF-CF3ppy)2(dtbbpy)]PF6, undergoes reductive quenching with Hantzsch ester (HE) and i-Pr2NEt and the generation of the low valent Co(I) species is afforded by the oxidation of Ir(II) to restore Ir(III). A proton donor, which can be H2O or the oxidized HE or i-Pr2NEt, forms the Co-hydride species that coordinates the acrylate 14. This species reacts further with the alkyne via carbometalation and after protonation the product is eliminated. When the alkyne 13 is 3-hexyne, or any other symmetrical aliphatic alkyne, the stereoselectivity of the syn-carbometalation affords the syn product 15 in good yield (up to 84%) (Scheme 6). By broadening the scope of the alkynes, not only could the other diastereomer be observed due to E/Z isomerization, but also the products originated from the different regioselectivities induced by unsymmetrical alkynes. Nevertheless, the regiochemistry is controlled by the cobalt catalyst. The reaction was also tested with FeBr2 or NiCl2 as the catalyst giving the moderate yields of 41% and 38% respectively, therefore the scope was not extended with these salts.
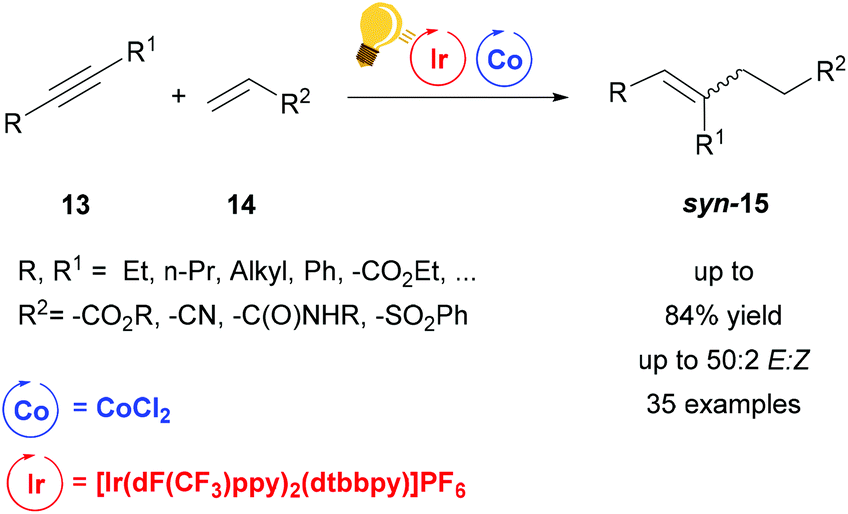 | ||
| Scheme 6 Visible-light-driven Co catalysis for alkene–alkyne coupling. Reagents and conditions: alkyne 13 (1.00 equiv.), alkene 14 (1.20 equiv.), Ir-photocat. (1 mol%), Co salt (10 mol% equiv.), dppe (20 mol%), i-Pr2NEt:HE (1:2, 1.50 equiv.), H2O (1 equiv.) blue LED, 1.5–18 h. (dppe = diphenylphosphino ethane).50 | ||
Tandem photoredox and Co catalysis was also employed by Zhao's and Wu's groups for a hydrocarboxylation reaction of alkynes with CO2 gas (1 atm).51 Only the E-isomers were obtained, when bis-aliphatic substituted alkynes were used as educts (Scheme 7). When the two substituents on the alkyne were sterically differentiated, CO2 insertion occurred to the less sterically hindered carbon with regioselectivity up to 3.5:1. ZnBr2 was used as the reductant and the hydrogen atom source arose from i-Pr2NEt. The key intermediate was a five-membered cobaltacycle (Scheme 7), generated by the concomitant insertion of CO2 and the alkyne to the in situ formed Co complex with dcype (bis(dicyclohexylphosphine)ethane). E/Z isomerization was possible via energy transfer from the Ir(III)* to the acrylate. In this way, the authors could have access to diverse heterocycles from one-pot alkyne hydrocarboxylation/alkene isomerization and final cyclization via this synergistic Ir/Co catalysis.
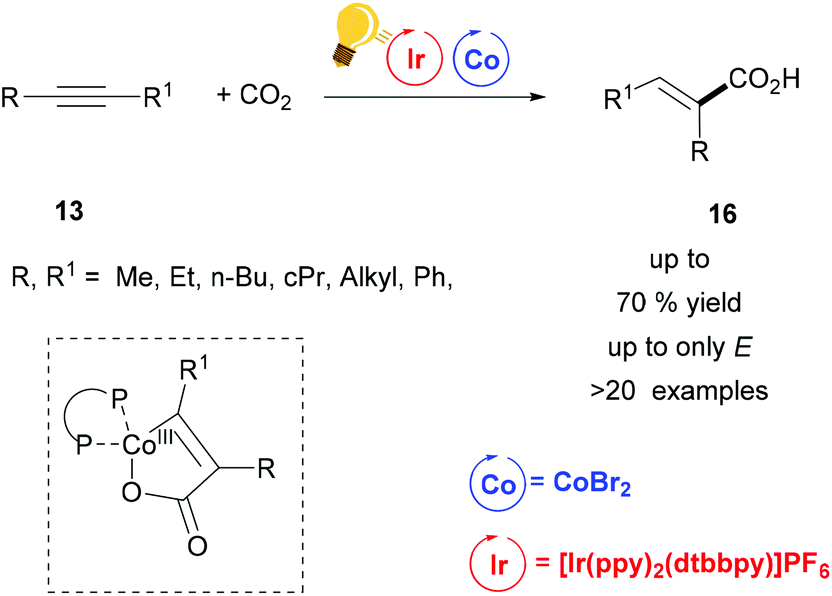 | ||
| Scheme 7 Alkyne Carboxylation via synergistic Ir(III)-photoredox/Co catalysis. Reagents and conditions: alkyne 13 (1.00 equiv.), CO2 (1 atm), Ir-photocat. (1 mol%), CoBr2 (10 mol%), dcype (10 mol%), ZnBr2 (1 equiv.), i-Pr2NEt (3.00 equiv.), in CH3CN, blue LED, 24 h.51 | ||
Other examples of tandem catalysis with cobaloximes and organic or metal photoredox catalysts were also employed in a variety of other reactions,52,53 also stereoselective,54 but these are beyond the scope of this perspective.
Nickel(II) complexes
Nickel(II) complexes have received great attention due to their success in catalytic processes, especially with regard to replacing the expensive palladium in C–C cross-coupling reactions and in reductive coupling.55–57 Nickel behaves very differently from palladium. Thus, consistent deviations in the mechanisms of catalysed reactions are present, as for example radical pathways are more favoured.55 The pivotal studies of Molander,58 Doyle and MacMillan59 merged the use of photoredox catalysis in the single-electron transmetalation for the cross-coupling catalysed by nickel.60 Molander and coworkers exploited visible-light irradiation of an Ir(III) complex for the homolytic C–B bond cleavage of an organoboron reagent 17, which occurs via a SET process with a concomitant reductive quenching of the Ir(III) photosensitizer.58The so-generated alkyl radical is trapped by the nickel catalyst Ni(COD)2, (COD: cyclooctadiene). In their successive study,61 the authors explained that stereoconvergence does not arise from facial-selective addition to the Ni center, but rather from the reductive elimination step (Scheme 8). In fact, the addition of the second radical leads to the formation of two diastereomeric Ni(III) complexes that are in equilibrium. The major enantiomer is produced by that diastereomer, which eliminates faster, giving rise to a dynamic kinetic resolution process. An enantioenriched product (Scheme 8) could be obtained when (4R,4′S)-4,4′-dibenzyl-2,2′-bis(oxazoline) (BnBox) was added to the Ni(COD)2 catalyst.
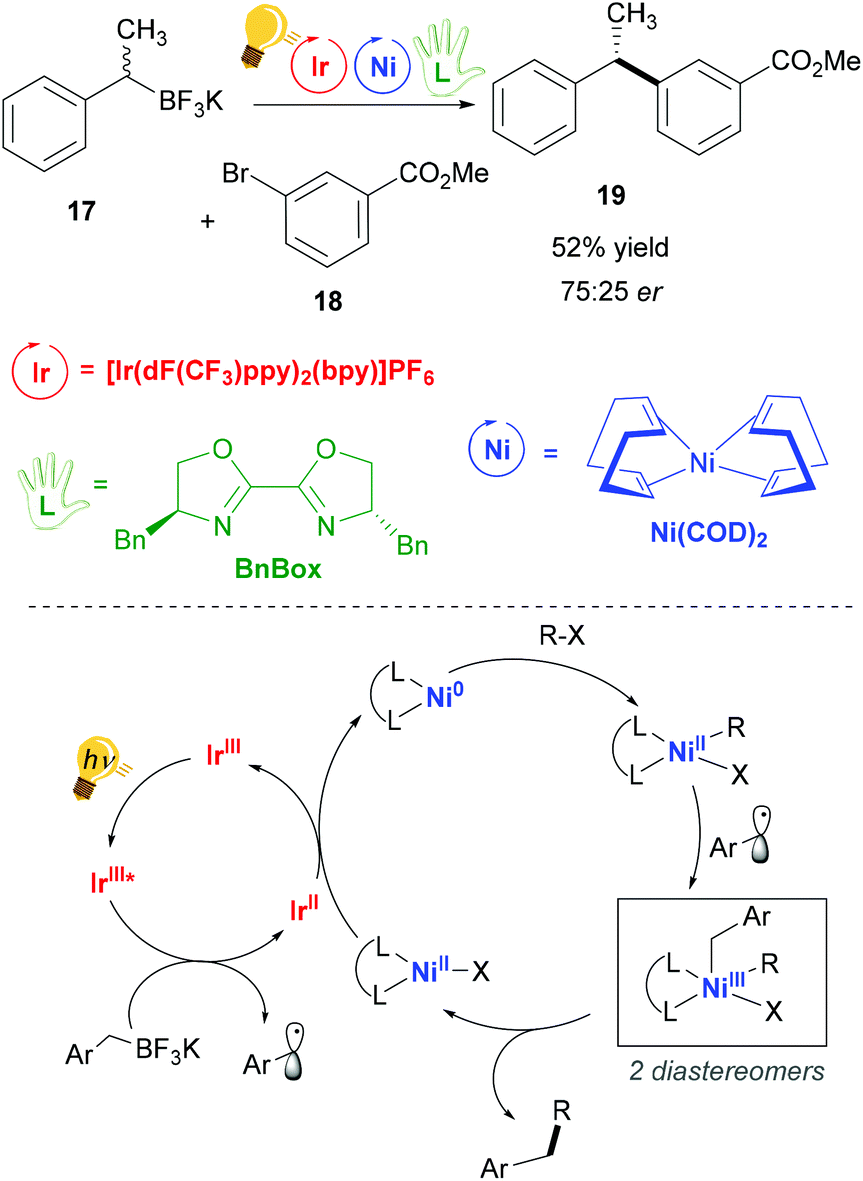 | ||
| Scheme 8 Photoredox/Ni dual catalytic cross-coupling (top) and mechanism (bottom). Reagents and conditions: organoboron racemic mixture 17 (1.20 equiv.), aryl bromide 18 (1.00 equiv.), 2,6-lutidine (3.50 equiv.), Ir-photocat. (2.0 mol%), Ni-cat. (3.0 mol%), chiral ligand (3.0 mol%), THF/MeOH (95/5), blue LED, 24 h.58 | ||
Further studies were conducted in the same group in order to widen the scope of alkyltrifluoroborates as starting materials, but not always enantiomerically enriched products could be obtained.62 Benzylic α-trifluoromethylated alkylboron reagents 20 were used with heteroarylbromides 21 in order to prepare diaryltrifluoroethanes via a photoredox/nickel dual catalysis (Scheme 9).63 In this case, a limited enantioselectivity was observed in the final products (up to 65% ee). In the same group, the exploitation of α-amino acids with N-trifluoroboratomethyl functionalization 23 was also investigated in the dual Ir-photoredox/Ni cross-coupling catalysis, to generate enantiopure aminomethylarenes.64 The strong oxidant Ir* undergoes a single-electron transfer, generating the reactive C(sp3) amino radicals, which is captured by the Ni(0) complex. In the following step, the addition of the aryl bromide generates the Ni(III) species and the final cross-coupling affords the desired product 25 (Scheme 10). The chirality of the starting material is retained under these mild reaction conditions, tolerating a broad range of functional groups and large molecular fragments.
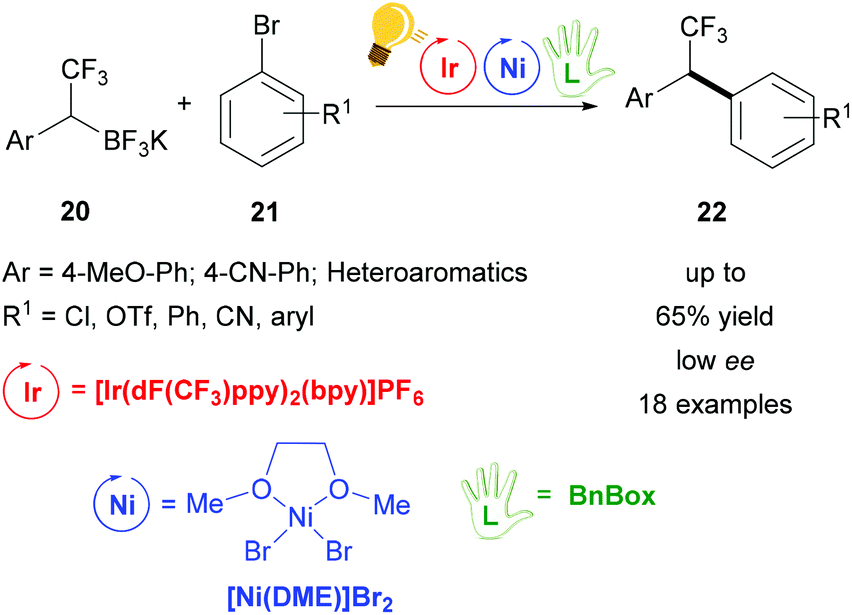 | ||
| Scheme 9 Photoredox/Ni dual catalysis for the synthesis of 1,1,-diaryl-2,2,2-trifluoroethanes. Reagents and conditions: trifluoroborate 20 (2.00 equiv.), aryl bromide 21 (1.00 equiv.), Ir-photocat. (3.0 mol%), Ni-cat. (5.0 mol%), chiral ligand (7.5 mol%), 1,4-dioxane, rt, two fluorescent lamps (26 W), rt, 48 h.63 | ||
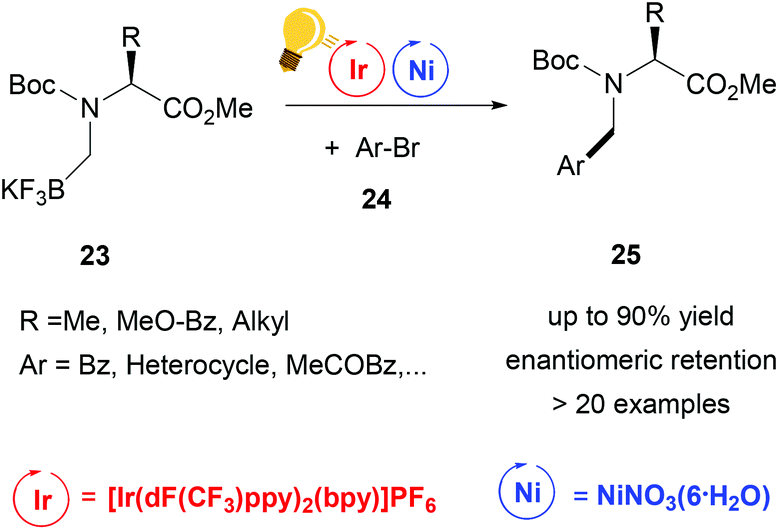 | ||
| Scheme 10 Alpha-arylation of chiral α-aminoacid by Ir-photoredox/Ni catalytic cross-coupling. Reagents and conditions: Boc-N-trifluoroboratomethyl aminoacid 23 (1.00 equiv.), aryl bromide 24 (1.00 equiv.), 2,6-lutidine (3.00 equiv.), Ir-photocat. (2.0 mol%), Ni-cat. (5.0 mol%), bioxazole (10 mol%) in EtOAc, 26 W CFL, rt, 5–8 h.64 | ||
Non-traditional leaving groups, such as carboxylic acid, were exploited in the synergistic photoredox/nickel catalysis for the cross-coupling with aryl and vinyl halides by MacMillan and coworkers.59,65–67 With this protocol, a great number of commercially available feedstock chemicals, such as alkyl-, α-amino-, and α-oxy-carboxylic acids, are used in order to form new C–C bonds under mild conditions. The suggested mechanism depicts the formation of the alkyl radical after decarboxylation via a SET to the excited photosensitizer, and this radical is trapped by the nickel core.
