
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium–arene complexes bearing naphthyl-substituted 1,3-dioxoindan-2-carboxamides ligands for G-quadruplex DNA recognition†
Laura A.
Hager
a,
Stephan
Mokesch
a,
Claudia
Kieler
b,
Silvia
Alonso-de Castro
c,
Dina
Baier
b,
Alexander
Roller
a,
Wolfgang
Kandioller
 a,
Bernhard K.
Keppler
a,
Walter
Berger
b,
Luca
Salassa
cd and
Alessio
Terenzi
*ac
a,
Bernhard K.
Keppler
a,
Walter
Berger
b,
Luca
Salassa
cd and
Alessio
Terenzi
*ac
aInstitute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna, Waehringer Str. 42, A-1090 Vienna, Austria. E-mail: aterenzi@dipc.org
bDepartment of Medicine I, Institute of Cancer Research and Comprehensive Cancer Center, Medical University Vienna, Borschkegasse 8a, A-1090 Vienna, Austria
cDonostia International Physics Center, Paseo Manuel de Lardizabal 4, Donostia, 20018, Spain
dIkerbasque, Basque Foundation for Science, Bilbao, 48013, Spain
First published on 4th July 2019
Abstract
Quadruplex nucleic acids – DNA/RNA secondary structures formed in guanine rich sequences – proved to have key roles in the biology of cancers and, as such, in recent years they emerged as promising targets for small molecules. Many reports demonstrated that metal complexes can effectively stabilize quadruplex structures, promoting telomerase inhibition, downregulation of the expression of cancer-related genes and ultimately cancer cell death. Although extensively explored as anticancer agents, studies on the ability of ruthenium arene complexes to interact with quadruplex nucleic acids are surprisingly almost unknown. Herein, we report on the synthesis and characterization of four novel Ru(II) arene complexes with 1,3-dioxoindan-2-carboxamides ligands bearing pendant naphthyl-groups designed to bind quadruplexes by both stacking and coordinating interactions. We show how improvements on the hydrolytic stability of such complexes, by substituting the chlorido leaving ligand with pyridine, have a dramatic impact on their interaction with quadruplexes and on their cytotoxicity against ovarian cancer cells.
Introduction
In the last 15 years, there has been considerable interest in new DNA-targeting drugs able to recognize non-canonical motifs called G-quadruplexes (G4s).1 Such DNA structures are formed in G-rich sequences as a stacked arrangement of guanine tetrads which are connected by loops of varying length. Even though they share the same core structure, G4s exhibit a polymorphic nature. This in turn leads to motifs characterized by unique binding sites for the potentially selective binding of small molecules. Furthermore, G4s are not randomly distributed within the human genome, but are overrepresented in telomeres and in the promoter regions of cancer-related genes, with key roles in their regulation.2 For these reasons they have become popular biological targets in cancer research, and there might be the chance that G4s will be the protagonists of a new DNA-based targeted therapy era.1 Currently, there are two fluoroquinolone-based G4-binders which entered clinical trials as G4-targeting anticancer drug candidates.1Complexes of different metals (e.g. Pt, Ni, Au) can effectively interact with G4 structures.3–5 Many of these compounds have also been tested against cancer cell lines both as potential drugs and/or probes. In contrast, reports on ruthenium-based compounds capable of binding (selectively) to G4s are only starting to emerge. This is somehow surprising considering that Ru complexes such as NAMI-A and IT-139 (formerly NKP-1339) have been considered as promising metal-based alternatives to clinically used Pt drugs.6–9 The ruthenium-based G4 binders reported so far are mostly substitutionally inert polypyridyl complexes where one of the N,N-ligands has an extended aromatic moiety which allows for partial stacking with the G4 tetrads.10–19 Although extensively explored as anticancer agents,20–22 only one family of Ru–arene complexes has been designed and investigated as G4-binding drugs to the best of our knowledge. Liu and Mei developed Ru(II)–arene complexes bearing phenanthroimidazole ligands which proved to stabilize the quadruplex formed in c-Myc oncogene via a groove binding mechanism and to inhibit the proliferation, migration, and invasion of breast cancer cells.23–25 Overall, the scarce use of Ru-based scaffolds for G4-binding can principally be ascribed to the preferred octahedral or piano-stool geometries, which are not ideal for stacking on top of the guanine tetrads of the G4 structure.
In recent years, we developed different classes of metal-based compounds to selectively target G-quadruplexes, including Ni(II) salen-type derivatives,26 Pt(II) supramolecular coordination complexes,27,28 and Ru(II) N-heterocyclic carbenes.29,30 The aim of this article is to explore a new strategy for the design of Ru–arene complexes possessing distinct G4 binding abilities that can positively affect their anticancer activity. This approach could pave the way for utilizing this promising class of compounds and their structural versatility for G-quadruplex targeting purposes.
In this work, we present the synthesis of four novel half-sandwich Ru(II) complexes attached to the 1,3-dioxoindan-2-carboxamide ligand scaffold bearing a pendant naphthyl-group. O,O-Dioxoindane-based ligands were selected among our toolkit of ligands since they show important biological activity via topoisomerase inhibition when coordinated to the organometallic Ru(II) cym (cym = p-cymene) moiety.31 Herein, we modified the dioxoindane scaffold with alkyl linkers (of different length) attached to a pendant naphthyl group able to π-stack on top of the G-quadruplex tetrads, thereby conferring G4 binding activity to the final compounds. Furthermore, we explored the effect of substituting the chlorido leaving ligand with pyridine, which has been shown to significantly increase hydrolytic stability.32–36 This approach was adopted considering that the interplay between stability and dynamic behavior (ligand release via hydrolysis) is assumed to be a key step in the activation of most organometallic Ru(II) complexes.6,9,20 We consequently investigated the ability of the resulting compounds to bind G4 motifs in vitro through spectroscopy and high resolution mass spectrometry and evaluated their anti-proliferative activity against ovarian cancer cells. Overall, we have found that small improvements in the hydrolytic stability of the Ru scaffold by pyridine coordination led to dramatic enhancements in both G4 binding properties and cytotoxicity.
