
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of base stabilized fluoroborylene and its radical cation†
Samir Kumar
Sarkar
 a,
Mujahuddin M.
Siddiqui
a,
Subrata
Kundu
a,
Munmun
Ghosh
a,
Johannes
Kretsch
a,
Peter
Stollberg
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
*ad,
A. Claudia
Stückl
a,
Brigitte
Schwederski
b,
Wolfgang
Kaim
*b,
Sagar
Ghorai
c,
Eluvathingal D.
Jemmis
*c and
Herbert W.
Roesky
*a
a,
Mujahuddin M.
Siddiqui
a,
Subrata
Kundu
a,
Munmun
Ghosh
a,
Johannes
Kretsch
a,
Peter
Stollberg
a,
Regine
Herbst-Irmer
a,
Dietmar
Stalke
*ad,
A. Claudia
Stückl
a,
Brigitte
Schwederski
b,
Wolfgang
Kaim
*b,
Sagar
Ghorai
c,
Eluvathingal D.
Jemmis
*c and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Universität Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de; hroesky@gwdg.de
bInstitute für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: kaim@iac.uni-stuttgart.de
cInorganic and Physical Chemistry Department Indian Institute of Science, Bangalore-560012, India. E-mail: jemmis@iisc.ac.in
dSolid State and Structural Chemistry Unit Indian Institute of Science, Bangalore-560012, India. E-mail: dstalke@chemie.uni-goettingen.de
First published on 10th May 2019
Abstract
Herein, we report the synthesis and characterization of the metal free low valent fluoroborylene [(Me-cAAC)2BF] (1) stabilized by cyclic (alkyl)(amino) carbene (cAAC). The fluoroborylene 1 is obtained by the reductive defluorination of Me-cAAC:BF3 with 2.0 equivalents of KC8 in the presence of 1.0 equivalent of Me-cAAC. Due to its highly electron rich nature, 1 underwent one-electron oxidation with 1.0 equivalent of lithium tetrakis(pentafluorophenyl)borate [LiB(C6F5)4] to form the radical cation [(Me-cAAC)2BF]˙+[B(C6F5)4]− (2). DFT studies suggested that the lone pair of electrons is localized on the boron atom in 1, which explains its unprecedented reactivity. Both compounds 1 and 2 were characterized by X-ray crystallography. The radical cation 2 was studied by EPR spectroscopy.
The boron analogues of carbene are known as borylenes which are isolobal to CO and N2. With only one substituent, two vacant orbitals and the presence of a lone pair of electrons, borylenes are highly electron rich reactive transient species and thus more difficult to stabilize as compared to carbenes.1 Consequently, free borylenes (R–B:) have never been obtained as stable compounds and can only be isolated by chemical trapping. In 1998, Braunschweig and co-workers were successful in isolating the first stable terminal borylene complex [(OC)5WBN(SiMe3)2] stabilized with a metal carbonyl fragment.2a Later, N-heterocyclic carbenes (NHCs) and cyclic (alkyl)(amino) carbenes (cAACs) were used for the stabilization of different amino, cyano, aryl and alkyl borylene derivatives.2
Free diatomic borylenes such as :BH, :BX (X = F, Cl, Br, I) are highly unstable as compared to aryl/amino borylenes and have been detected by microwave spectroscopy.3 In 2007, Robinson and co-workers reported the isolation of NHC-stabilized neutral diborene (NHC)2B2H2 by the reduction of (NHC)BBr3 with KC8 (Scheme 1).4a In 2011, Bertrand and co-workers isolated the first hydroborylene (cAAC)2BH (B) by the reduction of (cAAC)BBr3 with an excess of KC8 in the presence of cAAC (Scheme 1).4b The yield of the reaction is very low and the synthetic route of the formation of (cAAC)2BH is still unclear.