When vinyl halides are used, their stereochemistry is transferred to the products without E/Z isomerization (Scheme 11).66 Nevertheless, in respect of the substrate, the racemic α-oxy acid 26, the stereochemistry remains unclear. Excellent diastereoselectivity with the retention of configuration in a ribose derivative 26a (Scheme 11 – bottom) shows the potential utility of this vinylation protocol for pharmacologically active agents.
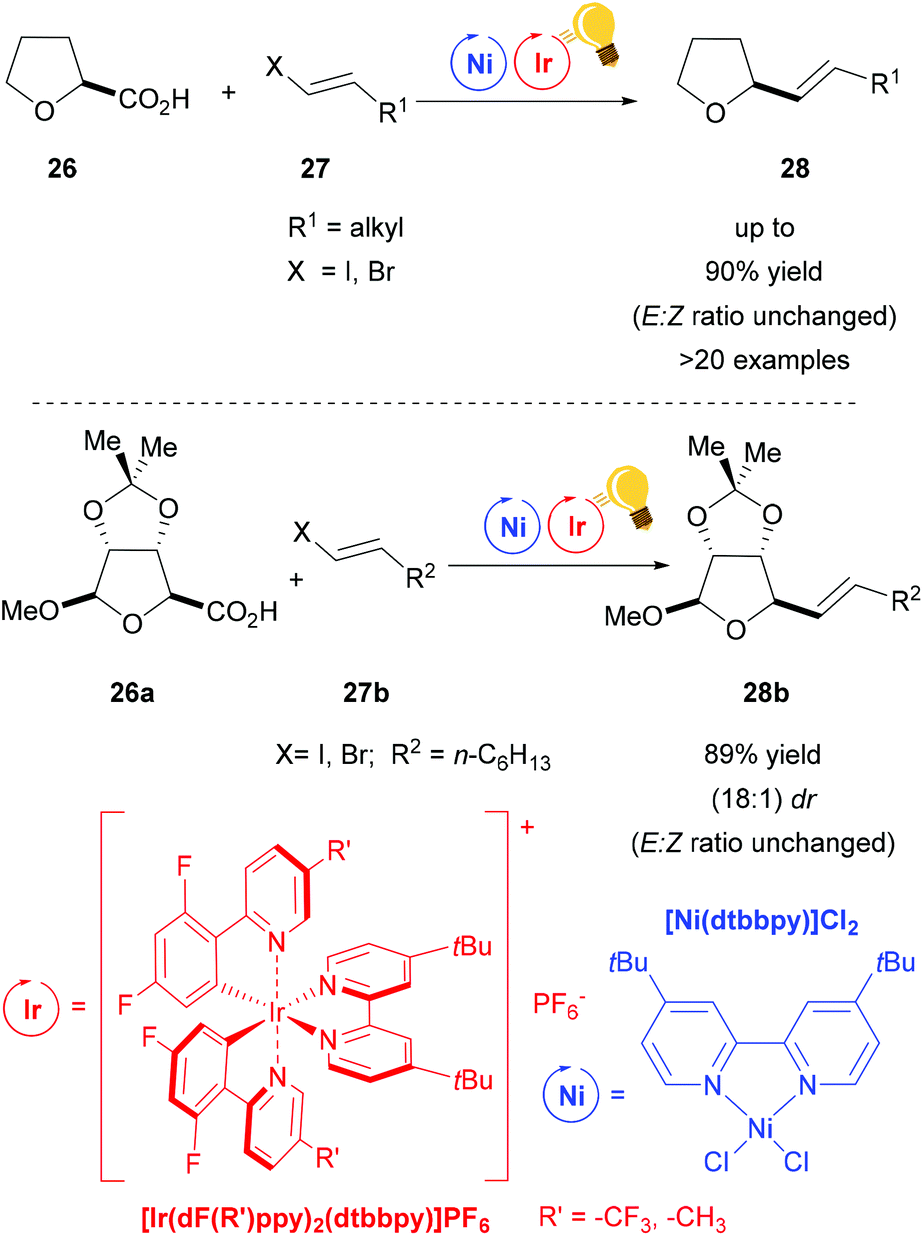 | ||
| Scheme 11 Decarboxylative olefination via photoredox/Ni dual catalysis. Reagents and conditions: Ni-cat. (2.0 mol%), Ir-photocat. (1.0 mol%), DBU, DMSO, 25 °C, N2, blue LED (34 W), 18–72 h.66 | ||
In the groups of Fu and MacMillan, the synergistic use of photoredox and decarboxylative cross-coupling with a chiral Ni(II) complex enabled the preparation of enantioenriched benzylic amines 30, starting from racemic α-amino acids 29 (Scheme 12).65 The chiral Ni complex was formed in situ by the addition of a chiral ligand (bis[(4S)-4-(4-tert-butylphenyl)-4,5-dihydro-2-oxazolyl]acetonitrile) that determines the enantioselectivity (82–92% ee). The asymmetric arylation was demonstrated with a wide scope of N-Boc protected α-amino acids 29 and many functional groups were tolerated.65 Thus, this protocol could be used for the preparation of important pharmaceuticals, starting from inexpensive racemic mixtures.
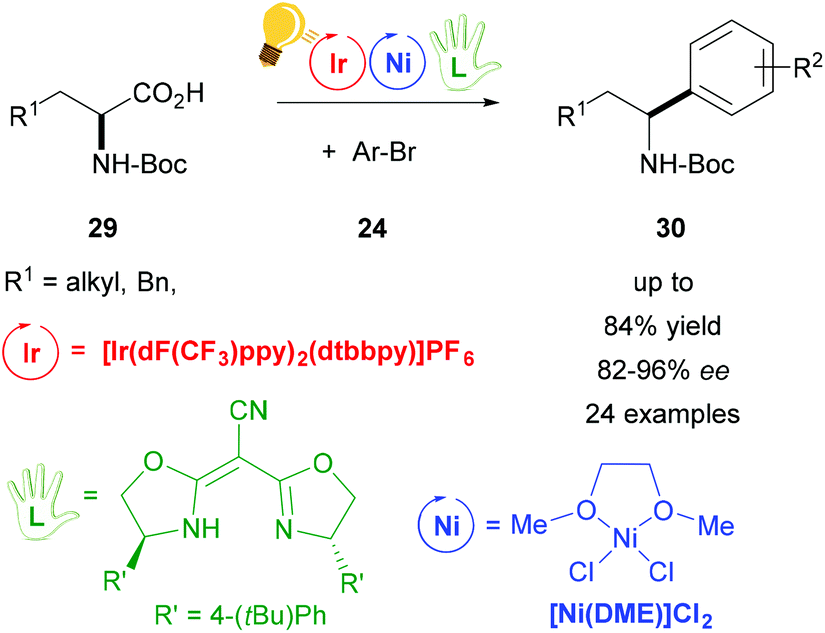 | ||
| Scheme 12 Enantioselective decarboxylative arylation of α-amino acids. Reagents and conditions: Ir-photocat. (2.0 mol%), Ni-cat (2.0 mol%), chiral ligand (2.2 mol%), TBAI, Cs2CO3, DME/toluene, rt, N2, blue LED.65 | ||
The synergy of Ir-photoredox and Ni catalysis has been recently applied to prepare α,β-unsaturated sulfones in high yields (up to 96%) from vinyl bromides and sodium sulfonates.68 The products present a selectivity for the trans isomer (trans:cis from 5:1 up to only trans).
Dual photoredox/Ni catalysis was also successfully applied in C–O coupling.69 Exploiting this method, the groups of Wang and Xiao achieved good (8:1) to high α-stereoselectivity (α only) in the synthesis of phenolic glycosides 33.70 Diverse pyranoses and furanoses, with Bn protected OH, were converted to the corresponding phenyl glycoside 33 upon reaction with an aryl bromide 32. Visible light irradiation of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 generated the excited state that underwent reductive quenching by an aryl/Ni(II)/sugar-adduct. The α/β ratio showed a selectivity towards the α-product, however no explanation on steric control was given (Scheme 13).
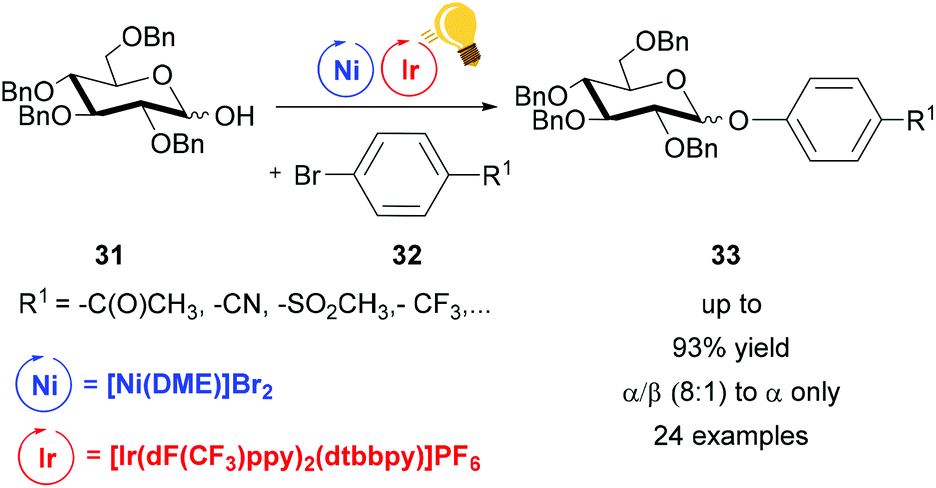 | ||
| Scheme 13 Synthesis of phenolic glycosides. Reagents and conditions: Ir-photocat. (1.0 mol%), Ni (5.0 mol%), dtbbpy (5.0 mol%), DABCO (10.00 equiv.), K2CO3, MeCN, 40 °C, Ar, two blue LED (3 W), 40 °C, Ar.70 | ||
Dual visible-light photoredox/Ni catalysis was also applied to prepare chiral 1,4-benzodioxanes 35via an intramolecular asymmetric C–O coupling reaction by Xiao and coworkers.71 The prochiral substrates are derivatives of 2-(2-iodophenoxy)propane-1,3-diol 34 that undergo an oxidative addition to the Ni(0) catalyst, which is coordinated in situ by the axially chiral bipyridine ligand (L in Scheme 14). The proposed enantio-determining step is the Ni(II)–alkoxide bond formation from one of the two hydroxyl groups of the substrate. Meanwhile, the photo-excited Ir(III)* undergoes reductive quenching, generating Ni(III) aryl alkoxides, which eliminate reductively the final product (35 in Scheme 14). The optimized reaction conditions lead to a maximum enantiomeric ratio of 87:13 with an overall yield of 87%.
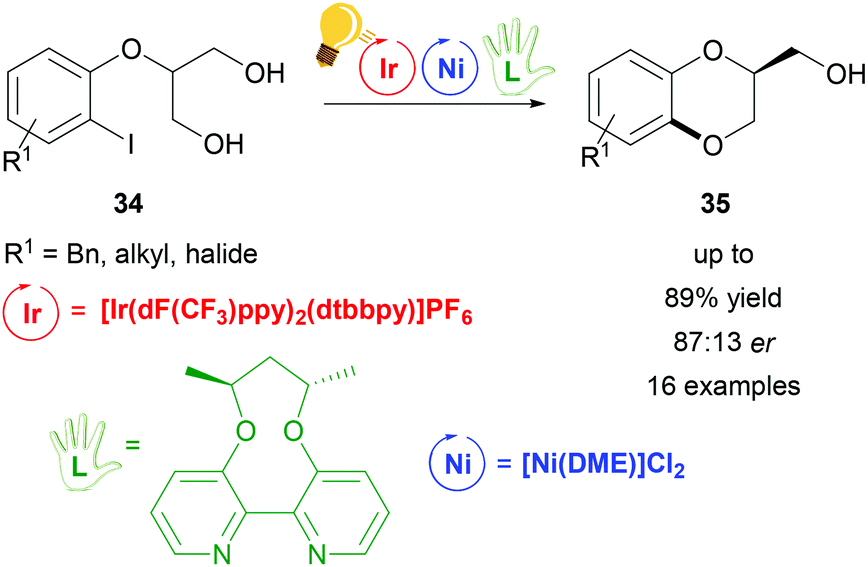 | ||
| Scheme 14 Enantioselective synthesis of 1,4-benzodioxanes. Reagents and conditions: Ir-photocat. (3.0 mol%), Ni-cat. (5.0 mol%), chiral ligand (5.0 mol%), K2CO3, quinuclidine (20.0 mol%), THF, rt, Ar, blue LED (30 W), 24 h.71 | ||
The same working group of Xiao obtained di- and perfluoroalkylation of β-ketoesters 36 with a high enantiomeric ratio (up to 94:6 er) by dual photoredox/Ni catalysis.72 The photocatalyst was again the [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 complex, irradiated with 7 W blue LEDs, but preliminary tests were done using fac-Ir(ppy)3, which also gave good results although in moderate yields (up to 48%). Stereoselectivity was induced by the presence of the chiral bis(oxazoline) ligand BnBox (Scheme 15), which coordinated the nickel core in situ. The highest yield (67%) and the best er (94:6) was obtained when 1,2-dimethoxyethane (DME) was used as the solvent and 20.0 mol% of Ni and chiral ligand were employed.
