
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Click chemistry as a route to the synthesis of structurally new and magnetically interesting coordination clusters: a {NiII8} complex with a trapezoidal prismatic topology†
Parisa
Abbasi
a,
Angeliki A.
Athanasopoulou
a,
Eleni C.
Mazarakioti
a,
Kevin J.
Gagnon
b,
Simon J.
Teat
 b,
Albert
Escuer
c,
Melanie
Pilkington
*a and
Theocharis C.
Stamatatos‡
*a
b,
Albert
Escuer
c,
Melanie
Pilkington
*a and
Theocharis C.
Stamatatos‡
*a
aDepartment of Chemistry, 1812 Sir Isaac Brock Way, Brock University, L2S 3A1 St. Catharines, Ontario, Canada. E-mail: mpilkington@brocku.ca; Tel: +19056885550 ext. 3403
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
cDepartment de Quimica Inorgànica i Orgànica, Secció Inorgànica and Institut de Nanociència i Nanotecnologia (IN2UB), Universitat de Barcelona, Martí Franqués 1-11, 08028 Barcelona, Spain
First published on 27th June 2019
Abstract
The synthesis of a new {Ni8} cluster bearing tetrazolate- and azido-bridging ligands, and supported by chelating α-methyl-2-pyridine-methanol (mpmH) groups, is described herein. The reported compound has a unique trapezoidal prismatic topology, resulting from an unexpected in situ click reaction between the MeCN reaction solvent and the N3− ions under mild, room-temperature conditions. Such a click chemistry approach to the preparation of 0-D compounds is relatively unexplored and represents a fruitful strategy for the synthesis of new coordination clusters and molecule-based magnetic materials.
The search for new structural types of molecular magnetic materials is currently driven by a number of considerations, including the selection of the metal ion, its oxidation state(s), the coordination environment, the electronic, structural and geometrical properties, and the cooperative ability of the employed organic and/or inorganic ligands to facilitate the formation and crystallization of the targeted species.1 Undoubtedly, one of the most challenging synthetic aspects in the quest for structurally novel and magnetically interesting polynuclear 3d-metal compounds, or coordination clusters, is the in situ generation of new and unpredictable ligand types which are capable of bridging many metal ions and propagating appreciable magnetic interactions between the spin carriers.2
Although click chemistry has been used in almost all key areas of synthetic organic chemistry,3 there are only few examples of polynuclear metal complexes resulting from ligands that were derived in situ by click reactions.4 In fact, a convenient synthetic route based on the metal-ion assisted [2 + 3] cycloaddition of nitriles with azides was explored by Sharpless and coworkers,5 and developed by Xiong6 and others7 to prepare coordination polymers via in situ generated 5-substituted 1H-tetrazolate bridging ligands. End-on (EO) bridging azides are an important class of ligands in polynuclear 3d-metal cluster chemistry and molecular magnetism due to their versatile binding modes and known capability to promote ferromagnetic exchange interactions between the metal ions they bridge.8
In addition, tetrazoles have been found to adopt at least nine distinct types of coordination modes with metal ions in the construction of metal–organic frameworks.6 Therefore, tetrazoles have attracted increasing attention in molecular chemistry and crystal engineering due to the excellent coordination ability of the four nitrogen atoms of the functional group, acting as either a multidentate or a bridging building block in various supramolecular assemblies.6,9 Interestingly, all previously reported coordination compounds (clusters and polymers) resulting from click reactions and bearing in situ generated tetrazolate ligands have been prepared from hydro/solvothermal methods.6,7,9,10 The only exception, to the best of our knowledge, is a {CoII10}/azide/tetrazolate cluster reported by Zhang and Sato,11 which was prepared and crystallized by a conventional solution-based synthetic methodology.