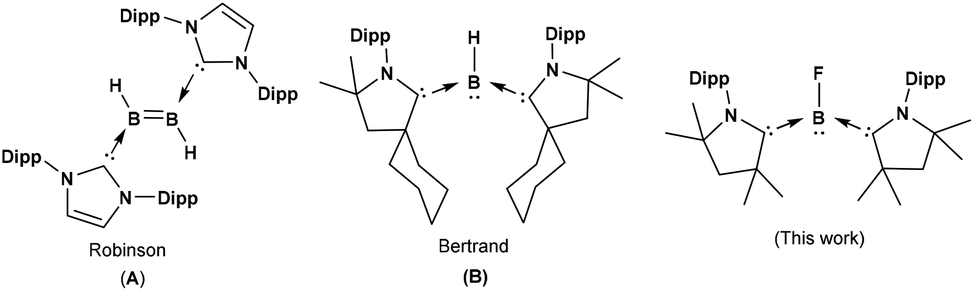 | ||
| Scheme 1 Lewis base-stabilized borylenes. | ||
The next higher analogues of parent hydroborylene (:BH) are the haloborylenes (:BX). Haloborylenes are low-valent boron compounds where the boron atom is in the formal +I oxidation state which is in sharp contrast with diarylboron halides (R2BX) wherein the boron atom possesses the +III oxidation state. Haloborylenes have the potential in unraveling hitherto unexplored aspects of borylene chemistry as they are the key intermediate compounds or species to synthesize unique and diverse borylene derivatives. Therefore, there is a need to synthesize and isolate such species. Synthesis of free fluoroborylene (:BF) was attempted by passing BF3 over crystalline boron at 2000 °C per 1 mm in the gas phase but it formed a green uncharacterized polymeric material even at −196 °C.3a Very few metal BF complexes such as Fe(BF)(CO)4,5a Fe(PF3)4(BF),5b Cp2Ru2(CO)4(μ-BF)5c and Fe(BF)(CO)2(CNArTripp2)2 [ArTripp2, 2,6-(2,4,6-(i-Pr)3C6H2)2C6H3; i-Pr, iso-propyl]5d were reported. The first two were characterized spectroscopically and the last two were structurally characterized by X-ray crystallography. To date no synthesis or isolation of fluoroborylenes without a metal sphere has been reported and it still remains a challenging task. Very recently, Xie and co-workers reported bromoborylene (:BBr) using a carborane-bridged bis(silylene) ligand6a,b and Braunschweig and co-workers have isolated base stabilized chloroborylene (:BCl).6c
Cyclic (alkyl)(amino) carbenes (cAACs) which are stronger σ-donors (nucleophilic) and better π-acceptors (electrophilic) compared to the classical N-heterocyclic carbenes (NHCs) have gained much interest owing to their versatile applications.7 Cyclic (alkyl)(amino) carbenes (cAACs) have been judiciously used for the stabilization of transition metals as well as p-block elements in low oxidation states.7 We have shown that cAACs can be utilized as excellent ligands for the stabilization of different low oxidation states of aluminum, germanium, silicon and phosphorus and their different radicals.8 Herein, we report the synthesis and isolation of cyclic alkylamino carbene (cAAC) stabilized fluoroborylene (:BF) and its radical cation. The isolated compounds have been fully characterized by X-ray crystallography, multinuclear-spectroscopy, cyclic voltammetry and electron paramagnetic resonance (EPR) studies. Also, quantum-chemical studies have been performed to understand the electronic structure and bonding.