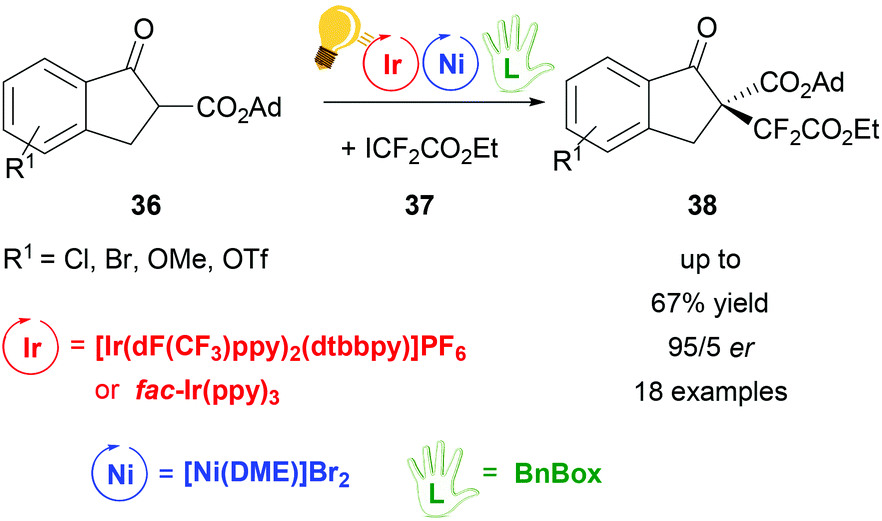 | ||
| Scheme 15 Synthesis of enantioselective perfluoroalkylation of β-ketoesters. Reagents and conditions: ICF2CO2Et 36 (2.00 equiv.), Ir-photocat. (3.0 mol%), Ni-cat. (20.0 mol%), BnBox (20.0 mol%), NaHCO3, DME, rt, blue LED (7 W), 30 h. Ad = adamantyl.72 | ||
Copper complexes
Copper has been widely considered for general application in catalysis and for photoredox catalysis. Recently two reviews about photoredox catalysis were published by Zysman-Colman and coworkers and Reiser, König et al. in which copper was reported as a photoredox catalyst in larger parts of these studies.22,23 Although the use of copper catalysts in stereoselective photoredox catalysis is limited, there are already several examples ranging from arylation and acetylation to decarboxylative elimination reactions. Many copper catalyst species are formed in situ from inexpensive copper salts (CuX, CuX2, (X = Cl, Br); Cu(OTf)2 and [Cu(MeCN)4]PF6) and a (chiral) ligand. These systems have shown good performance, often with more than 90% yield and up to 99% ee under very mild reaction conditions, varying from −20 °C to room temperature. The reactions are environmentally friendly with all of them working with visible light. So far, only mononuclear copper species have been described for stereoselective photoredox catalysis whereby chiral bisoxazoline ligands are dominantly represented, as well as chiral phosphine ligands and the (R,R)-2,6-bis(4-phenyl-2-oxazolinyl)pyridine (PhPyBox) ligand. For some reactions Cu(II) salts have been applied directly, whereas for most of the reactions Cu(I) salts have been used, which have been oxidized to Cu(II) within the photoredox process, representing the actual active catalyzing species in the catalysis cycle of the reaction.One of the first reactions using copper in a stereoselective photoredox catalysis was published in 2015 by Perepichka et al. (Scheme 16). For the asymmetric alkynylation of tetrahydroisoquinoline (THIQ) 39[Ir(ppy)2(dtbbpy)]PF6 was used in combination with copper(I) bromide and the chiral ligand (R)-(+)-1-(2-diphenylphosphino-1-naphthyl)isoquinoline ((R)-QUINAP), obtaining the product 41 in yields of up to 90% and 96% ee. The proposed mechanism consists of a photoredox cycle of the iridium photocatalyst generating the active radical cation species of N-aryl-tetrahydroisoquinoline 39 through the energy of visible light, which is then transformed into an iminium ion by hydrogen abstraction that reacts with nucleophiles in the second cycle. In this copper cycle CuBr and (R)-QUINAP form the active catalyst, binding the alkyne substrate 40 and forming the product 41 with the corresponding iminium ion (Scheme 16 – bottom).73
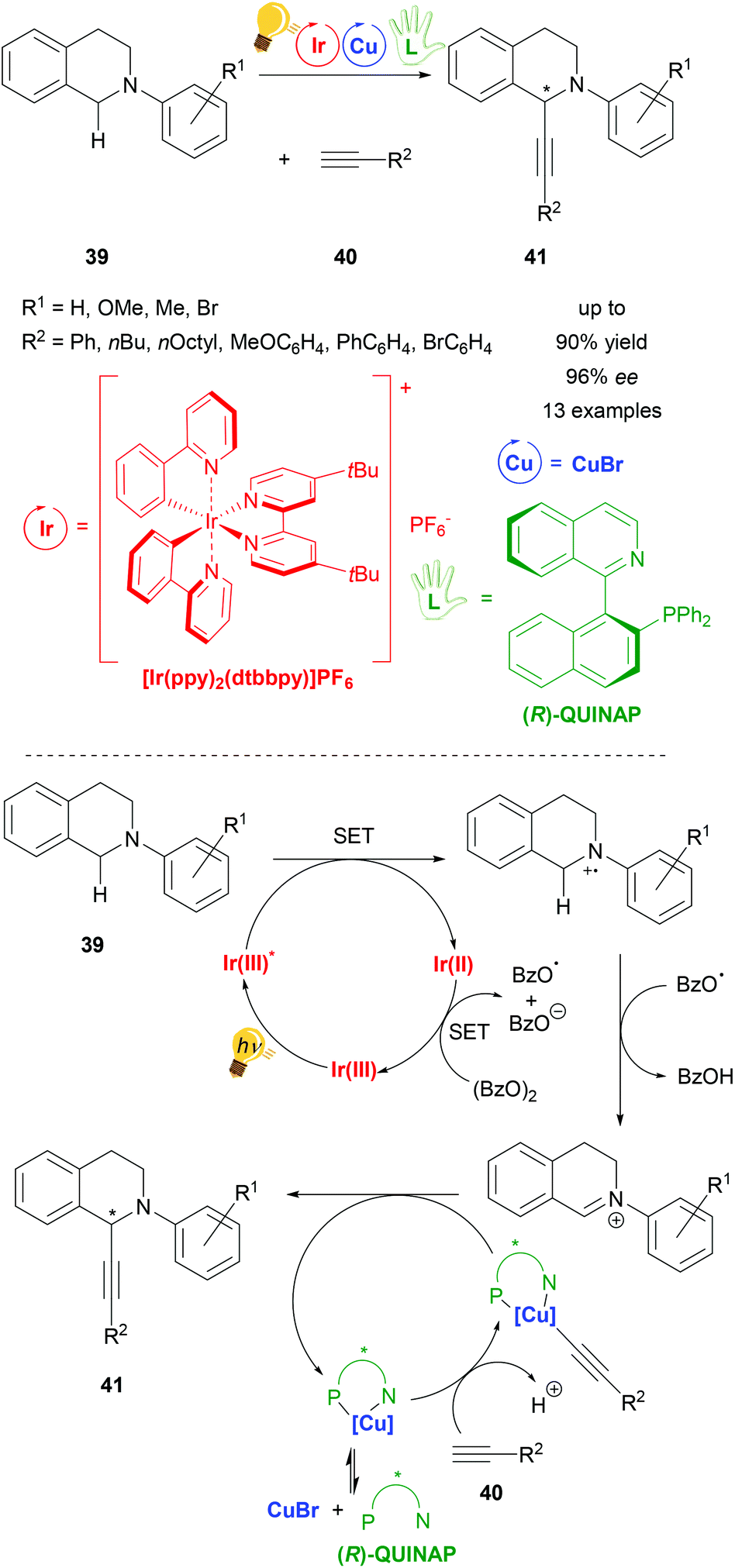 | ||
| Scheme 16 Asymmetric alkynylation of tetrahydroisoquinoline (THIQ) via photoredox catalysis, overview (top) and mechanism (bottom). Reagents and conditions: alkine 40 (1.50 equiv.), Ir-photocat. (1.0 mol%), Cu salt (10.0 mol%), (R)-QUINAP (15.0 mol%), (BzO)2 (1.20 equiv.), THF/MeCN (1/1), −20 °C, Ar, visible light (26 W), 2 d.73 | ||
A complementary study of the functionalization of N-aryl-tetrahydroisoquinoline 39 was accomplished by the same group in 2016 (Scheme 17).74 The THIQ 39 was arylated via a stereoselective photoredox catalysis by also including [Ir(ppy)2(dtbbpy)]PF6 as a photosensitizer and a combination of copper(I) bromide and the chiral PhPyBox ligand as catalysts. The arylated THIQs 43 were obtained in yields of up to 85% and with ee values of up to 90%. The proposed mechanism also shows the formation of a radical cation species of the tetrahydroisoquinoline via SET, which then binds to the active chiral copper complex and is transformed to an iminium ion. The coupling with the corresponding boronic acid 42 leads to the desired product 43.74 It is noteworthy that the reaction without the iridium catalyst was already described in 2008 by Baslé and Li. The product with hydrogen atoms for the substituents R1 and R2 was obtained in yields up to 75% and 44% ee.75
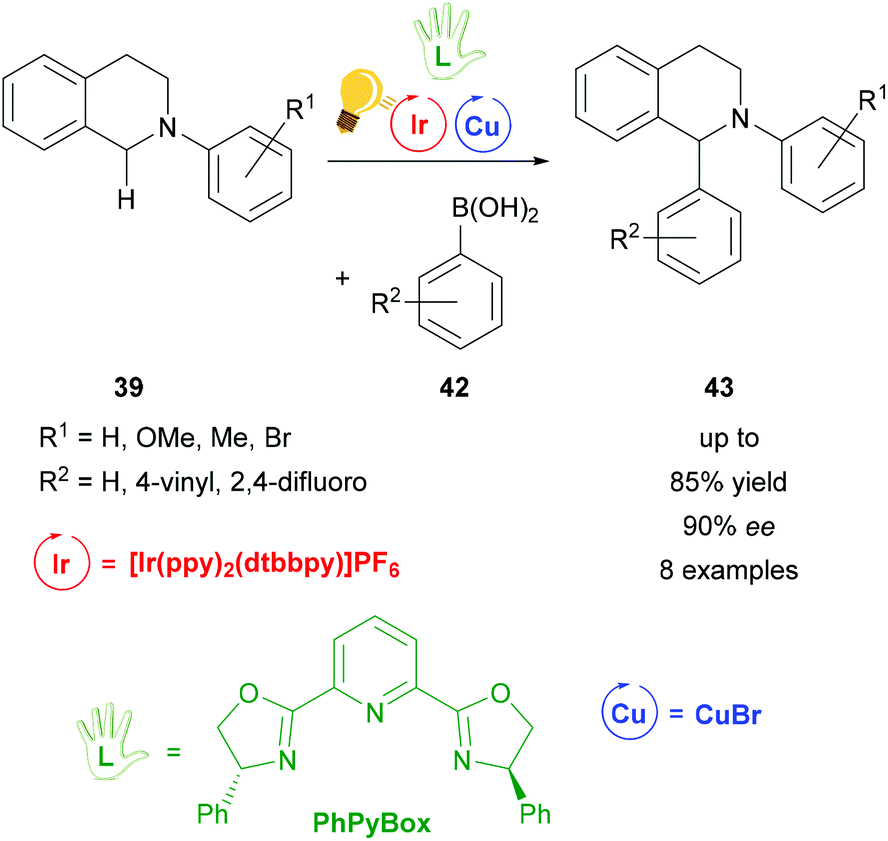 | ||
| Scheme 17 Photoredox-catalyzed arylation of THIQ. Reagents and conditions: boronic-acid 42 (3.00 equiv.), Ir-photocat. (1.0 mol%), Cu salt (10.0 mol%), TBHP (2.00 equiv.), PhPyBox (12.0 mol%), DCE, 4 °C, Ar, visible light, 48 h.74 | ||
A similar reaction mechanism via an iminium intermediate can be found for the alkylation reaction of secondary amines 45 already described by Gao et al. in 2013 (Scheme 18). In contrast to the examples described above, the reaction was carried out at room temperature without cooling, yielding 46 in up to 93(86)% and up to 3:1 dr.76
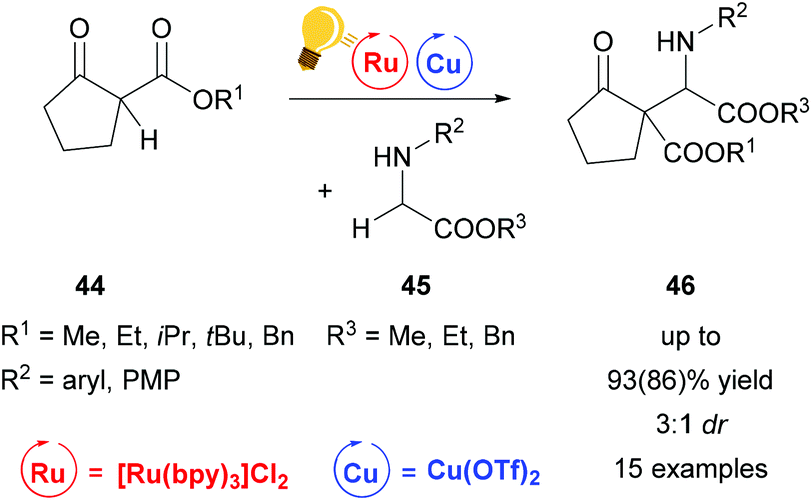 | ||
| Scheme 18 Alkylation of secondary amines via Ru/Cu photoredox catalysis. Reagents and conditions: secondary amine 45 (0.2 mmol) Ru-photocat. (1.0 mol%), Cu salt (10.0 mol%), toluene, rt, air, blue LEDs, 12 h.76 | ||
In 2016, Lin et al. described a reaction to insert one CF2H group stereoselectively into different substituted alkenes 47 based on dual iridium and copper catalysis (Scheme 19). For this purpose the corresponding alkenes 47 were reacted in a one-pot synthesis with the difluoromethyltriphenylphosphonium bromide ([Ph3PCF2H]Br) 48 using Ir(ppy)3 with the addition of copper(II) bromide as photoredox catalysts. For the four examples of this study, yields of up to 83% and E/Z values of up to 97:3 were reported for 49. The mechanism was proposed to be of radical nature.77
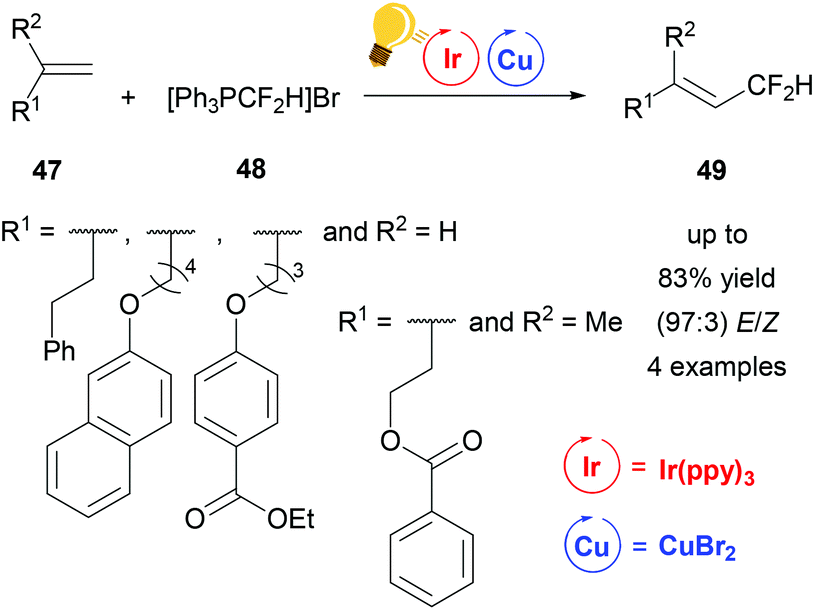 | ||
| Scheme 19 Overview of the difluoromethylation of alkenes via photoredox catalysis. Reagents and conditions: [Ph3PCF2H]Br 48 (3.00 equiv.), Ir-photocat. (0.0015 mmol), Cu salt (0.10 mmol), DBU (6.00 equiv.) after 10 h, DMF, rt, N2, visible light, 20 h.77 | ||
Fluorinated compounds have gained increased attention because of their possible pharmaceutical applications. In 2016 Liu and coworkers presented the decarboxylative difluoroacetylation reaction of α,β-unsaturated carboxylic acids 50 with iododifluoroacetate 37 by a photoredox catalysis using [Cu(MeCN)4]PF6 and [Ru(bpy)3]Cl2 as photoredox catalysts (Scheme 20)78 in addition to Lin et al.77 Within this work a series of substituted phenyl rings, bearing one or more electron-withdrawing and -donating groups as well as furyl, pyridine and thiophene derivatives were transformed to the corresponding difluoroalkylated alkenes 51 with extremely high E/Z values of up to 99%. The optimal reaction conditions were found by initial test reactions with (E)-cinnamic acid and were very mild, at room temperature and without any extra ligand. The yields of the isolated products were measured and the ratio of the E- and Z-isomer was calculated according to the corresponding 19F-NMR of the reaction mixture. The proposed mechanism shows an oxidation of copper(I) salt initiated by the photoactivated ruthenium catalyst to the actual catalyzing copper(II) species, which is regenerated to copper(I) by the elimination of carbon dioxide and the formation of the desired product 51.78