To facilitate the formation and crystallization of 0-D cluster compounds, the employment of chelating and/or bridging organic ligand(s) appears as a necessary synthetic parameter which prevents the extensive aggregation/polymerization of metal ions that would otherwise lead to multidimensional coordination polymers. Thus, we have recently initiated a program targeting the synthesis of new and potentially chiral pyridyl-alkoxide based chelating/bridging ligands and their use in the formation of high-nuclearity 3d-metal cluster compounds with interesting magnetic properties, such as high-spin molecules and single-molecule magnets (SMMs). To this end, the employment of α-methyl-2-pyridine-methanol (mpmH; Scheme 1, left) in Mn carboxylate chemistry has afforded a {Mn31} SMM with an unprecedented nanosized structure and a large energy barrier of ∼60 K for reversal of the magnetization.12 As a part of our ongoing studies on understanding the chemistry–structure–magnetism relationships within a library of coordination 3d-metal clusters based on mpmH, we have herein been able to synthesize – under mild conditions – and characterize a structurally unique {Ni8} cluster (1), which possesses a trapezoidal prismatic topology resulting from the coordination of both end-on bridging azides (N3−) and 5-methyltetrazolates (mtz−). The mtz− ligands are unexpectedly present in the structure of 1 due to the in situ, metal-assisted click reaction between the N3− ions and the MeCN reaction solvent (Scheme 1, right). The dual presence of both bridging azido and mtz− groups in 1 resulted in the presence of both ferro- and antiferromagnetic exchange interactions between the NiII centers, respectively.
 | ||
| Scheme 1 Structural formula and abbreviation of the chelate ligand α-methyl-2-pyridine-methanol (mpmH) used in this work (left), and the click reaction that leads to the formation of the crystallographically established μ4-bridging mtz− groups in 1 (right). | ||
Racemic mpmH (rac-mpmH) was prepared via the NaBH4 reduction of 2-acetylpyridine according to literature reports (see ESI†).13 The reaction of Ni(ClO4)2·6H2O, rac-mpmH and NaN3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio in MeCN gave a green solution which, upon evaporation at room temperature for two months, afforded turquoise plate-like crystals of [Ni8(N3)8(mtz)4(rac-mpm)4(rac-mpmH)4]·2.2H2O (1·2.2H2O) in 30% yield.† The best estimate of the amount of disordered lattice H2O based on the crystallographic analysis (2.2H2O) was in good agreement with elemental analysis on an air-dried sample (2H2O), and the molecular weight used for analysis of the magnetic data was based on the dihydrate. Interestingly, similar reaction schemes, albeit in the presence of commercially available 5-methyl-1H-tetrazole, failed to give any crystalline products. Complex 1 (Fig. 1, top) crystallizes in the polar, orthorhombic space group Fddd with one quarter of the molecule in the asymmetric unit. The cluster has virtual D2h symmetry and can be described as a trapezoidal prism of eight NiII ions arranged into four, symmetry-related, dinuclear {Ni2} units (Fig. 1, bottom). An alternative way to describe the {Ni8} metal topology is that of a saddle-like conformation.
:1:2 ratio in MeCN gave a green solution which, upon evaporation at room temperature for two months, afforded turquoise plate-like crystals of [Ni8(N3)8(mtz)4(rac-mpm)4(rac-mpmH)4]·2.2H2O (1·2.2H2O) in 30% yield.† The best estimate of the amount of disordered lattice H2O based on the crystallographic analysis (2.2H2O) was in good agreement with elemental analysis on an air-dried sample (2H2O), and the molecular weight used for analysis of the magnetic data was based on the dihydrate. Interestingly, similar reaction schemes, albeit in the presence of commercially available 5-methyl-1H-tetrazole, failed to give any crystalline products. Complex 1 (Fig. 1, top) crystallizes in the polar, orthorhombic space group Fddd with one quarter of the molecule in the asymmetric unit. The cluster has virtual D2h symmetry and can be described as a trapezoidal prism of eight NiII ions arranged into four, symmetry-related, dinuclear {Ni2} units (Fig. 1, bottom). An alternative way to describe the {Ni8} metal topology is that of a saddle-like conformation.
