
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the supramolecular isomerism of MOF-74 by controlling the synthesis conditions†
Andreea
Gheorghe
 a,
Inhar
Imaz
b,
Jarl Ivar
van der Vlugt
c,
Daniel
Maspoch
bd and
Stefania
Tanase
*a
a,
Inhar
Imaz
b,
Jarl Ivar
van der Vlugt
c,
Daniel
Maspoch
bd and
Stefania
Tanase
*a
aHeterogeneous Catalysis and Sustainable Chemistry, Van‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands. E-mail: s.grecea@uva.nl
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and Barcelona Institute of Science and Technology, Campus UAB, Bellaterra 08193, Barcelona, Spain
cBioinspired, Homogeneous & Supramolecular Catalysis, Van‘t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
dICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 21st May 2019
Abstract
Supramolecular isomerism of metal–organic frameworks (MOFs) is known for several MOF structures, having direct implications on the properties of these materials. Although the synthesis of MOF isomers is mainly serendipitous in nature, achieving controlled formation of a target framework is highly relevant for practical applications. This work discusses the influence of additives and synthesis conditions on the formation of porous isomers containing Zn2+ as nodes and 2,5-dihydroxy-1,4-benzenedicarboxylate (dobdc4–) as a linker. Using solvent mixtures containing strongly coordinated molecules, e.g. N,N′-dimethylformamide (DMF) and N-methylpyrrolidone (NMP), facilitates the formation of porous structures of type [Zn2(dobdc)(S)x]·yS (S = DMF, NMP) which are built from dinuclear Zn2(O)2(CO2)3 secondary building units (SBUs) consisting of two different edge-sharing polyhedra with the Zn2+ ions in a unsaturated coordinative environment. In the presence of water, the Zn2+ dimers are converted to one-dimensional infinite Zn2+ chains, in which the number of Zn2+-linker bonds increases, therefore giving a hydrolytically more stable coordination environment. The full characterization of the isomers as well as their conversion to the most stable isomer is presented.
Introduction
Metal–organic frameworks (MOFs) are porous coordination polymers formed using di- or multitopic organic linkers and metal ions or clusters of metal ions. Currently, more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF structures are known.1 The increased focus on the targeted design and tailor-made synthesis of MOFs emerges from their potential applications in gas storage,2–4 molecular separations3,5,6 and sensing7–9 as well as catalysis.10–12 Such broad applications arise from the ability of chemists to fine-tune these materials, even at the atomic level.13 Tailoring MOF properties for specific applications can be achieved by making specific topologies11 and/or introducing key functional groups within the framework.14 Important parameters in controlling the network connectivity are not only the nature of the metal ions and organic linkers used but also the synthesis parameters, e.g. temperature, pressure, reaction time and type of reaction vessel.13,15 At the same time, the compositional parameters, i.e. molar ratio, solvents or solvent mixtures, the addition of modulators, the pH and the reactants’ concentrations also play key roles.13,15
000 MOF structures are known.1 The increased focus on the targeted design and tailor-made synthesis of MOFs emerges from their potential applications in gas storage,2–4 molecular separations3,5,6 and sensing7–9 as well as catalysis.10–12 Such broad applications arise from the ability of chemists to fine-tune these materials, even at the atomic level.13 Tailoring MOF properties for specific applications can be achieved by making specific topologies11 and/or introducing key functional groups within the framework.14 Important parameters in controlling the network connectivity are not only the nature of the metal ions and organic linkers used but also the synthesis parameters, e.g. temperature, pressure, reaction time and type of reaction vessel.13,15 At the same time, the compositional parameters, i.e. molar ratio, solvents or solvent mixtures, the addition of modulators, the pH and the reactants’ concentrations also play key roles.13,15
The main goal in MOF synthesis is the formation of molecular frameworks in a controlled manner, which is often challenging because of the many synthetic parameters involved. An enticing approach to directing the structure is to use modulators. The most common types of modulators are coordination modulators that are added in excess directly to the reaction mixture. They are usually monocarboxylic acids (e.g. formic,16–18 acetic,16,18–20 and benzoic acids17,21,22 as well as aminoacids23,24). They act as competitive coordination agents, regulating the pore size and the morphology of the crystals. Modulators play a key role in the synthesis of Zr-MOFs, facilitating the formation of Zr6O4(OH)4 clusters and therefore the growth of the crystals.22,25 They also slow down the crystal growth rate, therefore avoiding the fast precipitation of amorphous phases.20,26,27 Kaskel et al.28 reported that the amount of acetic acid used as modulator in the synthesis of C72H36O32Zr6 (DUT-52) has a structure-directing effect because it reduces the connectivity of the [Zr6O4(OH)4]12+ entity from 12 to 8 or 6, leading to frameworks with different structural topologies.28 Modulation can also induce the formation of defects that are responsible for enhanced gas uptake,29,30 proton conduction31 or catalytic activity.32–34 Shafir et al.24 showed that L-proline is an efficient modulator in the synthesis of Zr MOFs with increased particle size.