The synthetic route of (Me-cAAC)2BF (1) is shown in Scheme 2. Me-cAAC:BF3 was isolated as a white solid by a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of Me-CAAC and BF3·OEt2 in hexane at room temperature. A 1:1:2 molar ratio of Me-cAAC:BF3, cAAC: and KC8 was reacted in toluene at −78 °C, and the resultant suspension was slowly warmed up to room temperature to obtain a red violet solution. The reaction mixture was filtered and the resulting solution was reduced to 3 ml under vacuum. Red violet block-shaped crystals of (Me-cAAC)2BF (1) were obtained in 78% yield at 0 °C after 24 h. (Me-cAAC)2BF (1) was fully characterized by NMR (1H, 13C, 11B, 19F) and mass spectrometry (ESI, LIFDI). The composition of 1 was confirmed by single crystal X-ray diffraction and elemental analysis. Haloborylenes can be efficient precursors for the synthesis of a B(I) cation by the treatment with halide scavengers. In our attempt at synthesizing the boryl cation [(Me-cAAC)2B]+ (Scheme 3) by the reaction of 1 with one equivalent of LiB(C6F5)4, we observed an unprecedented 1 e− oxidation of 1 to yield the radical cation [(Me-cAAC)2BF]˙+[B(C6F5)4]− (2). The radical nature of 2 was deduced from broad resonances in the 1H NMR spectrum. Compound 2 was isolated as brown color crystals at 0 °C after three days. The molecular structure of the radical cation (2) was unambiguously determined by single crystal X-ray analysis.
:1 reaction of Me-CAAC and BF3·OEt2 in hexane at room temperature. A 1:1:2 molar ratio of Me-cAAC:BF3, cAAC: and KC8 was reacted in toluene at −78 °C, and the resultant suspension was slowly warmed up to room temperature to obtain a red violet solution. The reaction mixture was filtered and the resulting solution was reduced to 3 ml under vacuum. Red violet block-shaped crystals of (Me-cAAC)2BF (1) were obtained in 78% yield at 0 °C after 24 h. (Me-cAAC)2BF (1) was fully characterized by NMR (1H, 13C, 11B, 19F) and mass spectrometry (ESI, LIFDI). The composition of 1 was confirmed by single crystal X-ray diffraction and elemental analysis. Haloborylenes can be efficient precursors for the synthesis of a B(I) cation by the treatment with halide scavengers. In our attempt at synthesizing the boryl cation [(Me-cAAC)2B]+ (Scheme 3) by the reaction of 1 with one equivalent of LiB(C6F5)4, we observed an unprecedented 1 e− oxidation of 1 to yield the radical cation [(Me-cAAC)2BF]˙+[B(C6F5)4]− (2). The radical nature of 2 was deduced from broad resonances in the 1H NMR spectrum. Compound 2 was isolated as brown color crystals at 0 °C after three days. The molecular structure of the radical cation (2) was unambiguously determined by single crystal X-ray analysis.
 | ||
| Scheme 2 Synthesis of 1 (Dipp: 2,6-diisopropylphenyl). | ||
 | ||
| Scheme 3 Synthesis of 2. | ||
Compounds 1 and 2 are highly stable in the solid state as well as in solutions at room temperature under an inert atmosphere for months. Also, 1 is stable in open air for a couple of weeks in solid form and in dry solution for about 10 to 15 hours and slowly changes its intense violet color to colorless. Compound 2 immediately loses its brown color upon exposure to air. The UV/vis spectra of 1 and 2 were recorded in dry n-hexane and THF, respectively. Compound 1 shows absorption maxima at 516 nm (Fig. S12†) and compound 2 at 450, 518 and 738 nm, respectively (Fig. S13†).
To check the unprecedented formation of 2, we performed the cyclic voltammogram experiments of 1 in both CH3CN and THF at room temperature with 0.1 M and 0.2 M nBu4NPF6 respectively as an electrolyte (Fig. S14–16†). In both solvents a well-defined one-electron reversible redox couple was observed at −0.99 V (−0.92 V in THF vs. Fc/Fc+) suggesting the radical stability under the operative conditions (Fig. 1). The reversible cyclic voltammogram and single electron activation suggest that compound 1 is catalytically active. Also this gives an indication that 1 can be utilized as an excellent single electron transfer catalyst for many catalytic reactions. Furthermore, in THF an irreversible two-electron oxidation peak (Epc) showed up at −0.01 V but in CH3CN it was not clearly resolved, perhaps due to the instability of the oxidized species in CH3CN (Fig. S15†).