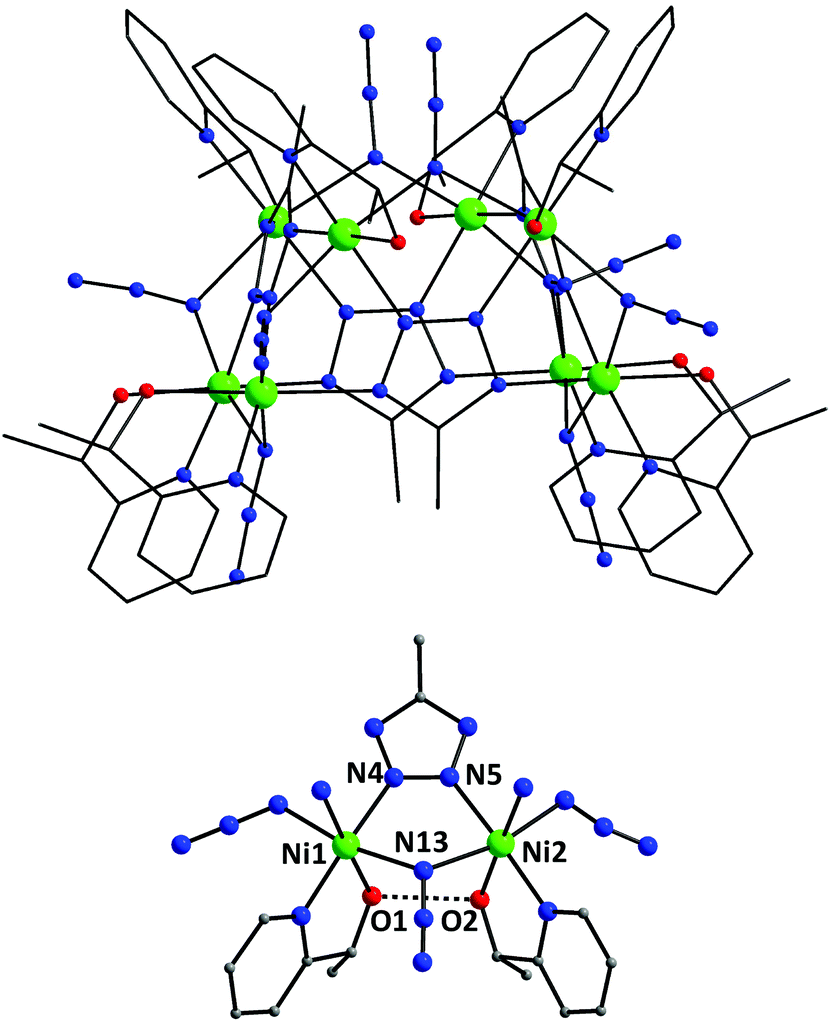 | ||
| Fig. 1 The molecular structure of 1 (top) and its partially labelled asymmetric unit (bottom). The dashed line represents one of the four symmetry-related H-bonding interactions between adjacent mpm−/mpm(H) pairs of ligands. Colour scheme: NiII green, N blue, O red, C gray, H light gray. H atoms are omitted for clarity. | ||
Each NiII ion in 1 is bridged to its neighboring metal ions through a μ-1,1 end-on N3− and a diazine-part of the mtz− ligands; the latter groups adopt an overall η1:η1:η1:η1:μ4 mode, each of them linking four NiII atoms in a nearly planar conformation (mean deviation of Ni atoms from the Ni4 plane is 0.016 Å). The μ4-bridging mode of the mtz− ligands in 1 accommodates the maximum number of metal ions that this group can potentially bind to, thus representing the second example of a 0-D molecular compound (the first in NiII chemistry) bearing μ4-mtz− ligands.11 The intramolecular Ni⋯Ni separations and Ni–(μ-N3)–Ni angles span the range 3.417(2)–6.255(2) Å and 108.0(4)–114.4(3)°, respectively, whereas the Ni–N–N–Ni torsion angles are within the range 1.0(9)–15.2(8)°. Peripheral ligation about the [Ni8(μ-N3)8(μ4-mtz)4]4+ core (Fig. 2, top) is provided by a total of eight N,O-bidentate chelating mpm−/mpmH groups, each of them capping a different NiII ion. The charge neutrality of the {Ni8} cluster requires four of the bidentate chelating mpm− ligands to be protonated, i.e. in the mpmH form. Indeed, this agrees with the short O1⋯O2 contact (2.51(1) Å) between two adjacent mpm−/mpm(H) ligands, thus implying a relatively strong H-bonding interaction (dashed lines in Fig. 1, bottom) and consequently the presence of a H-atom between the pair of these alkoxido groups (an O1⋯H⋯O2 homo-synthon). All NiII ions in 1 are six-coordinate with near-octahedral geometries. The crystal packing of 1·2.2H2O revealed that the lattice H2O molecules occupy the voids between adjacent {Ni8} clusters (Fig. S1†). The shortest Ni⋯Ni distance between neighboring {Ni8} clusters in the crystal is 8.422(1) Å.