The isostructural MOFs known as M-MOF-7435 are extensively studied due to the presence of unsaturated coordination sites at metal centers,35,36 their versatility of being prepared with different divalent ions35 and their high stability under ambient conditions and in the presence of water.37,38 Among them, Zn-MOF-74 is a very effective material for gas storage36 and molecular separations39 and it is also an active catalyst in several organic transformations.40,41
Using modulators is not a common approach in the synthesis of the MOF-74 series. Only Li et al.42 reported that the size and inherent defects of the nanocrystals of Co-MOF-74 can be tuned by employing salicylic acid as the modulator. Our aim was to study the role of Cinchona alkaloid derivatives, (+)-cinchonine (CN) and (−)-cinchonidine (CD) as coordination modulators in the synthesis of Zn-MOF-74, with the final goal to design chiral frameworks. We hypothesised that the chiral molecules CN and CD would coordinate to the Zn2+ ions during the crystallisation of Zn-MOF-74, thereby directing the handedness of the molecular framework. Bu et al.43 used Cinchona alkaloids to prepare a homochiral In3+ dicarboxylate framework, (Me2NH2)[In(thb)2]·xDMF (known as ATF-1P, H2thb = thiophene-2,5-dicarboxylic acid), using CD as the chiral additive.44 The chiral alkaloid binds to the In3+ ion and directs the handedness of the framework.44,45 The framework does not belong to a chiral space group due to the racemic distribution of the helical chains. Based on the structural similarities between the chiral ATF-1P43 framework with large 1D channels of ca. 13 Å,43 which are formed upon the interaction with chiral Cinchona alkaloid modulators, and the MOF-74 framework, which consists of 1D channels of ca. 11 Å, we hypothesised that chirality transfer might be achieved upon the coordination of CN or CD via amine and hydroxyl groups to the Zn2+ ions of MOF-74. Furthermore, the alkaloids can also interact with the framework walls via hydrogen bonding interactions. Indeed, Prochowicz et al.46 reported that Cinchona alkaloids lead to chiral coordination polymers using noncovalent interaction driven self-assembly processes. However, we found that using Cinchona alkaloids facilitates the formation of a new Zn-MOF-74 isomer, namely, [Zn2(dobdc)(NMP)]·⅓NMP (termed HIMS-74 in this work), where dobdc is 2,5-dioxido-1,4-benzenedicarboxylate and NMP is N-methylpyrrolidone (Scheme 1). Here, we report the controlled assembly and crystallisation of HIMS-74 and compare its structural features with the previously reported isomers, e.g. MOF-745 and UTSA-74.5 The role of the solvent in the stabilisation of specific framework is also discussed.
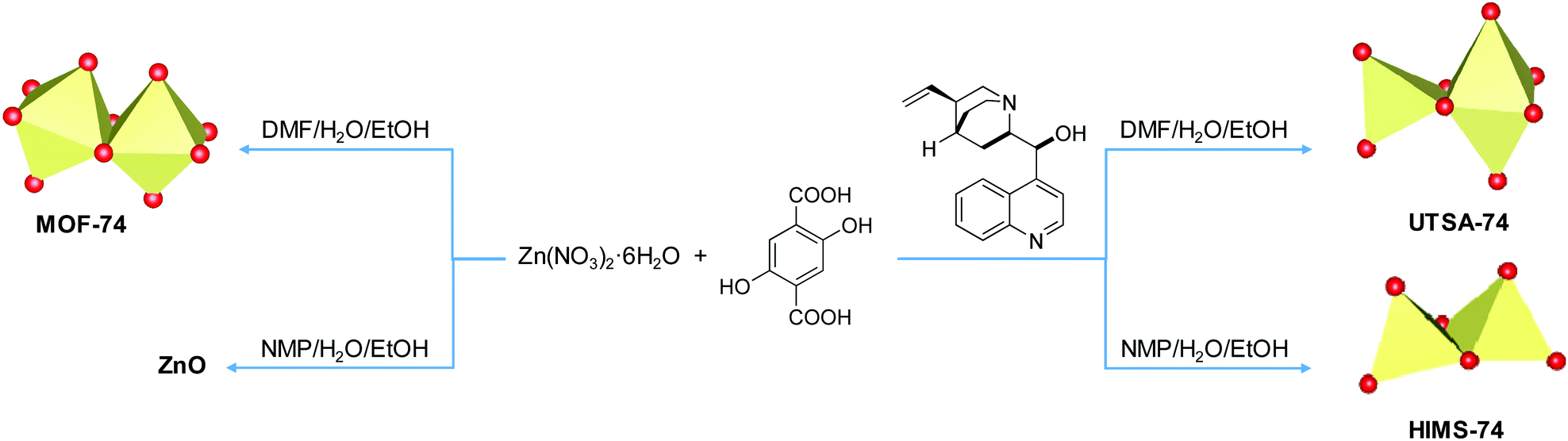 | ||
| Scheme 1 Influence of the synthetic conditions on the formation of secondary building units of porous molecular networks formed from Zn2+ as the nodes and 2,5-dihydroxy-1,4-benzenedicarboxylate (dobdc4–) as a linker. | ||
Results and discussion