 | ||
| Fig. 1 Scan rate dependent reversible cyclic voltammograms of 1 in acetonitrile (CH3CN) (0.1 M nBu4NPF6, 298 K) (scan rates: 50, 100, 200, 300, 500 mV s−1). | ||
To understand the radical nature of 2, EPR spectroscopy was performed. The EPR spectrum of the radical cation 2 at g = 2.0063 is dominated by a doublet of 23.2 G (Fig. 2), attributed to the coupling of the unpaired electron with one 19F nucleus. This large hyperfine splitting is comparable to the 16.5 G reported for the radical anion of 1,4-difluoro-2,3,5,6-tetramethyl-l,4-dibora-2,5-cyclohexadiene, an isoelectronic species to durosemiquinone9a or to the 16.8 G 19F coupling of SiF3 bonded to cAAC.9b Each of the two doublet EPR lines for 2 is partially resolved by spin coupling with 14N (I = 1, a = 4.1 G) and 11B (I = 3/2, 80%, a = 2.1 G) and 10B (I = 3, 20%, a = 0.7 G). The low values of 11,12B isotope coupling have been noted earlier.9a A 14N splitting of a few Gauss would agree with the typical values observed for cAAC stabilized paramagnetic species.9a,c
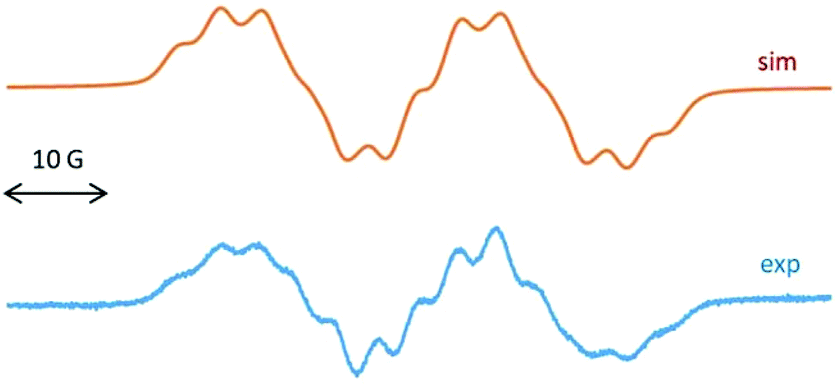 | ||
| Fig. 2 EPR spectrum of 2 (computer simulation, top) and (experimental, down) in THF at room temperature. | ||
Single crystal X-ray analysis of 1 (Fig. 3) reveal that the bond distances of B1–C1 (1.513 Å) and B1–C3 (1.527 Å) are almost equal and are average of the B–C single (1.59 Å) and double (1.44 Å) bonds. The B–C (∼1.52 Å) and C–N (∼1.38 Å) bond lengths of 1 are identical to the bond lengths of (cAAC)2BH (B).4b The longer B–C bonds and shorter C–N bonds in 2 (Fig. 3) compared to 1 suggest that the single electron in the oxidation from 1 to 2 is removed from a delocalized C–B–C moiety. Comparing with the (cAAC)2BH radical cation,4b relatively 0.04 Å shorter B–C bonds and 0.02 Å longer C–N bonds were observed for 2 which indicate the closer B-cAAC interaction. The B1–F1 (1.399 Å) bond length in 1 is comparable to the covalent radii for boron and fluorine (1.46 Å),10 and the longest among all the reported low-valent BF complexes,5c,d albeit a shorter B1–F1 bond was observed in 2 because of one electron oxidation (Table 1).