Results and discussion
Synthesis and X-ray crystal structures
Ru complexes 1a–b and 2a–b were prepared via a 5-step synthetic route summarized in Fig. 1. Commercially available 2-hydroxy-1,4-naphtoquinone (Lawsone) was converted to 2-oxido-3-phenyl-iodonio-1,4-naphthoquinone which served as a precursor for both ligands.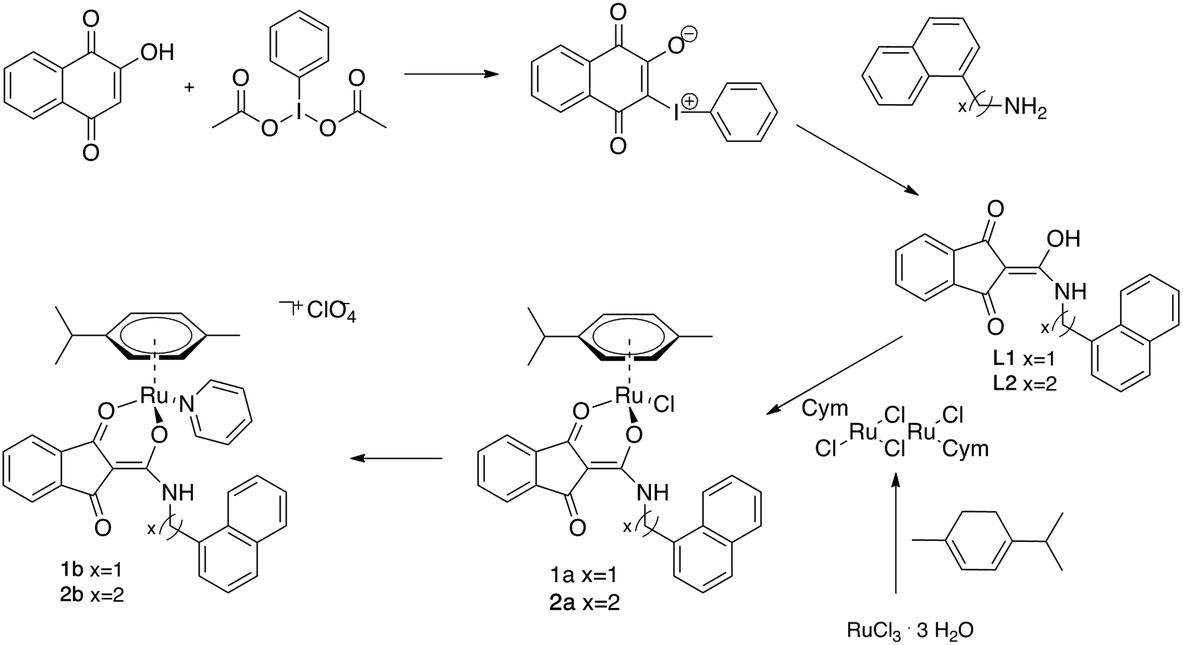 | ||
| Fig. 1 Synthetic route for the preparation of compounds 1a–b and 2a–b. | ||
The precursor, which undergoes a Wolff-type rearrangement to yield a (ring-contracted) ketocarbene intermediate, was then treated with the respective primary amine yielding the N-substituted 1,3-dioxoindan-2-carboxamides L1 and L2. Complexes were obtained by deprotonation of the respective ligand and subsequent reaction with the dimeric ruthenium species bis[dichlorido(cymene)ruthenium(II)]. In order to replace the leaving group by pyridine, the chlorido ligand was abstracted by AgNO3, followed by addition of pyridine and NaClO4. As the formed Ru–N bond could be cleaved under prolonged light irradiation, the last step of the reaction was performed under exclusion of light.
Complexes 1a and 2a were designed as multivalent G-quadruplex binders since they bear both a naphthyl moiety able to stack on top of the guanine tetrads as well as an exchangeable ligand directly coordinated to the ruthenium center, thereby providing a binding site for a guanine nitrogen (typically N7) after aquation of the metal center. Pyridine derivatives 1b and 2b were specifically designed to increase water solubility and to evaluate the role of the N-donor aromatic ligand on the G4 binding ability and the impact on the cytotoxicity of the complexes.
All the synthesized compounds were characterized by standard spectroscopic and analytical methods confirming the formation and purity of the desired complexes (see for instance Fig. S1† for 1H NMR of L1, 1a and 1b). UV-Vis spectra in buffered aqueous solutions of the precursors L1 and L2 and of the metal compounds 1a–b and 2a–b at different concentrations are reported in Fig. S2.† Ligands L1 and L2 display very similar spectral features with a broad band centered at 396 nm, a multi-peaked band in the range 290–320 nm and two intense peaks centered at 273 and 264 nm, respectively. Chlorido complexes 1a and 2a retain the broad band at about 400 nm but lack the narrow multi-peaked bands at lower wavelengths, exhibiting a single broad band at 277 nm. The same trend is followed by the pyridine complexes 1b and 2b, which, besides the band at 400 nm, show an additional band with two maxima at about 275 and 250 nm, respectively.