 | ||
| Fig. 2 A view of the [Ni8(μ-N3)8(μ4-mtz)4]4+ core of 1 (top) along with the trapezoidal prismatic topology of the eight NiII atoms (bottom). Colour scheme as in Fig. 1. Symmetry operations: a: 0.25 − x, y, 1.25 − z; b: 0.25 − x, 1.25 − y, z; c: x, 1.25 − y, 1.25 − z. | ||
Although there are many {Ni8} complexes reported in the literature with an open-shell structure, such as cubes14 and rings,15 the trapezoidal prismatic topology of complex 1 appears to be a new addition in NiII cluster chemistry. Given the structural novelty of 1, there are some geometrical features that deserve further discussion. A trapezoidal prism is a three-dimensional figure that consists of two trapezoids on opposite faces connected by four rectangles. A trapezoidal prism has 6 faces, 8 vertices and 12 edges. The two trapezoids within complex 1 (Fig. 2, bottom) are composed of Ni1–Ni2–Ni1b–Ni2c and Ni1a–Ni2a–Ni2b–Ni1c. The pair of parallel edges (i.e., the base) within each trapezoid consist of Ni1⋯Ni2 (3.591(2) Å)/Ni1b⋯Ni2c (6.255(2) Å) and Ni1a⋯Ni2a (3.591(2) Å)/Ni2b⋯Ni1c (6.255(2) Å), respectively. The non-parallel edges (i.e., the legs) of each trapezoid comprise the pairs Ni1⋯Ni1b (3.417(2) Å)/Ni2⋯Ni2c (3.419(2) Å) and Ni1a⋯Ni1c (3.417(2) Å)/Ni2a⋯Ni2b (3.419(2) Å), respectively. The four remaining faces serve to connect the opposite trapezoids, resulting in an overall trapezoidal prismatic topology for 1.
Based on detailed DFT calculations, Noodleman, Sharpless and co-workers have proposed several different mechanisms of tetrazole formation, including concerted cycloaddition and stepwise addition of neutral or anionic azide species to various nitriles at elevated temperatures.16 Given the reaction conditions employed for the synthesis of 1, it is very likely that an anionic, metal-assisted cycloaddition would be the predominant mechanism of the mtz− formation. This involves either a direct [2 + 3] cycloaddition or a two-step sequence wherein the azide acts as a nucleophile to attack the relatively electron-rich MeCN, followed by ring closure.16,17 A noticeable example of a {Mn16} cluster was recently reported by one of us, which was resulted from the cycloaddition of the azide anion to the nitrile functionality of the pyridylcyanoximate ligand (py)C(CN)NO−, yielding a new coordinating tetrazole-2-pyridylketone oxime ligand.18
Variable-temperature (2.0–300 K range), direct-current (dc) magnetic susceptibility measurements were performed on a freshly-prepared microcrystalline sample of 1·2H2O; a dc field of 0.3 T was applied from 30 to 300 K and a weak dc field of 0.03 T was used from 2 to 30 K to avoid saturation effects. The data are shown as χMT vs. T plot in Fig. 3. The χMT product slightly increases from a value of 9.93 cm3 mol−1 K at 300 K to 10.43 cm3 mol−1 K at 80 K, followed by a rapid decrease to a value of 1.49 cm3 mol−1 K at 2.0 K. The value of the χMT product at 300 K is slightly higher than the value of 9.68 cm3 mol−1 K (calculated with g = 2.2, as is usual in NiII cluster compounds14,15) expected for eight non-interacting NiII (S = 1) ions. The observed magnetic behaviour is consistent with the presence of both ferro- and antiferromagnetic exchange interactions between the eight NiII ions at the corresponding high- and low-T regions. The small χMT value at 2.0 K and its tendency to head to zero suggests that the antiferromagnetic component eventually dominates, thus fostering the stabilization of an S = 0 ground state for the octanuclear complex 1. Magnetization (M) vs. field (H) measurements were also performed for 1 at 2 K and the corresponding plot (Fig. 3, inset) shows a nearly linear increase up to a non-saturated value of ∼6 NμB at 5 T. This is consistent with an S = 0 ground state and a progressive population of low-lying spin states that are close in energy with S > 0.