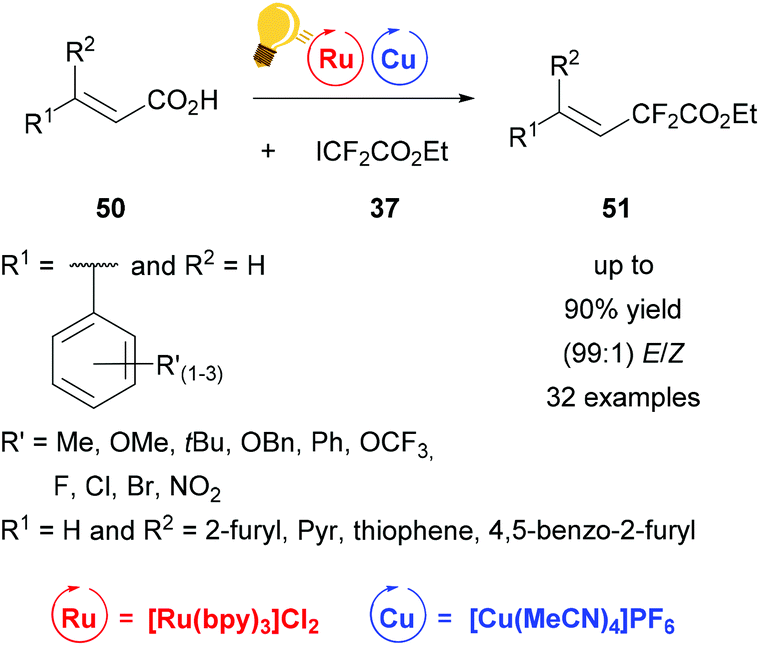 | ||
| Scheme 20 Photoredox catalysis for the synthesis of difluoroalkylated alkenes. Reagents and conditions: 37 (1.50 equiv.), Ru-photocat. (2.0 mol%), Cu salt (10.0 mol%), Et3N (1.50 equiv.), DCM, rt, Ar, blue LED (450 ± 15 nm, 3 W), 12 h.78 | ||
Also using a chiral (1R,2S)-bisoxazoline ligand, the first cyanation reaction catalyzed stereoselectively by a combination of Ir(ppy)3 and copper(I) bromide was reported in 2017 by Wang et al. (Scheme 21).79 Many examples were tested under these conditions. Products 54 were obtained in 99% yield with ee values of up to 99%. According to further test reactions the team of Wang suggested a radical decarboxylation process. The stereoinformation in the products 54 is introduced by the chiral copper complex formed out of copper bromide and the bisoxazoline ligand.79
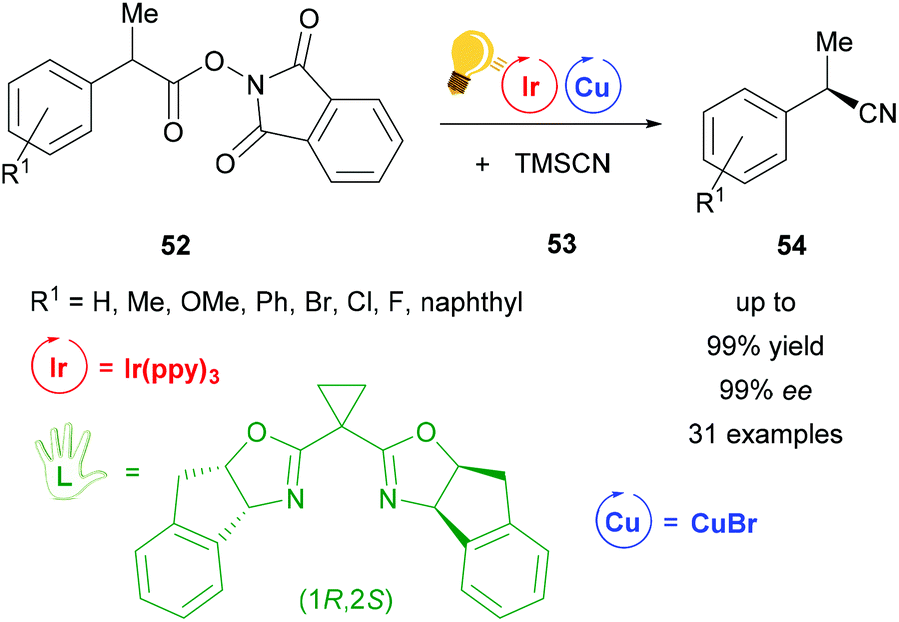 | ||
| Scheme 21 Cyanation by a photoredox catalyzed decarboxylation reaction. Reagents and conditions: Ir-photocat. (0.5 mol%), Cu salt (1.0 mol%), chiral ligand (1.2 mol%), DMF/p-xylene (4/6), rt, Ar, blue LED (12 W), 24 h.79 | ||
After the work of Wang,79 another cyanation reaction with additional alkylation was presented by Sha et al.80 The photoredox catalysis including Ir(ppy)3 as the photosensitizer and copper(I) bromide in combination with another bisoxazoline ligand, that was modified with a cyclopentylidene group, provides corresponding cyanoalkylated derivatives 58 in yields of 88% with ee values of up to 94% for reactions with R2 = cyclopentyl and ee values of up to 90% for reactions with R1 = H and variations of R2 (Scheme 22). In the proposed mechanism the chiral copper catalyst is oxidized to the active Cu(II) species by the intermediate Ir(IV), which is then reduced back to Ir(III). The chiral Cu(II) species reacts further with the benzylic radical and the cyano precursor 57, generating a chiral intermediate Cu(III). The products 58 are then formed via reductive elimination.80
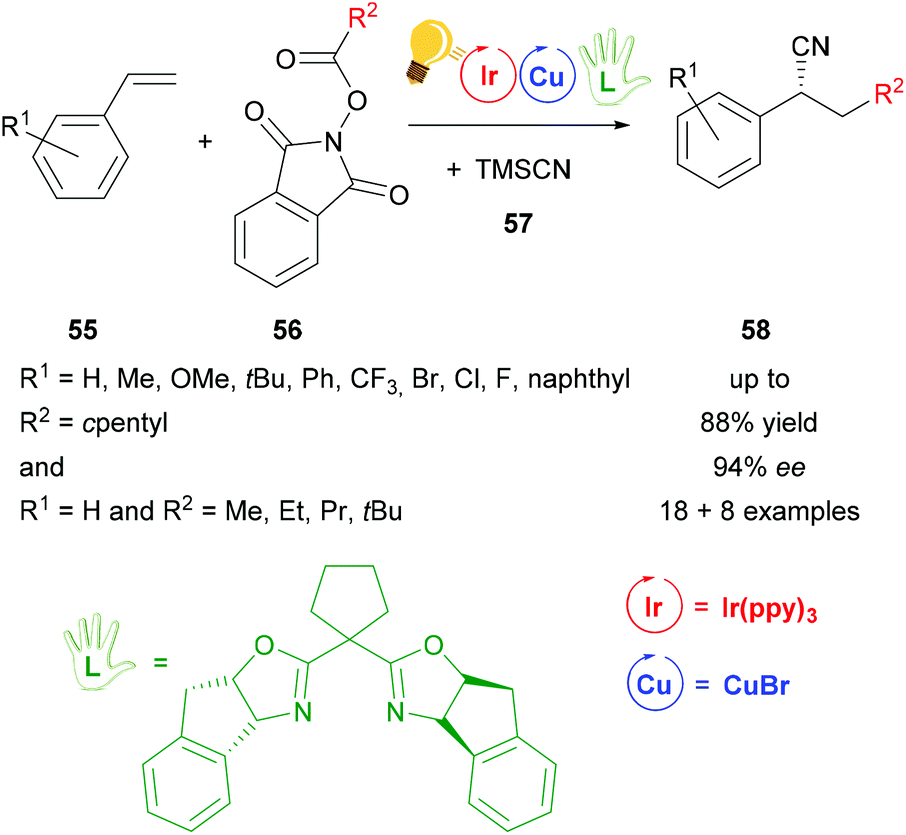 | ||
| Scheme 22 Photoredox catalyzed decarboxylative cyanoalkylation of a variety of styrenes. Reagents and conditions: 56 (0.20 mmol), TMSCN 57 (1.10 eq.), Ir-photocat. (0.5 mol%), Cu salt (1.0 mol%), chiral ligand (1.2 mol%), NMP/PhCl (4/6), rt, Ar, blue LEDs (5 W), 24 h.80 | ||
Zinc(II) complexes
Zinc complexes have successfully been used in important catalytic reactions, such as C–H activation, (asymmetric) hydrosilylation and hydroamination, as well as in ring-opening polymerization.81–83 Zinc alkoxides were used in stereoselective polymerization of D,L-lactide84,85 and β-lactones.11,85 Tong and coworkers reported on the synthesis of stereoregular poly(α-hydroxy acid) via a photoredox assisted ring opening polymerization of O-carboxyanhydride (OCA) 59, with the cooperative use of [(bpy)Ni(COD)] and zinc alkoxide complexes.12,13 In these photoredox polymerizations, the photosensitizer is an iridium(III) complex [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 that mediates the single electron transfer to the Zn/Ni adduct with the opened OCA. When the Zn cocatalyst is a hexamethyldisilazane complex Zn(HMDS)2, isotactic poly((α-hydroxy acid) 60 without epimerization is obtained (Scheme 23).13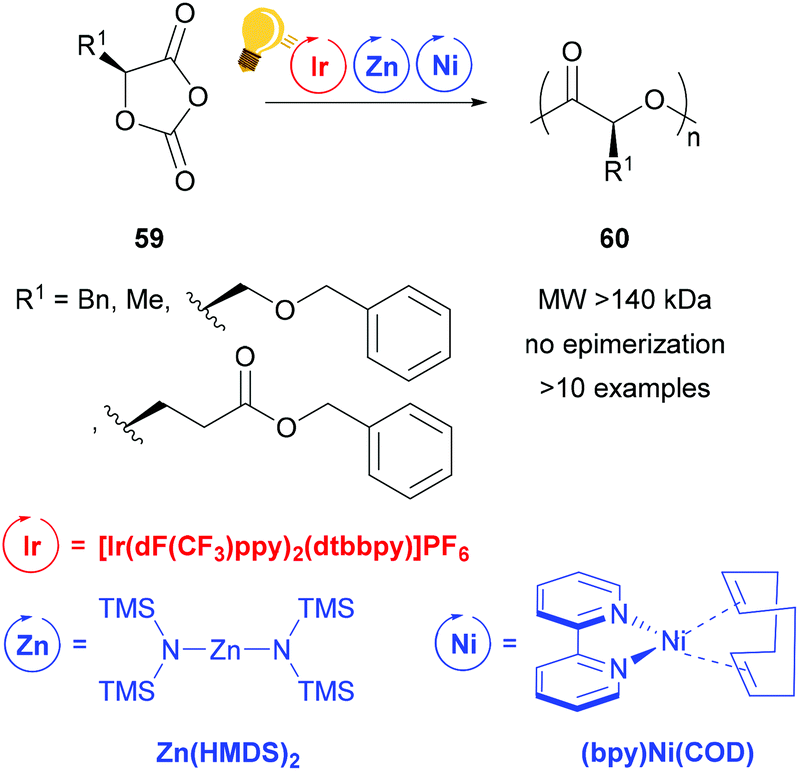 | ||
| Scheme 23 Photoredox supported ring-opening polymerization of O-carboxyanhydrides using a Zn/Ni cocatalyst. Reagents and conditions: 59 (200 equiv.), Ir-photocat. (0.10 equiv.), Zn and Ni (1.00 equiv. each), BnOH (1.00 equiv.), −15 °C, blue LED lamp (34 W), 4–8 h.13 | ||
In their following study, the group of Tong and Xie analysed the influence of a Zn complex with a sterically demanded Schiff-base in the photoredox ring opening polymerization of a racemic mixture of 59.12 Irradiation with a blue LED of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 improved the polymerization kinetic with the presence of (bpy)Ni(COD), but stereoselectivity is imposed by the zinc complex in Scheme 24. In fact the stereoregularity of the polymer is due to the difference in energy of the orientations that the two configurations of the monomer take in the transition state, affecting the direction of (rac)-59, while it approaches the metal core. The probability of a stereo-error leads to isoselective stereoblock polymers with high molecular weight.
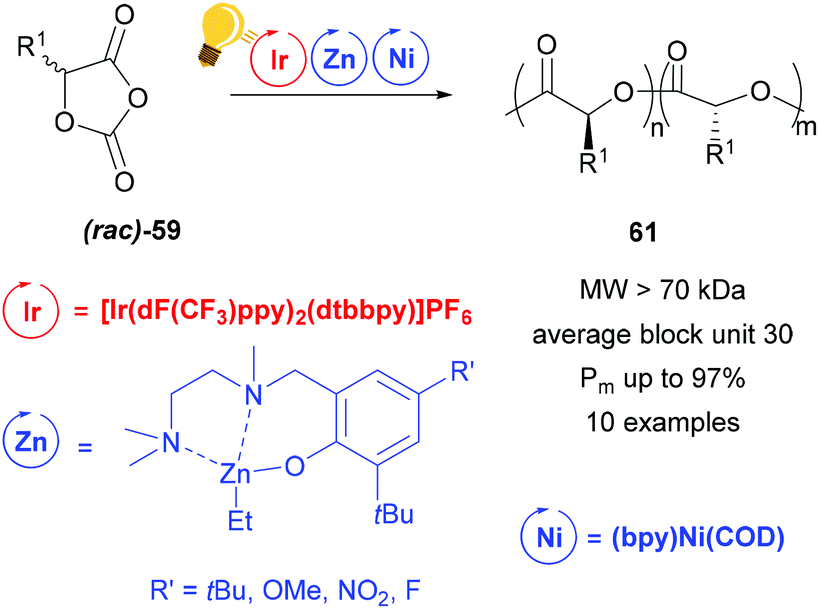 | ||
| Scheme 24 Photoredox stereoselective ring-opening polymerization of racemic O-carboxyanhydrides using a Zn/Ni cocatalyst. Reagents and conditions: 59 (200 equiv.), Ir-photocat. (0.10 equiv.), Zn and Ni (1.00 equiv. each), BnOH (1.00 equiv.), −5 °C, blue LED lamp (34 W), 4–7 h.12 | ||
Rhodium complexes
In 2014, Meggers et al. developed bis-cyclometalated iridium Lewis-acid complexes, which combine visible light induction and asymmetric catalysis in a single chiral catalyst.86 On this basis, the first rhodium complexes that were applied in asymmetric photoredox catalysis were obtained simply by replacing the iridium metal center of a well-known complex with rhodium.87,88 The chiral-at-metal, bis-cyclometalated rhodium complexes proved to be extremely versatile asymmetric photocatalysts. Even though the photophysical properties of the rhodium complexes compared to the iridium analogs were rather limited, their catalytic activity was found to be superior in many applications.27 In reactions where the photocatalyst merely serves as a “smart initiator”, the inferior photoredox properties of the rhodium system are counterbalanced by a much faster ligand exchange and hence a more efficient chain mechanism, which is required in the case of highly reactive intermediates with short lifetimes.89 In reactions where an efficient photoinduced electron transfer is required, an additional photoredox catalyst may be added for complementary dual catalysis.In this short review, only dual catalytic systems consisting of a photosensitizer and a chiral rhodium catalyst will be considered. For an extensive review discussing single and dual asymmetric photocatalysis with rhodium complexes, see Huang et al., 2019.27
With the metal as the stereogenic center, the bis-cyclometalated rhodium complexes RhO (X = O, R = H) and RhS (X = S, R = H) achieve chiral induction through helical chirality with left-handed (Λ-isomer) and right-handed (Δ-isomer) screw sense (Scheme 25). Upon release of the labile acetonitrile ligands, the substrate can bind in a bidentate fashion to the Lewis acidic center, by which the substrate is activated while providing a chiral environment for further reactions.