Single crystals suitable for X-ray diffraction studies were obtained for 1a, 2a and 1b and the molecular structures are shown in Fig. 2 while selected bond lengths and angles are given in Table 1.
 | ||
| Fig. 2 Representation of 1a, 2a, and 1b structures derived from X-ray analysis (for the anisotropic displacement ellipsoids see ESI†); hydrogen atoms and counter ions are omitted for clarity. Numbering and anisotropic displacement ellipsoids are reported in Fig. S3–S5 of the ESI† together with all crystal data given in Tables S2–S7.† | ||
| Bond length/angle | 1a | 2a | 1b |
|---|---|---|---|
| Ru–arene (centroid) | |||
| Ru(1)–O(1) | 2.107 | 2.098 | 2.069 |
| Ru(1)–O(2) | 2.095 | 2.085 | 2.099 |
| Ru(1)–Cl(1) | 2.404 | 2.410 | — |
| Ru(1)–N(2) | — | — | 2.109 |
| O(1)–Ru(1)–O(2) | 88.587 | 90.072 | 90.065 |
The synthesized complexes share similar structural features and adopt the typical three-legged piano-stool geometry commonly encountered in Ru(II) arene structures, with the Ru atom π-bonded to the p-cymene ligand, the anionic O,O-chelating 1,3-dioxoindan-2-carboxamide ligands coordinated in a bidentate fashion and the chlorido (1a and 2a) or the pyridine (1b) leaving groups completing the piano-stool configuration. The observed bond lengths are in good agreement with similar previously reported half-sandwich ruthenium complexes.31
Stability in solution and interaction with G-quadruplex structures by FRET
Before testing their G-quadruplex stabilizing capability, we checked the stability in solution of the synthesized metal-based compounds. Time dependent 1H-NMR spectra of 1a and 1b, selected as examples of chlorido and pyridine derivatives, respectively, showed that the compounds were stable after 24 h in DMSO and CH3CN, which were the solvents used for stock solutions (see Fig. S6–S9†). This control experiment was necessary in order to exclude possible ligand exchange of the complexes, like observed previously for other Ru–arene compounds in DMSO.37 Concerning the stability in aqueous solution, complexes 1a–b and 2a–b were stable in MES buffer at pH 6 (see UV-Vis and 1H-NMR spectra in Fig. S10–S12†). On the other hand, the same compounds showed reduced stability at higher pH values (Tris-HCl buffer supplemented with KCl, pH 7.8), with the pyridine derivatives being more stable towards ligand loss than the chlorido analogues (see time-dependent UV-Vis spectra in Fig. S13†). In particular, 1a readily dissociated ligand L1 upon dissolution, whereas 1b under the same conditions released L1 after 3 hours, indicating that coordination of a pyridine stabilized the binding of the 1,3-dioxoindan-2-carboxamides L1 and to the Ru center (Fig. S13†).G4 stabilization studies by FRET were carried out at pH 7.2, using potassium cacodylate 60 mM as buffer. MES and Tris-HCl buffers were not used since their pKa values change significantly with temperature, while cacodylate buffer does not suffer from this issue.38 Besides, fluorescent dyes used for the labeling of ODNs are protonated and quenched at acidic pH (e.g. in MES).39 At pH 7.2, compounds were expected to behave similarly to what observed in Tris-HCl, yet their stability in solution is sufficient for proving G4 binding in the FRET time scale (incubation 1 or 2 h). NMR analysis demonstrated, for instance, that a 60 mM cacodylate solution of 1b is stable for at least 2 h (Fig. S14†).
For the FRET assay, we incubated the metal compounds and the corresponding ligands with selected oligonucleotides (ODNs, see sequences in Table S8†) folded as G4s (from telomeric DNA and oncogene promoters) or duplex DNA. As shown by the ΔT1/2 values reported in Table 2, ligands L1 and L2 have a detrimental effect on G4 stability, decreasing the melting temperature of the ODNs in all studied cases. The chloride derivatives 1a and 2a do not exert any significant stabilization effect, except for 2a stabilizing only the B-DNA model ODN dsDNA. Interestingly, the pyridine derivatives 1b and 2b are the only ones which strongly affect the melting temperature of the selected ODNs, showing a preference for G4 models, in particular for the G4 formed in the promoter of the c-Kit gene.
| ΔT (°C) | |||||
|---|---|---|---|---|---|
| dsDNA | hTelo | c-Kit1 | Bcl2 | hTERT | |
| L1 | −3.5 | −2.4 | −7.6 | −2.4 | −4.0 |
| L2 | −3.4 | −2.1 | −7.1 | −2.3 | −2.9 |
| 1a | 4.0 | 0.9 | −0.1 | 1.2 | 2.7 |
| 1b | 10.2 | 11.0 | 20.9 | 5.1 | 9.5 |
| 3 + L1 | 6.3 | −0.4 | 10.1 | 5.7 | 2.4 |
| 2a | 10.4 | 3.0 | 1.8 | 3.7 | 2.6 |
| 2b | 9.7 | 6.5 | 15.7 | 3.8 | 9.4 |
| 3 + L2 | 8.6 | 1.8 | 10.5 | 5.4 | 0.7 |
| 3 | 4.4 | 0.4 | −0.3 | −0.6 | 1.1 |
Next, we performed a series of control experiments to better rationalize this behavior and understand the effect of ligand hydrolysis on the G4-stablilization of 1b and 2b. We treated the ODNs with complex 3 used as control (Fig. 3A) and in combination with ligands L1 and L2 as they are the constituents of compounds 1b and 2b, respectively. Remarkably, incubation of the ODNs with these combinations did not result in such a strong effect as observed for 1b and 2b, demonstrating that all the structural components of the complexes were needed to exert a distinct G4 stabilization (Table 2 and Fig. 3B).
 | ||
| Fig. 3 (A) Structures of 1b and of its constituents L1 and 3. (B) FRET melting profiles of the c-Kit1 G4 (0.2 μM) upon interaction with the indicated compounds (4 μM). (C) FRET melting profiles at different incubation times. | ||
Additionally, when we used a shorter incubation time (1 h), we observed an overall increase of the ODNs melting temperature (Fig. 3C and Table S9†). In this case, besides 1b and 2b, also the chlorido complexes 1a and 2a exhibited G4 stabilization, indicating that within this time window significant amounts of these compounds were still in their original form (i.e. not hydrolyzed). Furthermore, with the shorter incubation time of 1 h, the selectivity toward G4 over duplex DNA of compounds 1b and 2b increased significantly. Even though the exact fraction of stable compounds interacting with the selected G4s is not known, the ΔT1/2 values obtained in the described different conditions are strongly indicative of the importance of the complexes stability for the G4 stabilization.