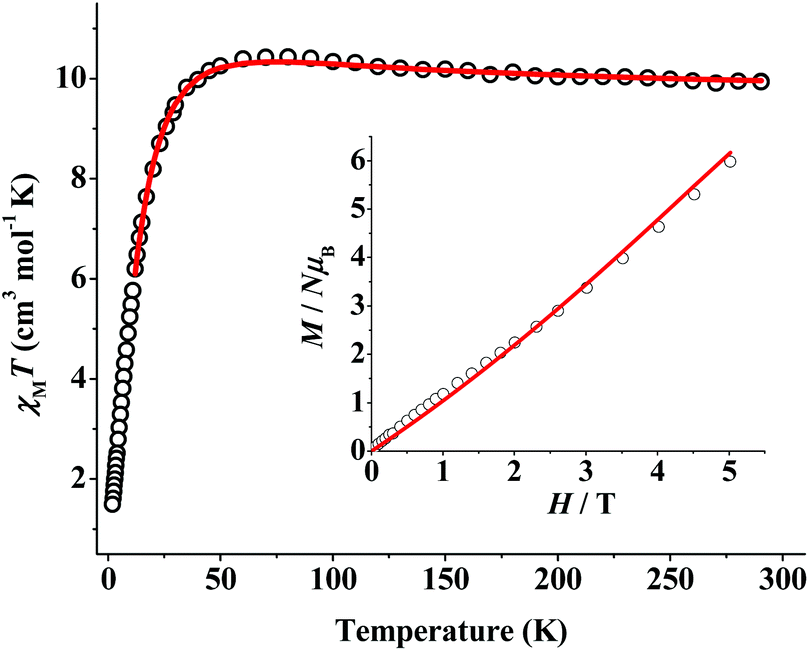 | ||
| Fig. 3 χ M T vs. T and M vs. H (inset) plots for 1·2H2O. The red solid lines correspond to the fit of the experimental data using a complete 4 − J spin Hamiltonian; see the text for the obtained fit parameters. | ||
There are four different types of superexchange interactions between the NiII ions in complex 1, which comprise the following magnetic pathways (Fig. 2 and S2†): (a) Ni–(NEO-azide)(NNmtz)–Ni (J1), (b) Ni–(NEO-azide)(NNmtz)2–Ni (J2), (c) Ni–(NNNNmtz)–Ni (J3), and (d) Ni–(NNNmtz)–Ni (J4). These bridges result in 20 superexchange pathways as illustrated in the complete 4 − J spin Hamiltonian of eqn (1). An excellent simultaneous fit of the magnetic susceptibility and magnetization data was obtained, and the resulting best-fit parameters were: J1 = +11.9 cm−1, J2 = +0.7 cm−1, J3 = −1.2 cm−1, J4 = −1.9 cm−1 and g = 2.18 [R(χMT) = 3.8 × 10−5 and R(M) = 2.0 × 10−3]. The obtained J constants agree with magnetostructural correlations previously reported for end-on azido- and diazine-bridged NiII complexes (vide infra), and they are consistent with the expected ferromagnetic interactions promoted by the N3−/2,3-tetrazolate combination (J1 and J2)19 and the weak antiferromagnetic interactions mediated solely by the long 1,4- or 1,3-tetrazolate pathways (J3 and J4),15,20 thus leading to an overall S = 0 ground state. The small J2 value, compared to J1, could be attributed to the co-presence of one EO-N3− (ferromagnetic coupler) and two diazine NN-bridges (antiferromagnetic couplers) from the mtz− groups.15,19,20
 | (1) |
Good fits of the experimental data can be also obtained with several sets of J values (see the corresponding discussion in ESI†), and therefore it is not meaningful to assign absolute values to each one of them. Considering the results derived from the different fits, the maximum ferromagnetic interaction should be attributed to the EO-azide/2,3-tetrazolate bridge, with a J value close to +11–12 cm−1, and weak interactions promoted by the remaining bridges. For most divalent 3d-metal complexes with EO bridging N3− ligands, the angle for switching from ferro- to antiferromagnetic coupling is typically >104°.8 Despite the fairly large average Ni–N–Ni angle of 110.6° in 1, and the co-presence of planar N,N-bridging mtz− ligand(s) with small torsion angles, which are known antiferromagnetic couplers, the ferromagnetic component induced by EO-N3− is still significant. This is not surprising as it has been previously observed in several examples of {Ni2} complexes bridged by μ-1,1 end-on N3− and pyrazolate-type bridging ligands with very large Ni–Nazide–Ni angles (>115°).19,21 This is also in line with DFT calculations, which predict that the coupling in NiII chemistry should be always ferromagnetic for all ranges of Ni–N–Ni angles, as has been experimentally confirmed by the ferromagnetically coupled polyoxometallate-based dinickel–azide compound which has a very large Ni–(μ-N3)–Ni angle of 129.3°.21