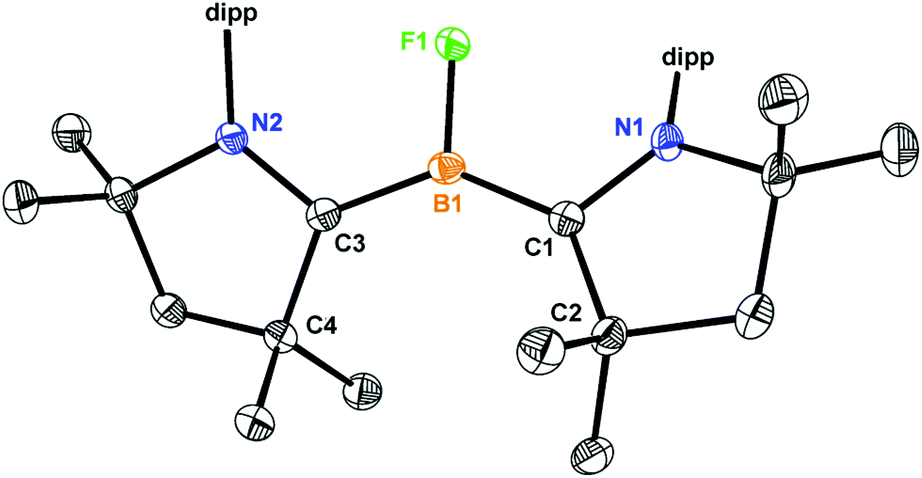 | ||
| Fig. 3 Crystal structure of 1 and 2 [in brackets], [B(C6F5)4]− is omitted for clarity. Anisotropic displacement parameters are depicted at the 50% probability level. Selected bond lengths (Å): B1–F1 1.3999(15) [1.369(6)], B1–C1 1.5137(19) [1.541(4)], N1–C1 1.3955(16) [1.376(4)], C1–C2 1.5462(17) [1.526(6)], B1–C3 1.5270(19) [1.541(4)], C3–N2 1.3787(16) [1.376(4)], C3–C4 1.5360(17) [1.526(6)]. | ||
| Parameter | Structure 1 | Structure 2 | ||
|---|---|---|---|---|
| X-ray | DFT | X-ray | DFT | |
| B1–F1 | 1.399 | 1.415 | 1.369 | 1.369 |
| B1–C1 | 1.514 | 1.521 | 1.541 | 1.563 |
| B1–C3 | 1.527 | 1.526 | 1.541 | 1.567 |
| C1–C2 | 1.546 | 1.533 | 1.526 | 1.534 |
| C3–C4 | 1.536 | 1.541 | 1.526 | 1.528 |
| N1–C1 | 1.396 | 1.380 | 1.376 | 1.348 |
| C3–N2 | 1.379 | 1.393 | 1.376 | 1.339 |
Density functional theory is used to provide insight into the electronic structure of 1 and 2 ([B(C6F5)4]− has been removed for simplicity). Calculations are carried out using the dispersion corrected functional BP86-GD3(BJ) with the def2-tzvp basis set.11 All the optimized geometrical parameters of 1 and 2 are in good agreement with the crystal structure data which are summarized in Table 1.
The ground state of 1 is singlet and 2 is doublet (Table T5, ESI†). The triplet state of 1 lies 21.3 kcal mol−1 higher in energy. The HOMO of 1 indicates that 1 is best described as borylene stabilized by Lewis bases where the lone pair at the B-atom is stabilized by the π back-bonding to both the cAAC ligands (Fig. S22†). The formation of 2 is accomplished by removing one electron from the delocalized π-orbital of 1, reflecting longer B–C1 and B–C3 bonds and shorter C1–N1 and C3–N2 bonds in 2 in comparison with 1. We have found that ΔE = −11.9 kcal mol−1 for the reaction which infers that the forward reaction is highly feasible (BP86-GD3(BJ) with the def2-tzvp basis set). Natural population analysis shows that the unpaired electron in 2 is delocalized over the boron atom and the ligand skeleton (Fig. 4a). The spin density plot (Fig. 4b) further substantiates the delocalized nature of the radical system, yet another example to show the crucial role of the cAAC ligand to stabilize the radical system.12 In order to further understand the nature of bonding in 1 and 2, we carried out an EDA-NOCV analysis by dividing the molecule into two fragments, BF and [cAAC]2 for 1, and BF+ and [cAAC]2 for 2.13 Various different electronic states of two different fragments were considered to identify the smallest orbital interaction energy (Eorb) term and hence the best fragments.14 Table T5 (ESI†) summarizes the results for the most favorable fragments. The deformation densities associated with the pairwise orbital interactions for the three strongest orbital interactions obtained from the EDA-NOCV analysis are shown in Fig. 5a and b.