Interaction with 9-ethylguanine and G4s by mass spectrometry
Incubation experiments (2 h in H2O, 20% MeOH) of 1a and 1b with 9-ethylguanine (9-EtG) followed by mass analyses demonstrated that both complexes are able to coordinate guanines at the N7 position (Fig. S15 and S16†). Further time-dependent experiments indicated a relatively fast exchange (within 30 min) between the pyridine of complex 1b and the nucleobase (Fig. S17†).We then used ESI-TOF mass spectrometry to evaluate the binding of 1a and 1b to two selected G-rich sequences, hTelo and c-Kit1 respectively. The two G4s contain stacks of three G-quartets and retain monovalent potassium ions in their folded structure (see 3D models of hTelo and c-Kit1 in Fig. 4A and B).40,41 Since non-volatile potassium salts are not compatible with their use in buffers for ESI mass spectrometry, we used ammonium acetate instead to ensure G4 formation, like previously reported.42
 | ||
| Fig. 4 (A, B) 3D structures of hTelo (A) and (B) c-Kit1 G4s, generated with Chimera using PDB 2HY9 and 2O3M, respectively. Guanines belonging to G-tetrads are highlighted in orange and unpaired guanines are shown in green. (C, D) Negative ESI-TOF-MS enlargements of the distribution of ammonium adducts for the 5-charge state of free 10 μM hTelo (C) and (D) c-Kit1 sequences in the absence and presence of equimolar amount of the compounds 1a and 1b. The three peaks in each spectrum correspond to the free oligodeoxynucleotide (orange circles), one NH4+ ion adduct, and two NH4+ ions adduct (one and two green diamonds, respectively). | ||
Mass spectra of the free G4s exhibited ions mainly in the charge states of −5 and −4. In each charge state, there were three peaks corresponding to the free oligodeoxynucleotide, one NH4+ ion adduct, and two NH4+ ions adduct (Fig. 4C and D).
After 1 h incubation of our compounds with the two G4s, we observed low-intensity peaks corresponding to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts (and their fragments) with 1a and 1b upon loss of chloride or pyridine, respectively (Fig. S18–S22†).
:1 adducts (and their fragments) with 1a and 1b upon loss of chloride or pyridine, respectively (Fig. S18–S22†).
Furthermore, we focused our attention to the G4 ammonium adduct distribution after incubation with 1a and 1b. At first, we compared the relative intensity of the ESI-MS peaks distribution of NH4+ adducts for the 5-charge of hTelo−5 and c-Kit1−5 with the one of their complexes (or their fragments) with 1a and 1b (Fig. S23†). For both G-quadruplexes, an intensity decrease of the peak corresponding to the ammonium free ODN occurred in the spectra of the adducts with 1a and 1b, indicating a lower tendency by the G4 to release NH4+ ions, hence a general stabilizing effect.43
Nevertheless, considering that the peaks corresponding to 1a/1b-G4 adducts have low intensity and, especially in the case of c-Kit1, different adduct fragments are present (see Fig. S20†), we also compared the ammonium distribution of free hTelo5− and c-Kit15−, before and after the interaction with our complexes. In this case, addition of 1a/1b to hTelo G4 (Fig. 4C) led only to a minor shift in ammonium adduct distribution with the peak corresponding to hTelo5− with one and two associated ammonium becoming slightly less abundant compared to the free G4. This could indicate a minor destabilization of the G4 structure with a small release of monovalent ions after the interaction with 1a and 1b.43 On the other hand, the interaction with c-Kit1−5 confirmed the stabilization effect (Fig. 4D): upon addition of 1a/b, the relative abundance of c-Kit15− with one or two coordinated ammonium ions clearly increased, indicating improved stability of the G4 structure. In other words, c-Kit15−, once bound to the metal compounds, held NH4+ ions between the G-tetrads more tightly than the free G4 when introduced into the gas phase.43 The observed stabilizing effect was stronger for compound 1b than for 1a.
The hTelo sequence contains no free guanines besides those participating in the G4 fold (Fig. 4A),44 whereas c-Kit1 features three non-stacked guanines (Fig. 4B), making them potentially accessible for easier ruthenation. In both MS experiments and FRET studies, c-Kit1 displays enhanced stabilization of the G4 fold by the pyridine derivative 1b as compared to hTelo, thus suggesting that the presence of unpaired guanines in the c-Kit1 DNA motif enables a dual binding mode, allowing for N7 ruthenation besides the stacking action of the naphthyl moiety.
Incubation experiments with 9-EtG and with the selected G4 models clearly indicated that both compounds 1a and 1b coordinate the N7 of guanines and simultaneously bind the G4s tetrad via stacking of the naphthyl group. The increased stability of 1b toward ligand exchange is key for the success of this dual mode of binding in our experimental conditions.