In summary, we have demonstrated herein that click chemistry is a useful synthetic route not only in organic chemistry and related fields, but also for the preparation of new coordination clusters with unprecedented structural motifs and magnetic properties related to the combination of bridging azides and in situ generated tetrazoles. The reported octanuclear NiII cluster was obtained under mild, room-temperature synthetic conditions and exhibits a fascinating trapezoidal prismatic topology, resulting from the presence of both end-on N3− and μ4-bridging mtz− groups; the latter serve to hold the vertices of the two {Ni4} trapezoids together and additionally link the two opposite trapezoids to the resulting prismatic conformation. We are currently trying to isolate the chiral forms of the {Ni8} compound from R- and S-mpmH in order to subsequently study the magnetic and electronic properties of the resulting chiral species. Work in progress also includes the in-depth investigation of click chemistry as a means of preparing magnetic coordination clusters with unique structural motifs, and the use of racemic and potentially chiral pyridine alkoxide ligands with other 3d-metal ions, as well as 4f-metals and 3d/4f-metal combinations. These results will be reported in the full paper of this work and other upcoming publications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC-DG (Th. C. S and M. P), ERA (Th. C. S), CFI (M. P) and the Ontario Trillium Fellowship (P. A). A. E. acknowledges financial support from the Ministerio de Economía y Competitividad, Project PGC2018-094031-B-100. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.Notes and references
- (a) C. Papatriantafyllopoulou, E. E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597–1628 RSC; (b) A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC; (c) C. J. Milios, S. Piligkos and E. K. Brechin, Dalton Trans., 2008, 1809–1817 RSC; (d) E. K. Brechin, Chem. Commun., 2005, 5141–5153 RSC; (e) M. Murrie, Polyhedron, 2018, 150, 1–9 CrossRef CAS.
- (a) Th. C. Stamatatos, C. G. Efthymiou, C. C. Stoumpos and S. P. Perlepes, Eur. J. Inorg. Chem., 2009, 3361–3391 CrossRef CAS; (b) S. Schmidt, D. Prodius, V. Mereacre, G. E. Kostakis and A. K. Powell, Chem. Commun., 2013, 49, 1696–1698 RSC; (c) G. Brunet, F. Habib, C. Cook, T. Pathmalingam, F. Loiseau, I. Korobkov, T. J. Burchell, A. M. Beauchemin and M. Murugesu, Chem. Commun., 2012, 48, 1287–1289 RSC; (d) A. G. Blackman, Eur. J. Inorg. Chem., 2008, 2633–2647 CrossRef CAS; (e) A. S. R. Chesman, D. R. Turner, B. Moubaraki, K. S. Murray, G. B. Deacon and S. R. Batten, Eur. J. Inorg. Chem., 2010, 59–73 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC; (c) M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- (a) C. Plenk, J. Krause, M. Beck and E. Rentschler, Chem. Commun., 2015, 51, 6524–6527 RSC; (b) W. P. Forrest, Z. Cao, W.-Z. Chen, K. M. Hassell, A. Kharlamova, G. Jakstonyte and T. Ren, Inorg. Chem., 2011, 50, 9345–9353 CrossRef CAS PubMed; (c) K. Xiong, F. Jiang, Y. Gai, Z. He, D. Yuan, L. Chen, K. Su and M. Hong, Cryst. Growth Des., 2012, 12, 3335–3341 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- H. Zhao, Z.-R. Qu, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC , and references therein.