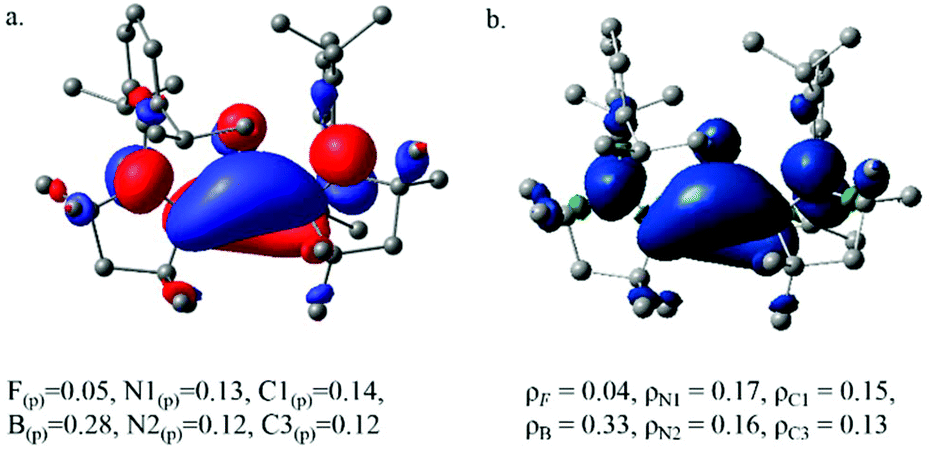 | ||
| Fig. 4 (a) SOMO of 2 with the percent of p orbital contribution at the BP86-GD3(BJ)/def2-tzvp level of theory. (b) Spin density plot of 2 with the Mulliken atomic spin densities at the BP86-GD3(BJ)/def2-tzvp level of theory. | ||
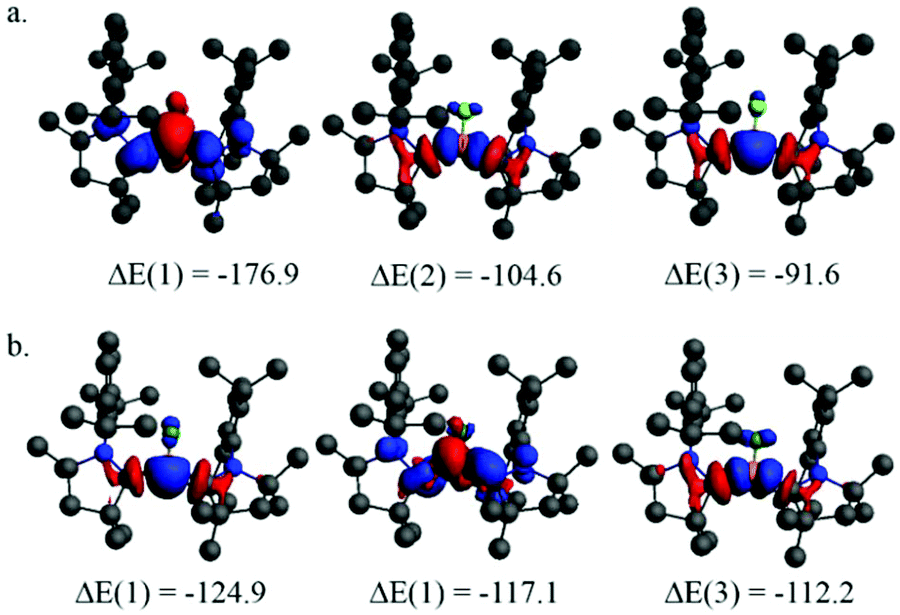 | ||
| Fig. 5 Plot of the deformation densities of the interactions for the pairwise orbital interactions of the three strongest orbital interactions with the associated interaction energies between (a) BF and [cAAC]2 in 1 and (b) BF+ and [cAAC]2 in 2. The direction of the charge flow is red → blue. | ||
The ΔE(1) in Fig. 5a shows that the strongest orbital interaction is the π-back donation from BF to [cAAC]2 and ΔE(2) and ΔE(3) correspond to the σ-donation from [cAAC]2 to BF in 1. Similarly, ΔE(1) and ΔE(3) in Fig. 5b correspond to the σ-donation from [cAAC]2 to BF+ and ΔE(2) gives the π-back donation from BF+ to [cAAC]2. The strength of σ-donation is stronger in 2 and the π-back donation is weaker compared to 1. Therefore, the formation of 2 takes place at the cost of π-delocalization and the benefit of stronger σ-bonding.