Cytotoxicity
We then tested the cytotoxic activity of 1a–b and 2a–b and their respective ligands against the ovarian cancer cell line A2780 (Fig. 5). The trends observed in cell-free experiments presented above have been surprisingly mirrored in cell culture. Complexes 1b and 2b were the most active compounds displaying IC50 values in the low micromolar range (Table 3), which can be explained by the increased stability of the pyridine derivatives while the chlorido complexes decompose faster under these conditions.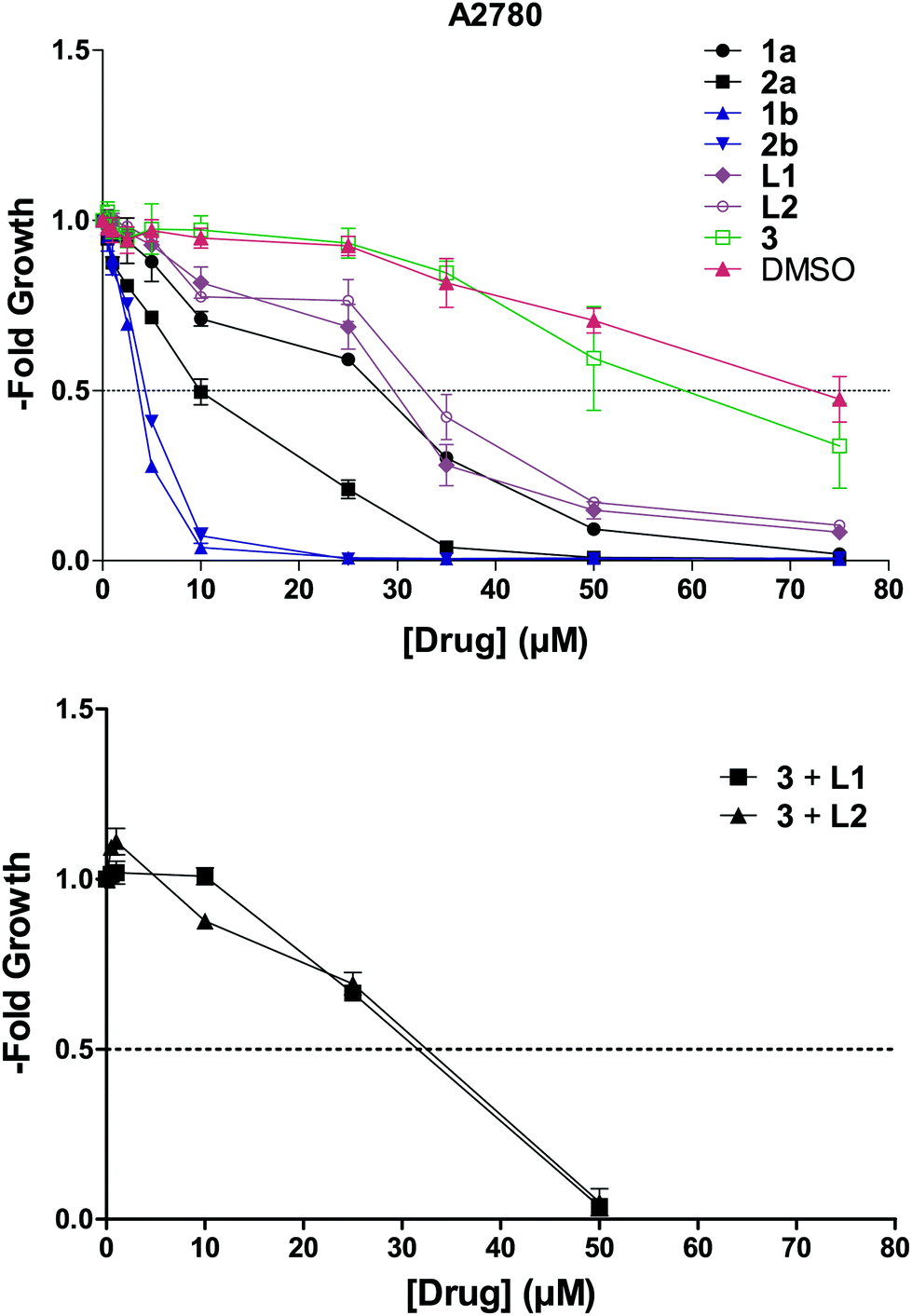 | ||
| Fig. 5 Concentration dependent effect of the synthesized compounds and 3 on A2780 cell viability. | ||
| L1 | L2 | 1a | 1b | 3 + L1 | 2a | 2b | 3 + L2 | 3 |
|---|---|---|---|---|---|---|---|---|
| 29.6 ± 1.1 | 33.1 ± 1.6 | 28.2 ± 0.3 | 3.7 ± 0.1 | 31.6 ± 0.7 | 10.4 ± 1.2 | 4.4 ± 0.1 | 32.4 ± 1.0 | 56.7 ± 8.1 |
As a matter of fact, neither the ligands or the chlorido complexes, nor the equimolar combination of the free ligands with the control complex 3 reached the activity of compounds 1b and 2b.
Considering that the cell viability is measured 72 h after drug exposure, it is worth pointing out that the main action of this class of compounds is most likely exerted during the first hours of exposure. This is consistent with the high activity of compounds 1b and 2b which proved to be the most stable among the synthesized molecules.
Conclusions
In summary, four novel Ru(II) arene complexes (two chlorido and two pyridine derivatives, respectively) with two 1,3-dioxoindan-2-carboxamides bearing naphthyl-groups separated by a spacer of different lengths have been synthesized and characterized, including the X-ray structure for three of the metal compounds. We proved through FRET and mass spectrometry that designing ruthenium arene compounds with the ability to stack on top of guanine tetrads and simultaneously bind free guanines is a viable strategy to obtain fairly good G4 binders. Besides, we showed that the G4 binding activity of these complexes correlates well with low-micromolar cytotoxicity towards ovarian cancer cells, unambiguously demonstrating that the whole complex is necessary for such events to occur, while neither the free ligands nor the combination of the ligands with the Ru(II)–arene pyridine fragment 3 were able to stabilize G4 folds or kill cancer cells to the same extent.This study is a first step toward the design of new metalloarene compounds with enhanced G4 binding activity. Currently, further studies carried out in our laboratory seek to replace the O,O-coordination motif with different donor atoms to evaluate the effect of an improved overall stability of the Ru–arene scaffold on G4 binding ability and correlated anticancer activity.