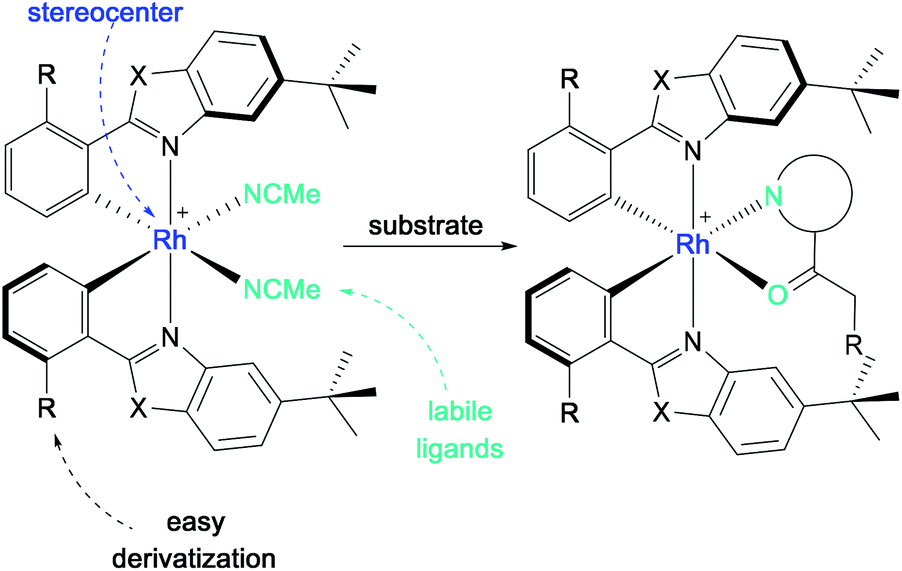 | ||
| Scheme 25 General structure of bis-cyclometalated rhodium(III) complexes for substrate activation in asymmetric photocatalysis (Λ-isomer shown). | ||
The asymmetric photocatalytic reaction modes that can currently be accessed by bis-cyclometalated rhodium complexes can be structured according to the intermediate rhodium-substrate complexes (Fig. 2).27 These are typically formed by N,O-bidentate coordination of 2-acyl imidazoles, N-acyl pyrazoles, or 2-acyl pyridines to the rhodium center forming either rhodium-enolate, rhodium-enone, or rhodium-ketone intermediate complexes, which can then react further either from their photo-excited state (not discussed) or from their ground state.
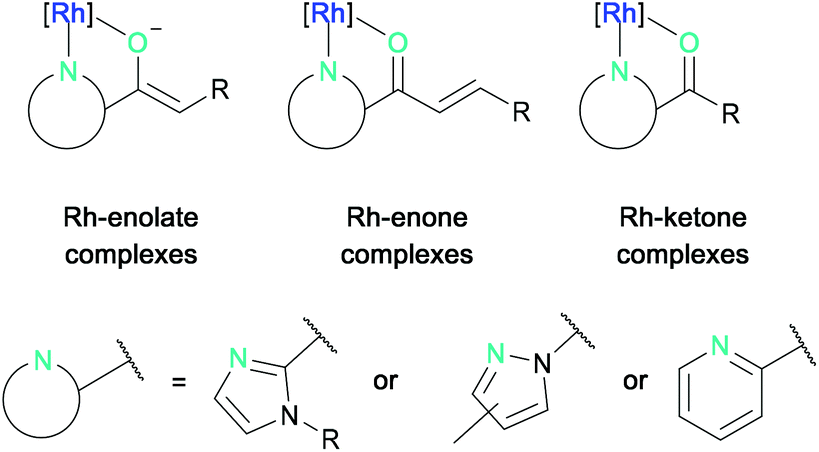 | ||
| Fig. 2 General reaction modes of bis-cyclometalated rhodium(III) complexes with N,O-chelating substrates. | ||
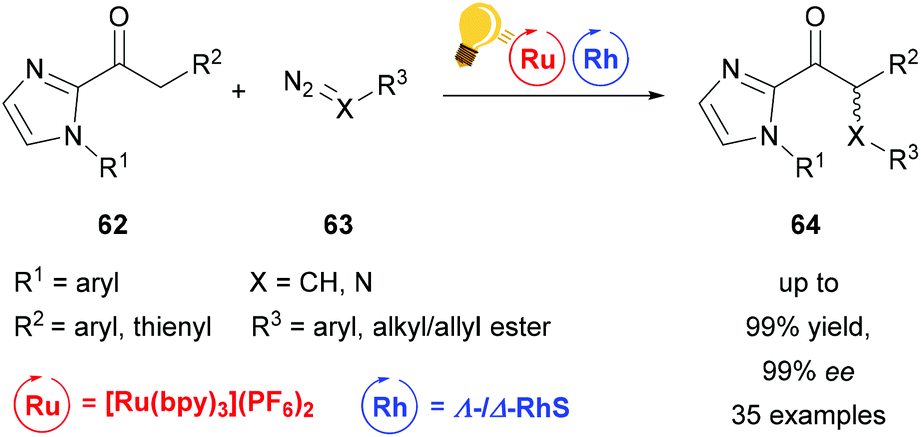 | ||
| Scheme 26 Visible-light activated asymmetric α-alkylation/amination of ketones. Reagents and conditions: Ru-photocat. (1.50–2.50 mol%), Rh-cat. (4.0 mol%), NaHPO4 (20.0 mol%), H2O (20.0 equiv.), acetone/DMSO (9/1), rt, Ar, CFL (21 W), 8–16 h.92 | ||
Building upon the success of merging visible light photocatalysis with rhodium as the chiral Lewis acid catalyst, Liang et al. developed an enantioselective difluoroalkylation of ketones 65 with bromodifluoroacetamides 66 to access fluorine containing γ-keto acid derivatives 67 (Scheme 27).94 First attempts using iridium as both LA and photocatalyst, which were previously successful in α-fluorination, failed when it was applied to difluoroalkylation.95 The combination of Λ-RhS with [Ir(ppy)2(dtbbpy)](PF6) gave the desired product 67 in satisfactory yield and excellent enantioselectivity. Under similar conditions, using [Ru(bpy)3](PF6)2 and replacing bromodifluoroacetamide with bromodifluoroacetate, 66a, the scope of the reaction could be extended to access α,β-unsaturated-γ-keto ester 68.
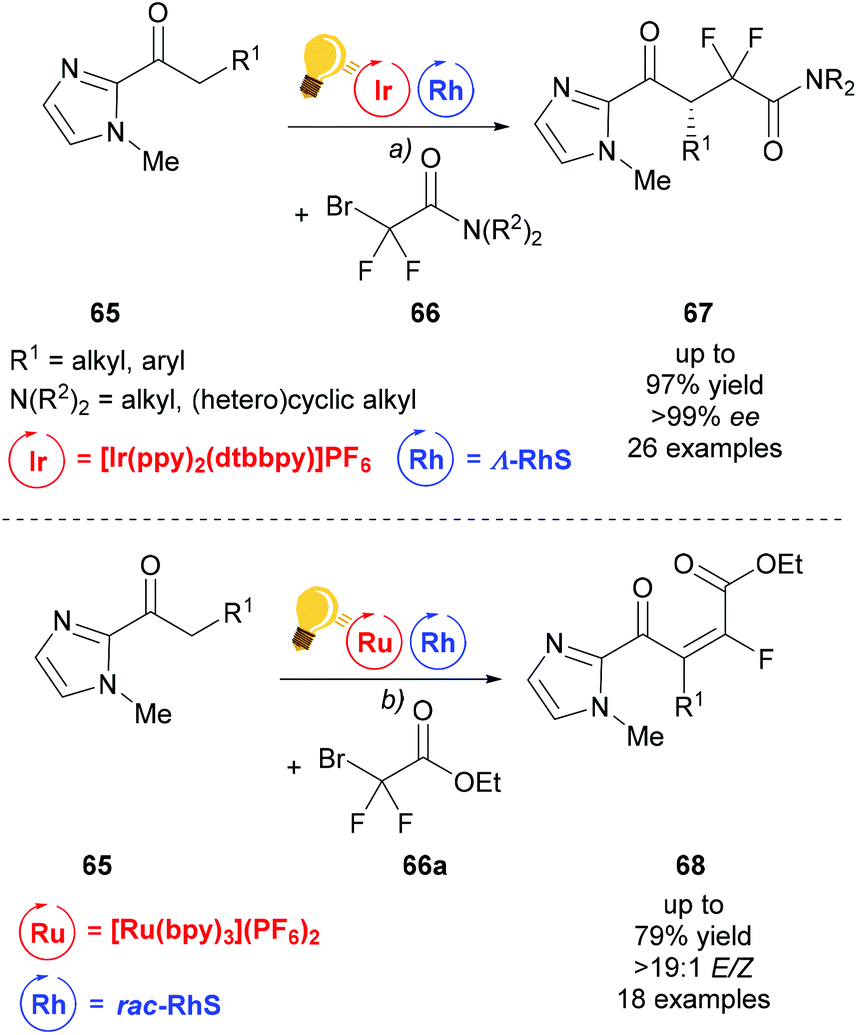 | ||
| Scheme 27 Visible-light activated, asymmetric difluoroalkylation (top) and trans-selective monofluoroalkylation (bottom). Reagents and conditions: (a) Ir-photocat. (1.0 mol%), Rh-cat. (4.0 mol%), DIPEA (2.00 equiv.), CH2Cl2, rt, N2, CFL (23 W); (b) Ru-photocat. (2.0 mol%), Rh-cat. (8.0 mol%), DIPEA (2.00 equiv.), acetone, rt, N2, CFL (23 W).94 | ||
 | ||
| Scheme 28 β-Amination through PCET-induced radical coupling. Reagents and conditions: Ir-photocat. (2.0 mol%), Rh-cat. (5.0 mol%), base (20.0 mol%), CH2Cl2, 4 Å molecular sieves, 22–25 °C, Ar, blue LED (24 + 36 W) 18 h.107 | ||
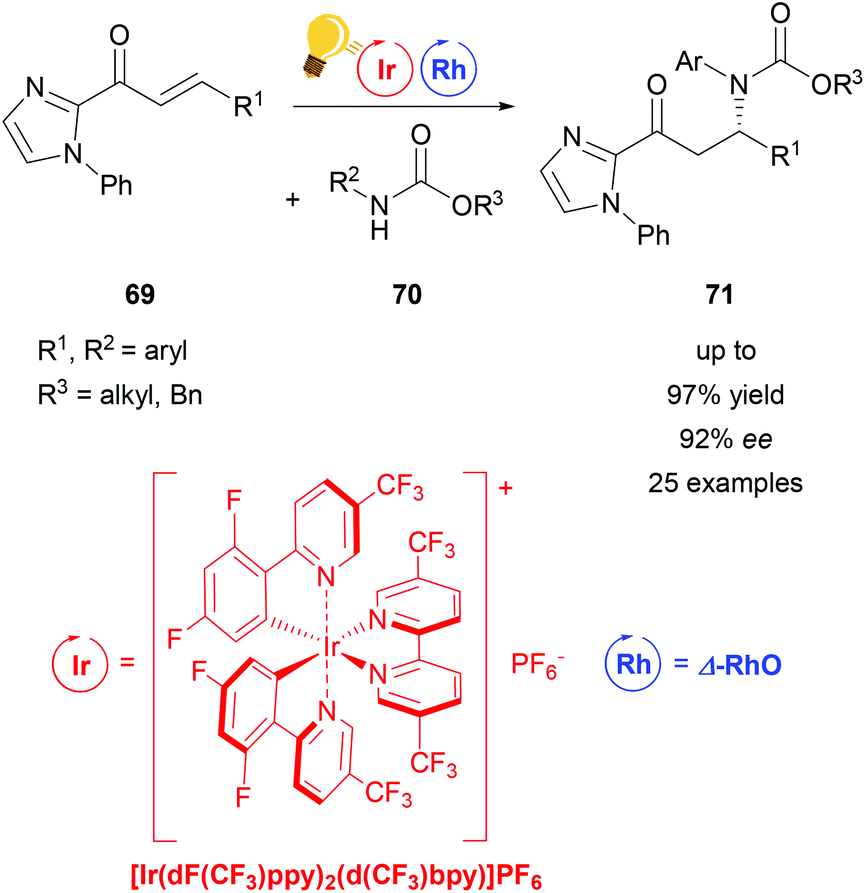
Direct functionalization of Csp3–H bonds is the most attractive way to form new carbon–carbon and carbon–heteroatom bonds.108,109 Building upon the tandem catalysis strategy, Wang et al. combined the Lewis acid catalyst Δ-RhS with fac-[Ir(ppy)3]PF6 as the photosensitizer to develop an enantioselective alkylation of unactivated Csp3–H bonds.110 For the generation of the oxygen-centered radicals, N-alkoxyphtalimides 73 are used as redox-active precursors, which are photocatalytically reduced in the presence of the Hantzsch ester. The key step in this reaction is a radical translocation (1,5-hydrogen atom transfer) from an oxygen centered to a carbon centered radical. Subsequent, stereocontrolled addition to an alkene 72 occurs with high enantioselectivity up to 97% and some diastereoselectivity. The high enantioselectivity, which can be achieved, demonstrates the acceleration of the radical addition by the rhodium-based Lewis acid, outcompeting background reactions (Scheme 29).