In summary, an efficient synthetic approach was developed to synthesize cAAC stabilized fluoroborylene and its radical cation. The one e− oxidation of 1 with one equivalent of LiB(C6F5)4 leads to the formation of a stable radical cation which is confirmed by EPR measurement. Compounds 1 and 2 were fully characterized by single crystal analyses. DFT computational studies infer that the electron(s) residing on the B-atom are delocalized over the cAAC ligands thereby providing stability for both complexes 1 and 2. Cyclic voltammetry experiments show that the two-electron oxidation of (cAAC)2BF is highly feasible to obtain a diradical dication, and the synthesis of a boryl cation [(Me-cAAC)2B]+ by removing the fluorine atom from 1 is possible under suitable reaction conditions. Both the studies are in progress. Also, we are investigating the catalytic activity of 1 as a single electron transfer catalyst.
Crystal Structure determination: Crystal handling and selection was carried out under an inert atmosphere and at low temperatures provided by the x-temp2 device.15 Subsequent data collection has been carried out on Bruker Smart Apex II systems with the enclosed software suit. The resulting experimental data have been integrated with SAINT16 and an additional multi-scan absorption correction and a 3λ correction17 was applied with SADABS.18 The structure was solved with SHELXT19 and refined with SHELXL20via the SHELXLE GUI.21
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. W. R. is thankful to the DFG for financial support (RO 224/68-1). D. S. is grateful to the DNRF (DNRF93) funded Center for Materials Crystallography (CMC). EDJ thanks the DST for funding through the J. C. Bose Fellowship. SKS thanks Alexander von Humboldt Foundation for a research fellowship. Dedicated to prof. Frank Edelmann.Notes and references
- (a) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed; (b) S. Bertsch, H. Braunschweig, B. Christ, M. Forster, K. Schwab and K. Radacki, Angew. Chem., Int. Ed., 2010, 49, 9517–9520 CrossRef CAS PubMed; (c) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC; (d) M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef CAS PubMed; (e) M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef CAS PubMed; (f) U. Flierler, M. Burzler, D. Leusser, J. Henn, H. Ott, H. Braunschweig and D. Stalke, Angew. Chem., 2008, 120, 4393–4397 ( Angew. Chem., Int. Ed. , 2008 , 47 , 4321–4325 ) CrossRef; (g) K. Götz, M. Kaupp, H. Braunschweig and D. Stalke, Chem. – Eur. J., 2009, 15, 623–632 CrossRef PubMed.
- (a) H. Braunschweig, C. Kollann and U. Englert, Angew. Chem., Int. Ed., 1998, 37, 3179–3180 CrossRef CAS; (b) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 CrossRef CAS PubMed; (c) M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grgne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464–14468 CrossRef CAS PubMed; (d) L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 CrossRef CAS PubMed; (e) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed.
- (a) P. L. Timms, J. Am. Chem. Soc., 1967, 89, 1629–1632 CrossRef CAS; (b) P. L. Timms, Acc. Chem. Res., 1973, 6, 118–123 CrossRef CAS; (c) M. Nomoto, T. Okabayashi, T. Klaus and M. Tanimoto, J. Mol. Struct., 1997, 413–414, 471–476 CrossRef CAS.