Materials and methods
General
All solvents were purchased from commercial sources and distilled prior use. Chloroform was dried over molecular sieves (3 Å) before use. All chemicals were purchased and used without further purification. Sodium perchlorate monohydrate (99.99%, Aldrich) was dried at 160 °C in vacuo prior to use. 2-Oxido-phenyliodonio-1,4-naphthoquinone was synthesized as described elsewhere.31 MilliQ water was used to prepare buffers and pH was measured using a Mettler Toledo pH meter. All oligonucleotides were purchased from IDT (Integrated DNA Technologies) in HPLC purity grade. The FRET probes used were FAM (6-carboxyfluorescein) and TAMRA (6-carboxy-tetramethylrhodamine).Melting points were determined with a Büchi Melting Point M-560. The solubility was determined by dissolving the compound in DMSO and subsequent dilution to a final concentration of 1% DMSO/MEM. The highest concentrated dilution, where no precipitation of the compound occurred, was the determined solubility. The NMR spectra were recorded at 25 °C using a Bruker FT-NMR spectrometer Avance IIITM 500 MHz. NMR spectra were measured in deuterated dimethyl sulfoxide (DMSO-d6) or chloroform (CDCl3) (see Fig. S24–S35† for the full spectra). CHN-elemental analyses were performed with a Eurovector EA3000 Elemental Analyzer in the microanalytical laboratory of the University of Vienna. Single crystals of 1a, 2a, and 1b suitable for X-ray diffraction analysis were grown by precipitation from DCM/Et2O at 4 °C. The X-ray intensity data were measured on Bruker D8 Venture and Bruker X8 Apex2 diffractometer equipped with multilayer monochromators, Mo K/α INCOATEC micro focus sealed tubes and Kryoflex cooling systems. The structures were solved by direct and patterson methods and refined by full-matrix least-squares techniques. Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were inserted at calculated positions and refined with riding model. The following software was used: Bruker SAINT v8.37A & V7.68A (Bruker AXS) using a narrow-frame algorithm for frame integration, SADABS (by George M. Sheldrick) for absorption correction, OLEX2 for structure solution,45 refinement, molecular diagrams and graphical user-interface, Shelxle for refinement and graphical user-interface,46 SHELXS-2015 for structure solution, SHELXL-2015 for refinement,47 Platon for symmetry check.48 Experimental data and CCDC-codes (available online at http://www.ccdc.cam.ac.uk/conts/retrieving.html) can be found in Table S1.† Crystal data, data collection parameters, and structure refinement details are given in Tables S2–S7.† Crystal structures visualized in Fig. S3–S5.†
Synthesis
m/z 328.14, mth: 328.10; elemental analysis calcd for C21H15NO3: C 76.58, H 4.59, N 4.25%; found: C 75.53, H 4.65, N 4.41%.
:1) for an hour at r.t., bis[dichlorido(cym)ruthenium(II)] (58.7 mg, 96 μmol, 0.9 eq.) was added and the mixture stirred for 22 h. The solvent was removed and residue dissolved in DCM, the resulting suspension was filtered and concentrated. The product was precipitated from DCM/n-hexane and dried in vacuo (72%, 83 mg, brown powder): m.p. >206 °C (decomp.); solubility 0.066 mg mL−1 ≡ 0.11 mM (MEM, 1%DMSO); 1H-NMR (500.10 MHz, CDCl3) δ: 1.19–1.27 (m, 6H), 2.18 (s, 3H), 2.67–2.74 (m, 1H), 4.94–5.24 (m, 2H), 5.24–5.30 (m, 2H), 5.42–5.53 (m, 2H), 7.40–7.62 (m, 8H), 7.97–7.83 (m, 1H), 7.88–7.91 (m, 1H), 8.05–8.08 (m, 1H), 9.00 (dd, 3J(H,H) = 6 Hz, 3J(H,H) = 6 Hz, 1H); 13C-NMR (125.75 MHz, CDCl3) δ 17.89 (cym-C10), 22.12 and 22.33 (cym-C8, -C9), 30.86 (cym-C7), 41.28 (C2′), 78.89 and 79.37 (cym-C3, -C5), 82.52 and 82.59 (cym-C2, -C6), 96.60 (cym-C4), 98.18 (C2), 99.56 (cym-C1), 120.69 and 120.92 (C4, C7), 123.35 (C7′), 125.07 (C4′), 125.48 (C5′), 126.02 (C9′), 126.65 (C8′), 128.19 (C6′), 128.88 (C10′), 131.31 (C6′a), 132.13 and 132.32 (C5, C6), 133.86 (C10′a), 136.41 (C3′), 137.78 (C3a, C7a), 165.92 (C1′), 190.94 (C3), 192.51 (C1) ppm. m/z 564.33, mth: 564.11; elemental analysis calcd for C31H28ClNO3Ru·0.25H2O: C 61.69, H 4.76, N 2.32%; found: C 61.77, H 4.75, N 2.39%.