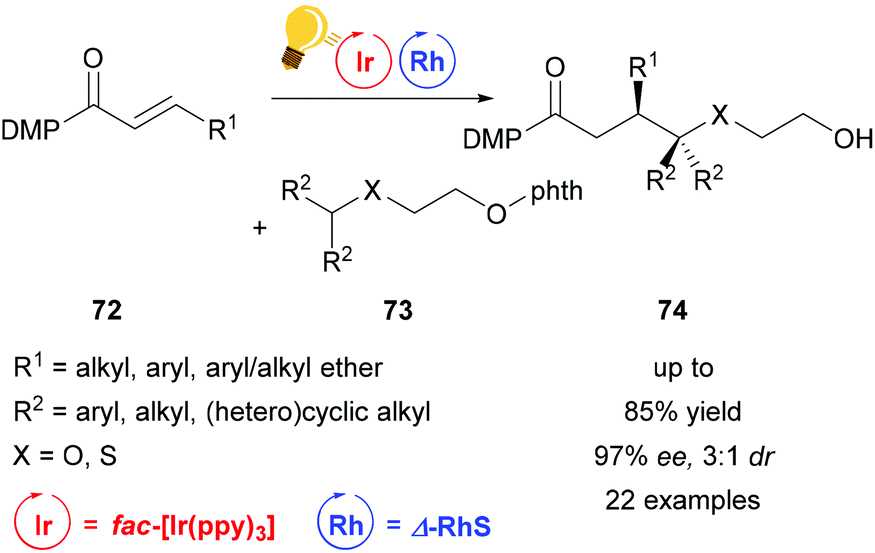 | ||
| Scheme 29 Asymmetric Csp3–H functionalization under photoredox conditions. Reagents and conditions: Ir-photocat. (1.0 mol%), Rh-cat. (8.0 mol%), Hantzsch ester (1.50 equiv.), THF, rt, N2, CFL (23 W), 40–65 h. DMP = 3,5-dimethylpyrazole, phth = N-phthalimide.110 | ||
Heteroatom-radical-enabled remote functionalization of unactivated alkyl C–H bonds is especially challenging due to the high reactivity of the intermediate radical species.111 Yuan et al. could achieve remote functionalization of unactivated Csp3–H of N-alkyl amides by combining asymmetric Lewis acid and photoredox catalysis.112 The chiral-at-metal Lewis acid Λ-RhO binds to the α,β-unsaturated 2-acylimidazole substrate 75, which can then be reduced to an enolate radical anion. The photoredox catalyst [Ir(dF(CF3)ppy)2-(d(CF3)bpy)]PF6 enables the single electron transfer to generate an amidyl radical, which translocates to a carbon centered radical in the δ-position after 1,5-hydrogen atom transfer. Through this pathway, a variety of δ-alkyl functionalized substrates 77 is accessible in good yields and high enantioselectivity (Scheme 30).
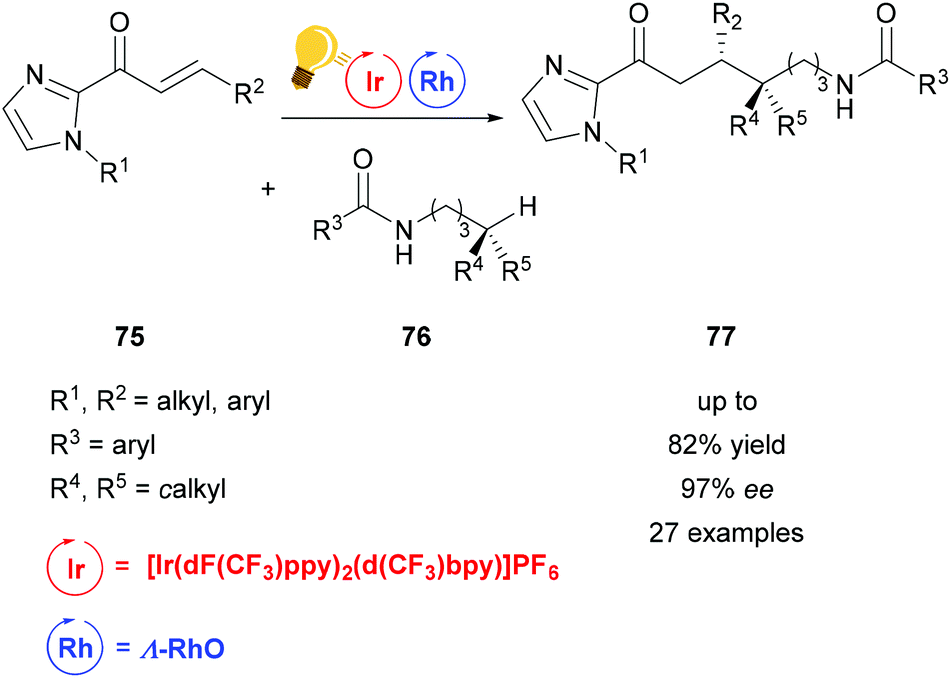 | ||
| Scheme 30 Asymmetric alkylation of remote Csp3–H bonds. Reagents and conditions: Ir-photocat. (4.0–8.0 mol%), Rh-cat. (6.0–8.0 mol%), base (5.0–8.0 mol%), CH2Cl2, 4 Å molecular sieves, 27 °C, Ar, blue LED (2 × 24 W), 38 h.112 | ||
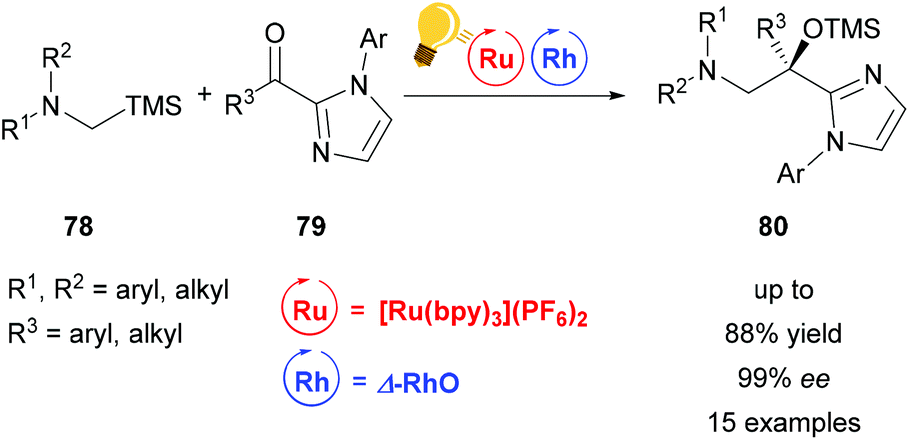 | ||
| Scheme 31 C–H activation with a chiral rhodium catalyst. Reagents and conditions: Ru-photocat. (1.0 mol%), Rh-cat. (4.0 mol%), MeCN/DMAC (4/1), rt, N2, CFL (23 W), 15 h.114 | ||
While the bis-cyclometalated rhodium complexes, which were developed by Meggers et al. work extremely well for a wide range of reactions with good yields and enantioselectivity, to date they have been the only rhodium-based catalysts used in combination with photoredox catalysis in order to achieve stereoselective transformations. A recent publication from Breit and coworkers describes a regiodivergent hydroaminoalkylation of alkynes and allenes by merging rhodium catalysis with a photoredox system in a non-stereoselective fashion (Scheme 32).115 By combining [Rh(COD)Cl2] with [Ir(ppy)(dtbbpy)]PF6 and (rac)-BINAP as a ligand, they could access the linear α-allylation product. By changing the ligand to DPEphos, which is bis[(2-diphenylphosphino)phenyl]ether, an inversion of selectivity was observed, favouring the branched α-allylation product. Due to the fact that no chiral ligand was used, no enantioselectivity but some substrate dependent diastereoselectivity could be observed.
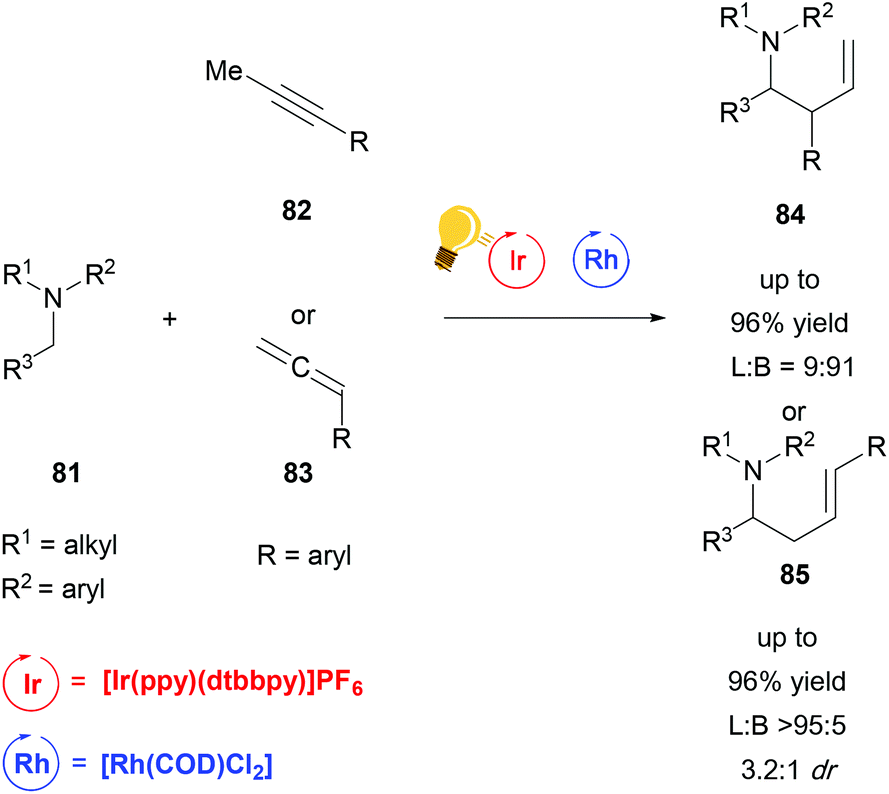 | ||
| Scheme 32 Reagents and conditions linear: Ir-photocat. (2.0 mol%), Rh-cat. (2.5 mol%), PPTS (20 mol%), (rac)-BINAP (10 mol%) DCE, r.t., Ar, blue LED (4.8 W), 3 d. Reagents and conditions branched: Ir-photocat. (0.5 mol%), Rh-cat. (5.0 mol%), PhMe2C2O2H (20 mol%), DPEphos (10 mol%) DCE/DMF (3:1), r.t., Ar, blue LED (4.8 W), 2 d. (L = linear; B = branched).115 | ||
The reaction generates synthetically useful homoallylic amines in an atom-economical and efficient manner. Particularly the selective access to branched allylic derivatives in a reaction involving radicals is remarkable. This work illustrates the power of cooperative catalytic systems and at the same time the current lack of methods to run such a reaction in an enantioselective fashion.
Palladium(II) complexes
With an ever increasing demand for greener chemistry, it is of great interest to improve traditional cross-coupling methodologies with the possibilities that are offered by visible-light driven photocatalysis. While improving existing procedures is one desirable goal, the synergistic cooperation of two metals enables otherwise inaccessible cross-coupling reactions under very mild conditions. In this section, palladium, one of the most widely used transition metals, is described as a catalyst that can benefit significantly from the merger with photoredox catalysis. Many efforts have been made to replace palladium altogether and move towards a metal-free organocatalysis. However, combined with photocatalysis, palladium-catalysis is drastically improved with regard to catalyst load, efficiency and selectivity and therefore remains a relevant option in synthetic chemistry.Huang and coworkers reported a significant acceleration of a palladium-catalyzed Heck reaction when adding visible-light mediated photo-catalysis to the reaction conditions (Scheme 33).116 As the Heck reaction was found to proceed only during periods of irradiation in a light-on/light-off experiment, it was concluded that the regenerating speed of the active palladium catalyst was enhanced by photocatalysis. Interestingly, this mild and ligand-free method provided cinnamates 88 exclusively in (E)-configuration, while stilbenes were obtained with high (Z)-selectivity. This was found to be due to a visible-light promoted isomerization of (E)-stilbene, as proved by the conversion of (E)-stilbene to (Z)-stilbene under reaction conditions.
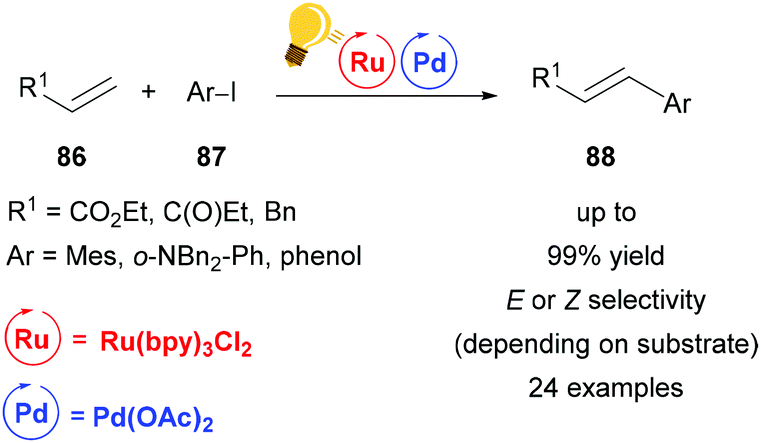 | ||
| Scheme 33 Ligand-free, visible-light promoted palladium-catalysed Heck cross-coupling. Reagents and conditions: olefin 86 (2.00 equiv.), aryl iodide 87 (1.00 equiv.), Ru-photocat. (1.0 mol%), Pd-cat. (1.0 mol%), Et3N (2.00 equiv.), DMF, rt, blue LED.116 | ||
An example of a higher ordered catalyst for stereoselective photoredox catalysis is presented by the homochiral ruthenium/palladium coordination cage by Su and coworkers (Scheme 34).117 A metal organic cage ([Pd6(RuL3)]28+) is used as a chiral confined photoredox catalyst to couple naphthols 89 in a regio- and stereoselective fashion. In contrast to conventional 1,1-coupling, the axially chiral product 90 is obtained by 1,4-coupling of naphthols. The mechanism proceeds through several experimentally confirmed radical species. The authors state the possibility of intramolecular charge separation between Ru and Pd. The interdependency of high yield/low ee or high ee/low yield reflects the cage character of the catalyst, as the host/guest ratio heavily affects both yield and selectivity.