- (a) Y. Z. Wang, B. Quillian, P. R. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. V. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed; (b) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed.
- (a) K. Kämpfer, H. Nöth, W. Petz and G. Schmid, Inorganica Chimica Acta, Padova, Italy, 1968 Search PubMed; (b) P. L. Timms, Acc. Chem. Res., 1973, 6, 118–123 CrossRef CAS; (c) D. Vidović and S. Aldridge, Angew. Chem., Int. Ed., 2009, 48, 3669–3672 CrossRef PubMed; (d) M. J. Drance, J. D. Sears, A. M. Mrse, C. E. Moore, A. L. Rheingold, M. L. Neidig and J. S. Figueroa, Science, 2019, 363, 1203–1205 CrossRef CAS PubMed.
- (a) H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680–13683 CrossRef CAS PubMed; (b) H. Wang, J. Zhang, H. K. Lee and Z. Xie, J. Am. Chem. Soc., 2018, 140, 3888–3891 CrossRef CAS PubMed; (c) M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Harterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019 10.1039/C9SC01039D.
- (a) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (b) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (c) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (d) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (e) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (f) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- (a) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (b) S. Kundu, P. P. Samuel, S. Sinhababu, A. V. Luebben, B. Dittrich, D. M. Andrada, G. Frenking, A. C. Stückl, B. Schwederski, A. Paretzki, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2017, 139, 11028–11031 CrossRef CAS PubMed; (c) B. Li, S. Kundu, A. C. Stückl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed; (d) S. Kundu, S. Sinhababu, A. V. Luebben, T. Mondal, D. Koley, B. Dittrich and H. W. Roesky, J. Am. Chem. Soc., 2018, 140, 151–154 CrossRef CAS PubMed; (e) S. Kundu, S. Sinhababu, M. M. Siddiqui, A. V. Luebben, B. Dittrich, T. Yang, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2018, 140, 9409–9412 CrossRef CAS PubMed; (f) M. M. Siddiqui, S. Sinhababu, S. Dutta, S. Kundu, P. N. Ruth, A. Münch, R. Herbst-Irmer, D. Stalke, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 11776–11780 CrossRef CAS PubMed; (g) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepotter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 801–1805 Search PubMed; (h) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepötter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed.
- (a) H. Bock, W. Kaim, P. L. Timms and P. Hawker, Chem. Ber., 1980, 113, 3196–3207 CrossRef CAS; (b) S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, I. Fernández, G. Frenking, A. C. Stückl, B. Schwederski, W. Kaim and H. W. Roesky, Chem. – Eur. J., 2018, 24, 1264–1268 CrossRef CAS PubMed; (c) S. Kundu, P. P. Samuel, S. Sinhababu, A. V. Luebben, B. Dittrich, D. M. Andrada, G. Frenking, A. C. Stückl, B. Schwederski, A. Paretzki, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2017, 139, 11028–11031 CrossRef CAS PubMed.
- J. Emsley, The Elements, Clarendon Press, Oxford, 1989 Search PubMed.
- See the ESI for the description of the computational details.†.
- (a) P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 CrossRef CAS PubMed; (b) J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß and E. Bill, Nat. Commun., 2018, 9, 1197 CrossRef PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- (a) R. Tonner and G. Frenking, Chem. – Eur. J., 2008, 14, 3260–3272 CrossRef CAS PubMed; (b) D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 12319–12324 CrossRef CAS PubMed.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC.
- Bruker AXS Inc., in Bruker Apex CCD, SAINT v8.30C, Bruker AXS Inst. Inc., WI, USA, Madison, 2013 Search PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data. Details of crystal structure refinements and crystal images, UV–Vis spectra, CV, EPR measurements, and theoretical investigations. CCDC 1900360 (1) and 1900361 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01899a |
| This journal is © The Royal Society of Chemistry 2019 |