:1), for an hour at r.t., bis[dichlorido(cym)ruthenium(II)] (60.2 mg, 98 μmol, 0.9 eq.) was added and the mixture stirred for 22 h. The solvent was removed and residue dissolved in DCM, the resulting suspension was filtered and concentrated. The product was precipitated from DCM/n-hexane and dried in vacuo. (75%, 90 mg, brown powder): m.p. >129 °C (decomp.); solubility 0.080 mg mL−1 ≡ 0.13 mM (MEM, 1%DMSO); 1H-NMR (500.10 MHz, CDCl3) δ: 1.28–1.32 (m, 6H), 2.18 (s, 3H), 2.78–2.85 (m, 1H), 3.20–3.51 (m, 2H), 3.71–3.97 (m, 2H), 5.06–5.09 (m, 1H), 5.15–5.23 (m, 2H), 5.42–5.44 (m, 1H), 7.36–7.60 (m, 8H), 7.76–7.79 (m, 1H), 7.88–7.91 (m, 1H), 8.13–8.15 (m, 1H), 8.70 (dd, 3J(H,H) = 6 Hz, 3J(H,H) = 6 Hz, 1H); 13C-NMR (125.75 MHz, CDCl3) δ 17.98 (cym-C10), 22.40 (cym-C8, -C9), 30.99 (cym-C7), 33.83 (C3′), 39.97 (C2′), 78.79 and 79.38 (cym-C3, -C5), 82.06 and 82.61 (cym-C2, -C6), 96.46 (cym-C4), 98.11 (C2), 99.54 (cym-C1), 120.65 and 120.83 (C4, C7), 123.91 (C8′), 125.77 (C10′), 125.90 (C5′), 126.17 (C9′), 127.25 (C7′), 127.35 (C6′), 129.05 (C11′), 132.05 (C5, C6), 132.07 (C7′a), 132.21 (C5, C6), 134.06 (C11′a), 135.37 (C4′), 136.39 and, 137.78 (C3a, C7a), 165.97 (C1′), 190.64 (C3), 192.47 (C1) ppm. m/z 578.38, mth: 578.13; elemental analysis calcd for C32H30ClNO3Ru·0.25H2O: C 62.23, H 4.98, N 2.27%; found: C 61.99, H 5.04, N 2.38%.
UV-Vis
UV-Vis absorption spectra were recorded on a PerkinElmer LAMBDA 35 double beam spectrophotometer, equipped with a Peltier temperature controller. Measurements were carried out at 25 °C using 1 cm path-length quartz cuvettes. Compounds were dissolved in acetonitrile and diluted in the respective working buffer to the desired concentration, with the final content of acetonitrile kept below 1%. Lambert–Beer extinction coefficients were determined in 2 mM MES buffer at pH = 6 by adding compound stock in acetonitrile in small increments.FRET
FRET experiments were performed in 96-well plates and run on an Applied Biosystems® 7500 Real-Time PCR cycler equipped with a FAM filter (λexc = 492 nm; λem = 516 nm). The lyophilized strands were first diluted in MilliQ water to obtain 100 μM stock solutions. These were diluted to a concentration of 400 nM in 60 mM potassium cacodylate buffer (pH 7.4) and then annealed to form G4 structures by heating to 95 °C for 5 min, followed by slowly cooling to room temperature overnight. Experiments were carried out in a 96-well plate with a total volume of 30 μL. Final concentration of the oligonucleotides was 200 nM. All compounds were previously dissolved in DMSO or ACN to give 1 mM stock solutions. These were further diluted using 60 mM potassium cacodylate and added to obtain the final concentration (with a total percentage of DMSO or ACN ≤ 0.8%). Ramp temperature program was set with a stepwise increase of 1 °C every 30 s starting from 25 °C to reach 95 °C, and measurements were acquired after each step. To compare different sets of data, FAM emission data were normalized (0 to 1).49T1/2 is defined as the temperature at which the normalized emission is 0.5. Measurements were made in duplicate. Analysis and plotting of the data were carried out using Origin 9.5 (OriginLab Corp.)Mass
000 FWHM; mass accuracy <5 ppm.
500 V; nebulizer 1.0 bar; dry gas flow 4.0 L min−1 at 120 °C; end plate offset voltage 500 V.43 Data were analyzed with the instrument software Bruker Daltonics DataAnalysis. 100 μM stock solutions of oligonucleotides hTelo and c-Kit1 were prepared in 100 mM ammonium acetate buffer and heated to 90 °C for 5 Min before being allowed to slowly cool to room temperature. Compounds 1a–b and 2a–b were prepared as 1 mM stock solutions in acetonitrile and diluted with MilliQ water to a concentration of 20 μM. An equimolar mixture of drug and G4 (10 μM, respectively) was injected containing 50 mM ammonium acetate. Experimental conditions were adjusted to allow ammonium ions coordinated between G-quartets to be retained,50 while ammonium ions associated to binding sites not specific for the G4 fold are mostly stripped during ionization. Peak patterns were interpreted as previously reported.43 Charge states of the oligonucleotide with the highest relative abundance (−5) were analyzed and interpreted.
Author contributions
A. T. conceived and directed the study in all its parts. L. A. H., S. M. and W. K. performed the synthesis and the structural characterization of the compounds. L. A. H., S. A. and A. T. carried out all the measurements in solution. A. R. resolved the crystal structures of the compounds. C. K., D. B. and W. B. performed the in-cell studies. A. T., L. S., W. B. and B. K. K. analyzed and interpreted the overall results. All the authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. T. has received funding from the Mahlke-Obermann Stiftung and the European Union's Seventh Framework Programme (grant agreement no. 609431) and from the European Union's Horizon 2020 Research and Innovation Programme under Marie Sklodowska-Curie Actions grant no. 746976.References
- S. Neidle, Nat. Rev. Chem., 2017, 1, 0041 CrossRef CAS.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef.
- R. Vilar, in Metallo-Drugs: Development and Action of Anticancer Agents, ed. A. Sigel, H. Sigel, E. Freisinger and R. K. O. Sigel, De Gruyter, Berlin, Boston, 2018, pp. 325–350 Search PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z.-W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS.
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- E. Alessio, Eur. J. Inorg. Chem., 2017, 2017, 1549–1560 CrossRef CAS.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- E. Wachter, D. Moyá, S. Parkin and E. C. Glazer, Chem. – Eur. J., 2016, 22, 550–559 CrossRef CAS.
- L. Xu, X. Chen, J. Wu, J. Wang, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 4008–4020 CrossRef CAS.