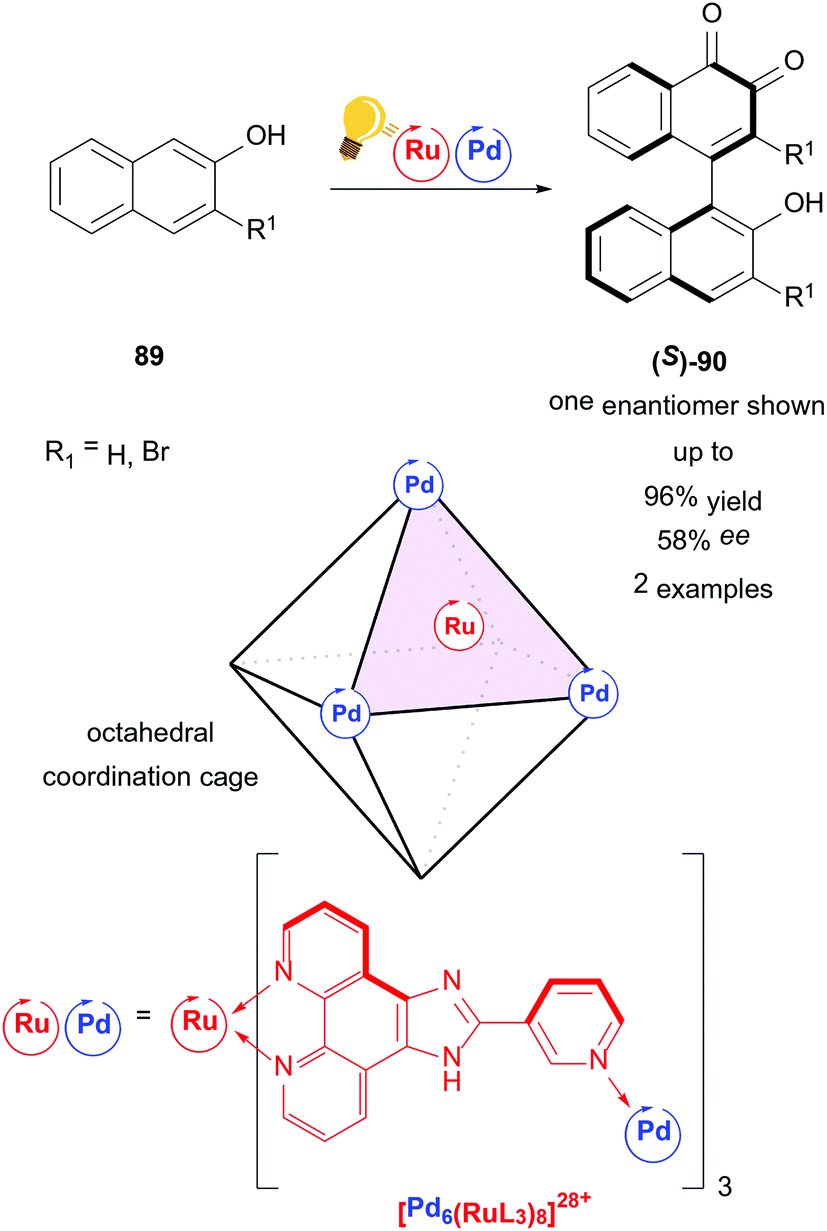 | ||
| Scheme 34 Coordination cage-catalyzed naphthol dimerization, photo-hydrogen-evolving unit shown as a catalyst. Reagents and conditions: naphthol 89 (1.00 equiv.), Ru/Pd-photocat. (10.0 mol%), D2O, rt, air, blue LED (8 W), 24 h.117 | ||
The powerful methodology of palladium-catalyzed asymmetric allylic alkylation is underdeveloped for “hard” or nonstabilized alkyl nucleophiles. By a combination of palladium- and photocatalysis using [Ir(ppy)2(dtbbpy)]PF6, Zhang and coworkers were able to achieve a highly regio- and enantioselective allylic alkylation with a range of such unstabilized alkyl nucleophiles (Scheme 35).118 Alkyl radicals generated from dihydropyridines (DHP) 92 serve as the coupling partners for π-allyl palladium complexes, thereby complementing the scope of previous palladium catalyzed asymmetric allylic alkylation protocols.
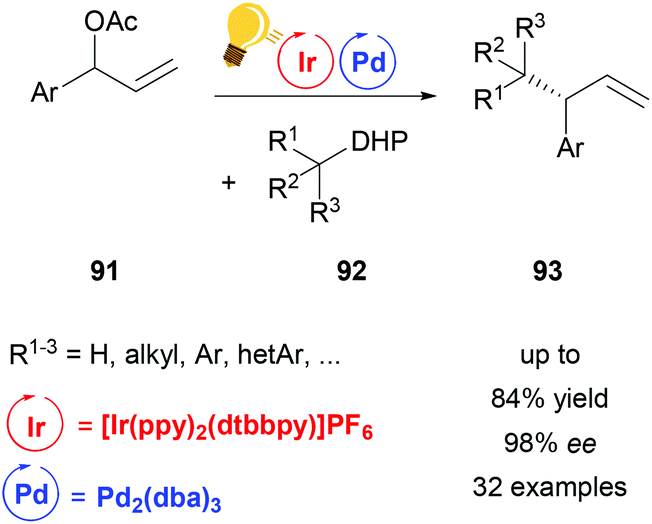 | ||
| Scheme 35 Palladium-catalysed asymmetric allylic alkylation. Reagents and conditions: allyl ester 91 (1.00 equiv.), dihydropyridine 92 (1.50 equiv.), Ir-photocat. (2.0 mol%), Pd-cat. (2.5 mol%), ligand (6.0 mol%), CH3CN, blue LED (45 W), 12 h.118 | ||
A last example of the increasing trend to merge palladium and photoredox catalysis is the decarboxylative alkenylation of aliphatic carboxylic acids 94 with vinyl arenes reported by Zheng and coworkers (Scheme 36).119 The β-alkylated styrenes 96 obtained by this protocol were found to be formed with high Z-selectivity. This is hard to achieve with classical ground-state transition metal catalysis due to the thermodynamically favorable E-alkenes and the high energy barrier for redox neutral decarboxylation. However, the use of an Ir-cocatalyst circumvented these issues. Remarkably, the procedure even permits the use of demanding substrates like amino acids while no external oxidant is required. The Ir-catalyst serves both as an electron-transfer and as an energy-transfer reagent in a mechanism believed to proceed through a reductive quenching of the excited Ir(III) by a carboxylate anion to generate the alkyl radical. The Pd(III) species, formed after the reaction of the radical with styrene, is reduced to Pd(II) by a second single electron transfer from Ir(II) regenerating the Ir(III), thus completing the photoredox cycle. The (E)-styrene formed after beta-hydride elimination is converted to the (Z)-isomer by subsequent uphill catalysis, mediated by energy transfer from the Ir-photocatalyst.
 | ||
| Scheme 36 cis-Selective decarboxylative alkenylation by photoredox/palladium/uphill triple catalysis. Reagents and conditions: carboxylic acid 94 (1.00 equiv.), Alkene 95 (2.50 equiv.), Ir-photocat. (2.0 mol%), Pd-cat. (5.0 mol%), TMP (6.0 mol%), K2HPO4 (2.50 equiv.), chlorobenzene, blue LED (36 W), rt, 24 h.119 | ||
Gold(I) complexes
In the last two decades, homogeneous gold catalysis has seen a surge of interest owing to its ability to activate C–C multiple bonds due to its soft, π-acidic character.120 However, the use of a Au(I)/(III) redox cycle by classical means (external oxidant) is mostly prevented by the high redox potential. Instead, proto-demetallation leads to hydrofunctionalized products.121,122 Recent efforts merge gold catalysis with visible-light mediated photo-catalysis, thus overcoming the Au(I)/(III) energy hurdle to generate elegant protocols for the mild cross-coupling of C–C multiple bonds.A Meyer–Schuster-rearrangement and subsequent arylation of propargyl alcohols was reported to proceed with excellent diastereoselectivity when a base (KH2PO4) was employed (Scheme 37).123 The reaction proceeds with blue light, green light and sunlight as well as with light from a CFL source in good yields. The same transformation was simultaneously investigated by the Shin group and the Luna group.124,125 The use of the catalytically inactive precatalyst [Ph3PAuCl] (as opposed to the cationic gold species) leads to high selectivity with nearly no side reactions like hydrofunctionalization or homocoupling. This further emphasizes the potential of visible light-mediated catalysis.
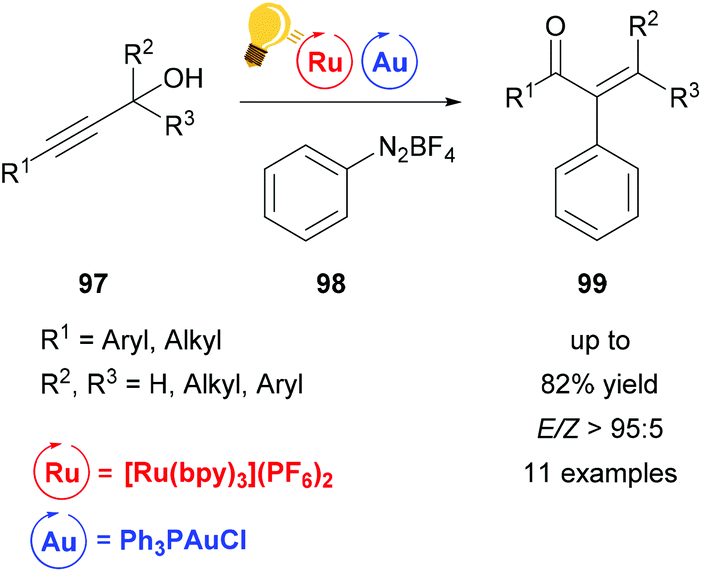 | ||
| Scheme 37 Tandem arylative Meyer–Schuster rearrangement. Reagents and conditions: propargyl alcohol (1.00 equiv.), PhN2BF498 (4.00 equiv.), KH2PO4 (4.00 equiv.), Ru-photocat. (2.5 mol%), Au-cat. (10.0 mol%), MeOH, rt, Ar, 5 W green LED, 16 h.123,126 | ||
Toste et al. used a combination of Au(I) and Ru(bpy)3(PF6)2 to establish a ring expansion-oxidative arylation protocol.126 This provides access to functionalized cyclic ketones 102 from alkenyl and allenyl cycloalkanols 100 and aryl diazonium salts 101 at ambient conditions. In contrast to previous work127,128 where the Au(I) interacts with the nucleophile first before the photoredox step, in this work photoredox catalysis occurs first, generating a cationic Au(III) intermediate that acts as a formal aryl electrophile (Scheme 38).
 | ||
| Scheme 38 Ring expansion–oxidative arylation protocol. Reagents and conditions: R1-PhN2BF4101 (3.00 equiv.), Ru-photocat. (2.5 mol%), Au-cat. (10.0 mol%), MeOH/CH3CN (3/1), rt, N2, visible light.126 | ||
The stereoselectivity of the reaction was examined by deuterium-labeling experiments. The reaction proceeds with a high degree of diastereoselectivity due to the previously described anti-migration129,130 and stereochemical retention commonly found with alkylgold(III)phenyl128 species (Scheme 39).
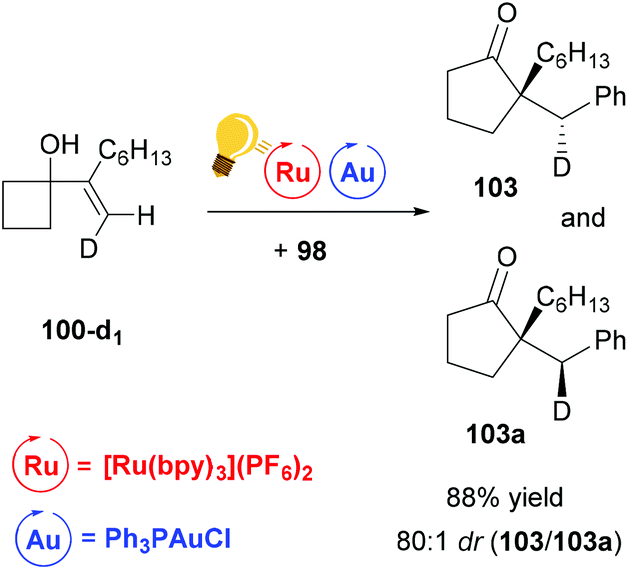 | ||
| Scheme 39 Ring expansion–oxidative arylation protocol. Deuterium-labelling experiment.126 Reagents and conditions: the same as described in Scheme 38. | ||
Ye et al. used the same catalyst system of Au(I) and [Ru(bpy)3](PF6)2 for the arylative cyclization of homopropargyl sulfonamides 104 with diazonium salts 105.131 This allows the facile synthesis of various enantioenriched 2,3-dihydropyrroles 106 in excellent enantioselectivities without any strong oxidants. The authors suggest that the reaction mechanism undergoes a Au(I)/(III) redox cycle, which is facilitated by stepwise SET oxidation in a Au(I)/Au(II)/Au(III) fashion (Scheme 40).
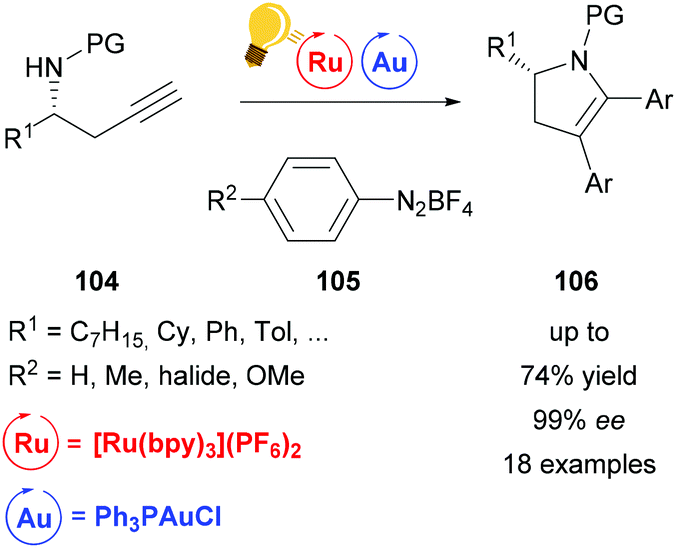 | ||
| Scheme 40 Arylative cyclization of homopropargyl sulfonamides. Reagents and conditions: Ru-photocat. (2.5 mol%), Au-cat. (20.0 mol%), MeOH/DMF (1/9), rt, white LED (13 W), 2 h.131 | ||
The difunctionalization of alkynes 107 can be realized by direct cyclization as in the example above. Another useful method is atom transfer radical addition.132 However, this approach proceeds via vinyl radicals, a highly reactive species, which makes stereoselectivity a difficult goal to achieve due to the facile E/Z-isomerization. Gold(I) however can be used to steer the highly reactive vinyl species towards stereoselectivity by generating a trans-vinyl gold intermediate (Scheme 41). Xu and coworkers reported a stereoselective thiosulfonylation reaction leading to thiofunctionalized vinyl sulfones.134 This reaction features 100% atom economy and excellent regioselectivity with a broad scope and very low catalyst loading. Employing this method, enyne systems could additionally be converted to carbo- and heterocycles. In contrast to the above examples, a cationic gold catalyst was necessary to achieve satisfactory yields.