- (a) T. Wu, B. H. Yi and D. Li, Inorg. Chem., 2005, 44, 4130–4132 CrossRef CAS PubMed; (b) M. Li, Z. Li and D. Li, Chem. Commun., 2008, 3390–3392 RSC.
- For some representative recent reviews, see: (a) A. Escuer, J. Esteban, S. P. Perlepes and Th. C. Stamatatos, Coord. Chem. Rev., 2014, 275, 87–129 CrossRef CAS; (b) Th. C. Stamatatos and E. Rentschler, Chem. Commun., 2019, 55, 11–26 RSC; (c) S. Mukherjee and P. S. Mukherjee, Acc. Chem. Res., 2013, 46, 2556–2566 CrossRef CAS PubMed.
- Y. Feng, X. Liu, L. Duan, Q. Yang, Q. Wei, G. Xie, S. Chen, X. Yang and S. Gao, Dalton Trans., 2015, 44, 2333–2339 RSC.
- H. Deng, Y.-C. Qiu, Y.-H. Li, Z.-H. Liu, R.-H. Zeng, M. Zeller and S. R. Batten, Chem. Commun., 2008, 2239–2241 RSC.
- Y.-Z. Zhang, S. Gao and O. Sato, Dalton Trans., 2015, 44, 480–483 RSC.
- P. Abbasi, K. Quinn, D. I. Alexandropoulos, M. Damjanović, W. Wernsdorfer, A. Escuer, J. Mayans, M. Pilkington and Th. C. Stamatatos, J. Am. Chem. Soc., 2017, 139, 15644–15647 CrossRef CAS PubMed.
- M. Kamitani, M. Ito, M. Itazaki and H. Nakazawa, Chem. Commun., 2014, 50, 7941–7944 RSC.
- (a) E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006–2018 RSC; (b) X.-P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062–16065 CrossRef CAS PubMed; (c) Y. Liu, V. Kravtsov, R. D. Walsh, P. Poddar, H. Srikanth and M. Eddaoudi, Chem. Commun., 2004, 2806–2807 RSC; (d) Z. Wang, Z. Jagličić, L.-L. Han, G.-L. Zhuang, G.-G. Luo, S.-Y. Zeng, C.-H. Tung and D. Sun, CrystEngComm, 2016, 18, 3462–3471 RSC.
- Z. Xu, L. K. Thompson, V. A. Milway, L. Zhao, T. Kelly and D. O. Miller, Inorg. Chem., 2003, 42, 2950–2959 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210–12216 CrossRef CAS PubMed.
- W. G. Finnegan, R. A. Henry and R. Lofquist, J. Am. Chem. Soc., 1958, 80, 3908–3911 CrossRef CAS.
- L. Alcazar, M. Font-Bardia and A. Escuer, Eur. J. Inorg. Chem., 2015, 1326–1329 CrossRef CAS.
- S. Demeshko, G. Leibeling, S. Dechert and F. Meyer, Dalton Trans., 2006, 3458–3465 RSC.
- (a) A. C. Fabretti, W. Malavasi, D. Gatteschi and R. Sessoli, J. Chem. Soc., Dalton Trans., 1991, 2331–2334 RSC; (b) G. A. van Albada, R. A. G. de Graaff, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1984, 23, 1404–1408 CrossRef CAS.
- (a) P. Mialane, A. Dolbecq, E. Rivière, J. Marrot and F. Sécheresse, Angew. Chem., Int. Ed., 2004, 43, 2274–2277 CrossRef CAS PubMed; (b) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Am. Chem. Soc., 1998, 120, 11122–11129 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data (CIF format), various synthetic, spectroscopic, structural, and magnetism data and figures. CCDC 1911866. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01998g |
| ‡ Present address: Chemistry Department, University of Patras, Patras 26504, Greece. E-mail: thstama@upatras.gr; Tel: +30-2610996008. |
| This journal is © The Royal Society of Chemistry 2019 |