- E. Wachter, B. S. Howerton, E. C. Hall, S. Parkin and E. C. Glazer, Chem. Commun., 2014, 50, 311–313 RSC.
- X. Chen, J.-H. Wu, Y.-W. Lai, R. Zhao, H. Chao and L.-N. Ji, Dalton Trans., 2013, 42, 4386–4397 RSC.
- Q. Yu, Y. Liu, J. Zhang, F. Yang, D. Sun, D. Liu, Y. Zhou and J. Liu, Metallomics, 2013, 5, 222–231 RSC.
- K. McQuaid, H. Abell, S. P. Gurung, D. Allan, G. Winter, T. Sorensen, D. J. Cardin, J. A. Brazier, C. J. Cardin and J. P. Hall, Angew. Chem., Int. Ed., 2019, 58, 9881–9885 Search PubMed.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet, A. Decottignies, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2018, 24, 19216–19227 CrossRef CAS.
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. MacNeil and J. A. Thomas, Chem. Sci., 2019, 10, 3502–3513 RSC.
- D. Bouzada, I. Salvadó, G. Barka, G. Rama, J. Martínez-Costas, R. Lorca, Á. Somoza, M. Melle-Franco, M. E. Vázquez and M. Vázquez López, Chem. Commun., 2018, 54, 658–661 RSC.
- J. Rubio-Magnieto, S. Kajouj, F. Di Meo, M. Fossépré, P. Trouillas, P. Norman, M. Linares, C. Moucheron and M. Surin, Chem. – Eur. J., 2018, 24, 15577–15588 CrossRef CAS.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- B. Therrien, Coord. Chem. Rev., 2009, 253, 493–519 CrossRef CAS.
- Q. Wu, K. Zheng, S. Liao, Y. Ding, Y. Li and W. Mei, Organometallics, 2016, 35, 317–326 CrossRef CAS.
- C. Fan, Q. Wu, T. Chen, Y. Zhang, W. Zheng, Q. Wang and W. Mei, MedChemComm, 2014, 5, 597–602 RSC.
- D. Sun, R. Zhang, F. Yuan, D. Liu, Y. Zhou and J. Liu, Dalton Trans., 2012, 41, 1734–1741 RSC.
- A. Terenzi, D. Lötsch, S. van Schoonhoven, A. Roller, C. R. Kowol, W. Berger, B. K. Keppler and G. Barone, Dalton Trans., 2016, 3, 7758–7767 RSC.
- O. Domarco, D. Lötsch, J. Schreiber, C. Dinhof, S. Van Schoonhoven, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Dalton Trans., 2017, 46, 329–332 RSC.
- O. Domarco, C. Kieler, C. Pirker, C. Dinhof, B. Englinger, J. M. Reisecker, G. Timelthaler, M. D. García, C. Peinador, B. K. Keppler, W. Berger and A. Terenzi, Angew. Chem., Int. Ed., 2019, 131, 8091–8096 CrossRef.
- W. Streciwilk, A. Terenzi, F. Lo Nardo, P. Prochnow, J. E. Bandow, B. K. Keppler and I. Ott, Eur. J. Inorg. Chem., 2018, 2018, 3104–3112 CrossRef CAS.
- W. Streciwilk, A. Terenzi, X. Cheng, L. Hager, Y. Dabiri, P. Prochnow, J. E. Bandow, S. Wölfl, B. K. Keppler and I. Ott, Eur. J. Med. Chem., 2018, 156, 148–161 CrossRef CAS.
- S. Mokesch, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller and B. K. Keppler, Organometallics, 2015, 34, 848–857 CrossRef CAS.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam and P. J. Sadler, Chem. Commun., 2009, 6622–6624 RSC.
- G. Ragazzon, I. Bratsos, E. Alessio, L. Salassa, A. Habtemariam, R. J. McQuitty, G. J. Clarkson and P. J. Sadler, Inorg. Chim. Acta, 2012, 393, 230–238 CrossRef CAS.
- E. Ruggiero, C. Garino, J. C. Mareque-Rivas, A. Habtemariam and L. Salassa, Chem. – Eur. J., 2016, 22, 2801–2811 CrossRef CAS.
- S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466–3479 CrossRef CAS.
- A. Habtemariam, C. Garino, E. Ruggiero, S. Alonso-de Castro, J. Mareque-Rivas and L. Salassa, Molecules, 2015, 20, 7276–7291 CrossRef CAS.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef CAS.
- H. Fukada and K. Takahashi, Proteins: Struct., Funct., Genet., 1998, 33, 159–166 CrossRef CAS.
- IDT, 5′ 6-FAM (Fluorescein), https://eu.idtdna.com/site/Catalog/Modifications/Product/1108.
- D. Wei, G. N. Parkinson, A. P. Reszka and S. Neidle, Nucleic Acids Res., 2012, 40, 4691–4700 CrossRef CAS.
- G. N. Parkinson, M. P. H. Lee and S. Neidle, Nature, 2002, 417, 876–880 CrossRef CAS.
- F. Balthasart, J. Plavec and V. Gabelica, J. Am. Soc. Mass Spectrom., 2013, 24, 1–8 CrossRef CAS.
- L. P. Bai, M. Hagihara, K. Nakatani and Z. H. Jiang, Sci. Rep., 2014, 4, 15–17 Search PubMed.
- J. Rodríguez, J. Mosquera, J. R. Couceiro, M. E. Vázquez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 15615–15618 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- D. Renčiuk, J.-L. Mergny, A. Guédin, J. Zhou, L. Beaurepaire and A. Bourdoncle, Methods, 2012, 57, 122–128 CrossRef.
- L.-P. Bai, J. Liu, L. Han, H.-M. Ho, R. Wang and Z.-H. Jiang, Anal. Bioanal. Chem., 2014, 406, 5455–5463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1870492–1870494. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02078k |
| This journal is © The Royal Society of Chemistry 2019 |