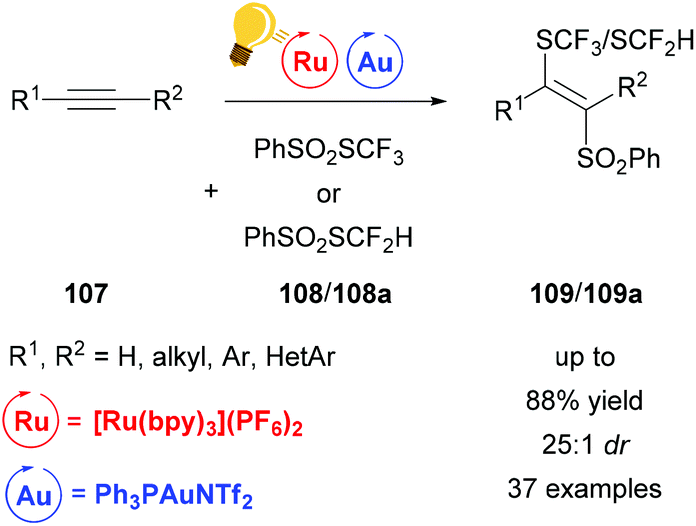 | ||
| Scheme 41 Atom transfer radical addition for a stereoselective thiosulfonylation. Reagents and conditions: alkyne 107 (1.00 equiv.), PhSO2SCF3108 or 108a (1.10 equiv.), Ru-photocat. (2.5 mol%), Au-cat. (0.5 mol%), MeOH/DMF (1/9), rt, white LED (13 W), 2 h.134 | ||
Conclusions and perspectives
Combining photochemistry and metal catalysis represents a fascinating move towards sustainable organic synthesis. The synergy between these two processes is an example of the cooperative use of photoredox and metal catalysts. In particular, in this perspective, we have drawn our attention to cooperative stereoselective catalysis. These reactions are ultimately suitable for the synthesis of highly functionalized materials and important agro- and pharmaceuticals. In fact the stereochemistries of these materials and molecules have a decisive role in their final physical and chemical properties. Moreover, it is not only desirable to start from inexpensive and abundant, sustainable substrates, but also the reaction conditions should be allowed to work at ambient temperature. Mild conditions are beneficial especially for those substrates with many functional groups, without the need for protection. Thanks to the photoredox catalyst, the generation of radicals occurs via electron transfer processes (usually SET), initiated by the absorption of photons. Light is emitted in all cases by artificial sources (e.g. LED, fluorescence lamp), but in principle, photons can be considered green reagents supplied by our unlimited source, the Sun.Photoredox catalysts based on metal complexes are in most cases robust and have usually reversible electrochemistry. Nevertheless, we observe a heavy dependence from Ru- or Ir-based coordination complexes. Even if these catalysts are never used in amounts higher than 5 mol%, scientists should consider employing photoredox catalysts based on more cost-effective metals,135 such as iron or copper. So far, only very few examples show the use of iron complexes as a photoredox catalyst136–138 and only rarely in combination with a chiral organocatalyst139 (thus, out of the scope of this perspective). Analogously, copper complexes take part in a variety of dual catalytic reactions, but mainly as a transition metal catalyst. As a matter of fact, although there are many studies on Cu-based photosensitizers, their use in combination with other catalysts (whether metal based or organic) is underrepresented. Of course, to choose an optimal photoredox catalyst, we should consider several elements. Besides their cost, one of them is their excitation energy, which should be preferentially at a long wavelength, such as visible light. In fact, excitation at a higher energy (e.g. UV) might cause absorption from organic reagents and other pathways might compete with the desired transformation. Recently, upconversion140via a triplet–triplet annihilation process was used to populate the excited state of the photocatalyst by near-infrared (NIR) irradiation.141,142 This strategy allows not only milder operating conditions, but also a higher penetration depth of the excitation light, which is useful especially in large-scale reactions. Furthermore, the redox property of the photoredox catalyst in its excited state has to be considered. The excited photoredox catalyst is at the same time a better oxidant (thus, an electron acceptor) and a better reductant (thus, an electron donor) than the ground state. Reductive or oxidative quenching processes are both potentially possible, therefore the same photocatalyst can mediate different transition metal catalysed pathways, on the condition that thermodynamic requirements are fulfilled.
Furthermore, the challenge to achieve new products with high stereoselectivity has been tackled by adopting several strategies in the cases herein reported. Asymmetric products were obtained with good to excellent enantiomeric excess when a chiral transition metal complex was used as a catalyst (e.g. Δ-RhS). Otherwise, the asymmetric transition metal catalyst was formed in situ by the addition of a chiral ligand to the reaction mixture. In this case the choice of the chiral ligand has a high impact on the final result. When the starting materials bear chiral centres, often we observe a conservation of stereochemistry, thanks to mild conditions induced by the photoredox activation. When racemic mixtures are used, enantioconvergence is very desirable and further efforts in this field are highly beneficial. The stereoselectivity and regiochemistry of unsaturated compounds can generally be steered by a transition metal catalyst and the geometry of addition in the catalytic cycle which is governed by sterical factors. By the careful choice of a suitable combination of the metal and the ligand sphere, a high degree of control can be achieved.
In conclusion, cooperative photoredox and transition metal catalysis offer a fast, stereoselective and sustainable access to a vast range of chiral products. In particular, this approach combines C–C and C–X bond formation with a broad functional group tolerance. In the perspective of a more sustainable chemistry, the use of visible light (or even NIR light) as energy for chemical transformation is highly desired and further studies on merging photocatalysis with stereoselective catalysis are very attractive.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DFG-funded Collaborative Research Centre (SFB) TRR 88/3MET “Cooperative Effects in Homo- and Heterometallic Complexes”Notes and references
- H. Rau, Chem. Rev., 1983, 83, 535–547 CrossRef CAS.
- Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- J. W. Beatty and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 10270–10273 CrossRef CAS PubMed.
- D. A. Di Rocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed.
- M. D. Karkas, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef CAS PubMed.
- J. Kalepu, L. T. Pilarski, P. Gandeepan and L. Ackermann, Chem. Sci., 2018, 9, 4203–4216 RSC.
- M.-A. Tehfe, L. Ma, B. Graff, F. Morlet-Savary, J.-P. Fouassier, J. Zhao and J. Lalevee, Macromol. Chem. Phys., 2012, 213, 2282–2286 CrossRef CAS.
- J. Xu, A. Atme, A. F. Marques Martins, K. Jung and C. Boyer, Polym. Chem., 2014, 5, 3321–3325 RSC.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- Q. Feng, L. Yang, Y. Zhong, D. Guo, G. Liu, L. Xie, W. Huang and R. Tong, Nat. Commun., 2018, 9, 1559 CrossRef PubMed.
- Q. Feng and R. Tong, J. Am. Chem. Soc., 2017, 139, 6177–6182 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- E. Paternò and C. Maselli, Chem. Zentralbl., 1912, 83(1), 1022–1023 Search PubMed.
- G. Ciamician, Science, 1912, 36, 385 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- W.-J. Zhou, Y.-H. Zhang, Y.-Y. Gui, L. Sun and D.-G. Yu, Synthesis, 2018, 50, 3359–3378 CrossRef CAS.
- M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144–4147 CrossRef CAS PubMed.
- M. Rueping, C. Vila and T. Bootwicha, ACS Catal., 2013, 3, 1676–1680 CrossRef CAS.
- X. Wang, G. D. Cuny and T. Noel, Angew. Chem., Int. Ed., 2013, 52, 7860–7864 CrossRef CAS PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef CAS PubMed.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392 CrossRef CAS PubMed.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed.
- R. Brimioulle and T. Bach, Science, 2013, 342, 840 CrossRef CAS PubMed.
- H. Guo, E. Herdtweck and T. Bach, Angew. Chem., Int. Ed., 2010, 49, 7782–7785 CrossRef CAS PubMed.
- M. L. Conner, Y. Xu and M. K. Brown, J. Am. Chem. Soc., 2015, 137, 3482–3485 CrossRef CAS PubMed.
- https://www.epa.gov/sites/production/files/2016-09/documents/chromium-compounds.pdf .
- J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed.
- P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- C.-x. Song, P. Chen and Y. Tang, RSC Adv., 2017, 7, 11233–11243 RSC.
- S. M. Thullen and T. Rovis, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS PubMed.
- P. Rai, K. Maji and B. Maji, Org. Lett., 2019, 21, 3755–3759 CrossRef CAS PubMed.
- J. Hou, A. Ee, W. Feng, J.-H. Xu, Y. Zhao and J. Wu, J. Am. Chem. Soc., 2018, 140, 5257–5263 CrossRef CAS PubMed.
- D. Kalsi, S. Dutta, N. Barsu, M. Rueping and B. Sundararaju, ACS Catal., 2018, 8, 8115–8120 CrossRef CAS.
- L. Niu, H. Yi, S. Wang, T. Liu, J. Liu and A. Lei, Nat. Commun., 2017, 8, 14226 CrossRef PubMed.
- L. Niu, S. Wang, J. Liu, H. Yi, X.-A. Liang, T. Liu and A. Lei, Chem. Commun., 2018, 54, 1659–1662 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- Y. Budnikova, D. Vicic and A. Klein, Inorganics, 2018, 6, 18 CrossRef.
- A. Klein, A. Sandleben and N. Vogt, Proc. Natl. Acad. Sci., India, Sect. A, 2016, 86, 533–549 CrossRef CAS.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195–2198 CrossRef CAS PubMed.
- D. Ryu, D. N. Primer, J. C. Tellis and G. A. Molander, Chemistry, 2016, 22, 120–123 CrossRef CAS PubMed.
- M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., 2016, 128, 262–266 CrossRef.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- A. Noble, S. J. McCarver and D. W. MacMillan, J. Am. Chem. Soc., 2015, 137, 624–627 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed.
- H. Yue, C. Zhu and M. Rueping, Angew. Chem., 2018, 130, 1385–1389 CrossRef.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed.
- H. Ye, C. Xiao, Q. Q. Zhou, P. G. Wang and W. J. Xiao, J. Org. Chem., 2018, 83, 13325–13334 CrossRef CAS PubMed.
- Q.-Q. Zhou, F.-D. Lu, D. Liu, L.-Q. Lu and W.-J. Xiao, Org. Chem. Front., 2018, 5, 3098–3102 RSC.
- J. Liu, W. Ding, Q. Q. Zhou, D. Liu, L. Q. Lu and W. J. Xiao, Org. Lett., 2018, 20, 461–464 CrossRef CAS PubMed.
- I. Perepichka, S. Kundu, Z. Hearne and C. J. Li, Org. Biomol. Chem., 2015, 13, 447–451 RSC.
- P. Querard, I. Perepichka, E. Zysman-Colman and C. J. Li, Beilstein J. Org. Chem., 2016, 12, 2636–2643 CrossRef CAS PubMed.
- O. Baslé and C.-J. Li, Org. Lett., 2008, 10, 3661–3663 CrossRef PubMed.
- X.-W. Gao, Q.-Y. Meng, M. Xiang, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Adv. Synth. Catal., 2013, 355, 2158–2164 CrossRef CAS.
- Q. Y. Lin, Y. Ran, X. H. Xu and F. L. Qing, Org. Lett., 2016, 18, 2419–2422 CrossRef CAS PubMed.
- H. R. Zhang, D. Q. Chen, Y. P. Han, Y. F. Qiu, D. P. Jin and X. Y. Liu, Chem. Commun., 2016, 52, 11827–11830 RSC.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 CrossRef CAS PubMed.
- W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489–7494 CrossRef CAS.
- N. V. Tzouras, I. K. Stamatopoulos, A. T. Papastavrou, A. A. Liori and G. C. Vougioukalakis, Coord. Chem. Rev., 2017, 343, 25–138 CrossRef CAS.
- M. J. González, L. A. López and R. Vicente, Tetrahedron Lett., 2015, 56, 1600–1608 CrossRef.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2004, 2504–2505, 10.1039/B405362A.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- C. Wang, L. A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094–1100 RSC.
- J. Ma, X. Zhang, X. Huang, S. Luo and E. Meggers, Nat. Protoc., 2018, 13, 605–632 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed.
- S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef CAS PubMed.
- T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed.
- X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
- R. P. L'Esperance, T. M. Ford and M. Jones, J. Am. Chem. Soc., 1988, 110, 209–213 CrossRef.
- H. Liang, G. Q. Xu, Z. T. Feng, Z. Y. Wang and P. F. Xu, J. Org. Chem., 2019, 84, 60–72 CrossRef CAS PubMed.
- G.-Q. Xu, H. Liang, J. Fang, Z.-L. Jia, J.-Q. Chen and P.-F. Xu, Chem. – Asian J., 2016, 11, 3355–3358 CrossRef CAS PubMed.
- H. J. Federsel, M. Hedberg, F. R. Qvarnstrom, M. P. Sjogren and W. Tian, Acc. Chem. Res., 2007, 40, 1377–1384 CrossRef CAS PubMed.
- H. J. Federsel, Drug News Perspect., 2008, 21, 193–199 CrossRef CAS PubMed.
- R. Edupuganti and F. A. Davis, Org. Biomol. Chem., 2012, 10, 5021–5031 RSC.
- S. Jones and C. J. Warner, Org. Biomol. Chem., 2012, 10, 2189–2200 RSC.
- M. A. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed.
- A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441–4557 CrossRef CAS PubMed.
- T. C. Nugent, Chiral amine synthesis: methods, developments and applications, John Wiley & Sons, 2010 Search PubMed.
- D. Enders, C. Wang and J. X. Liebich, Chemistry, 2009, 15, 11058–11076 CrossRef CAS PubMed.
- S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed.
- C. Cabrele, T. A. Martinek, O. Reiser and L. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655–663 CrossRef CAS PubMed.
- Z. Zhou, Y. Li, B. Han, L. Gong, E. Meggers and E. Meggers, Chem. Sci., 2017, 8, 5757–5763 RSC.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- R. Giri, B. F. Shi, K. M. Engle, N. Maugel and J. Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- X. Q. Hu, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2017, 56, 1960–1962 CrossRef CAS PubMed.
- W. Yuan, Z. Zhou, L. Gong and E. Meggers, Chem. Commun., 2017, 53, 8964–8967 RSC.
- C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS PubMed.
- J. Ma, K. Harms and E. Meggers, Chem. Commun., 2016, 52, 10183–10186 RSC.
- J. Zheng and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 3392–3397 CrossRef CAS PubMed.
- H. Zhang and X. Huang, Adv. Synth. Catal., 2016, 358, 3736–3742 CrossRef CAS.
- J. Guo, Y.-W. Xu, K. Li, L.-M. Xiao, S. Chen, K. Wu, X.-D. Chen, Y.-Z. Fan, J.-M. Liu and C.-Y. Su, Angew. Chem., Int. Ed., 2017, 56, 3852–3856 CrossRef CAS PubMed.
- H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef CAS PubMed.
- C. Zheng, W.-M. Cheng, H.-L. Li, R.-S. Na and R. Shang, Org. Lett., 2018, 20, 2559–2563 CrossRef CAS PubMed.
- M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed.
- H. A. Wegner and M. Auzias, Angew. Chem., Int. Ed., 2011, 50, 8236–8247 CrossRef CAS PubMed.
- A. Tlahuext-Aca, M. N. Hopkinson, R. A. Garza-Sanchez and F. Glorius, Chem. – Eur. J., 2016, 22, 5909–5913 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, E. Busto and A. Luna, Adv. Synth. Catal., 2016, 358, 1526–1533 CrossRef CAS.
- J. Um, H. Yun and S. Shin, Org. Lett., 2016, 18, 484–487 CrossRef CAS PubMed.
- X.-z. Shu, M. Zhang, Y. He, H. Frei and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5844–5847 CrossRef CAS PubMed.
- N. P. Mankad and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 12859–12861 CrossRef CAS PubMed.
- E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard III and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293–14300 CrossRef CAS PubMed.
- F. Kleinbeck and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 9178–9179 CrossRef CAS PubMed.
- J. P. Markham, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 9708–9709 CrossRef CAS PubMed.
- Z.-S. Wang, T.-D. Tan, C.-M. Wang, D.-Q. Yuan, T. Zhang, P. Zhu, C. Zhu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2017, 53, 6848–6851 RSC.
- J. M. Muñoz-Molina, T. R. Belderrain and P. J. Pérez, Eur. J. Inorg. Chem., 2011, 2011, 3155–3164 CrossRef.
- H. Li, C. Shan, C.-H. Tung and Z. Xu, Chem. Sci., 2017, 8, 2610–2615 RSC.
- H. Li, Z. Cheng, C.-H. Tung and Z. Xu, ACS Catal., 2018, 8, 8237–8243 CrossRef CAS.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS PubMed.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 2227–2233 CrossRef CAS.
- J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2247–2253 CrossRef CAS.
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- M. Freitag, N. Möller, A. Rühling, C. A. Strassert, B. J. Ravoo and F. Glorius, ChemPhotoChem, 2019, 3, 24–27 CrossRef CAS.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS PubMed.
Footnote |
| † We dedicated this to Prof. Annie Powell for wishing her a happy birthday. |
| This journal is © The Royal Society of Chemistry 2019 |