Influence of the reaction conditions on the synthesis of MOF-74 isomers
The Zn2+ ion in MOF-74 has one coordinatively unsaturated site, which is occupied by an exchangeable solvent molecule. This site is also available for the coordination of Cinchona alkaloid molecules. Therefore, we studied the role of both alkaloids and solvent in the hydrothermal reaction of Zn2+ with 2,5-dioxido-1,4-benzenedicarboxylic acid (H4dobdc). Fig. 1 shows the alkaloids used in this study.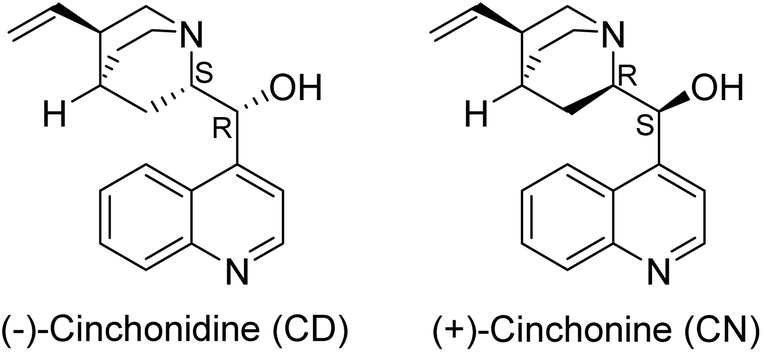 | ||
| Fig. 1 The structure of the Cinchona alkaloid modulators, indicating the different configurations of the chiral substituted carbon atom centres. | ||
Reacting Zn(NO3)2·6H2O, H4dobdc (molar ratio Zn2+:H4dobdc of 3:1) and CN or CD (1 or 3 equiv.) under hydrothermal conditions in two different solvent mixtures (DMF/EtOH/H2O or NMP/EtOH/H2O v/v/v ratio of 20/1/1) afforded six solid crystalline materials. Single-crystal XRD analysis performed on crystals isolated from DMF/EtOH/H2O reaction mixtures revealed the formation of the compound [Zn2(dobdc)(solvent)2]·xH2O·yDMF (solvent = H2O or DMF), which crystallises in the rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group and has a 3D porous structure. The asymmetric unit contains two crystallographically independent Zn2+ centres and one dobdc4– linker (Fig. 2, left). One Zn2+ coordination sphere is composed of four donor atoms from four fully deprotonated linkers, i.e. two phenolate oxygens and two carboxylate oxygens, forming a distorted tetrahedral geometry. The Zn–O bond lengths vary from 1.926(5) to 1.957(5) Å. Zn has an octahedral geometry, being surrounded by two phenolate oxygens, two carboxylate oxygens and two oxygen atoms from the solvent molecules (due to the high disorder of the molecular structure, it cannot be determined if they belong to water or DMF molecules). In this Zn2+, the Zn–O bond lengths vary from 1.959(5) to 2.120(6) Å. The organic linker binds one Zn2+via carboxylate oxygen, while it binds another Zn2+ as a chelate via carboxylate and phenolate oxygen, forming six-membered rings. All the crystallographic data indicate that this structure is identical to the UTSA-74 structure reported earlier.5
c space group and has a 3D porous structure. The asymmetric unit contains two crystallographically independent Zn2+ centres and one dobdc4– linker (Fig. 2, left). One Zn2+ coordination sphere is composed of four donor atoms from four fully deprotonated linkers, i.e. two phenolate oxygens and two carboxylate oxygens, forming a distorted tetrahedral geometry. The Zn–O bond lengths vary from 1.926(5) to 1.957(5) Å. Zn has an octahedral geometry, being surrounded by two phenolate oxygens, two carboxylate oxygens and two oxygen atoms from the solvent molecules (due to the high disorder of the molecular structure, it cannot be determined if they belong to water or DMF molecules). In this Zn2+, the Zn–O bond lengths vary from 1.959(5) to 2.120(6) Å. The organic linker binds one Zn2+via carboxylate oxygen, while it binds another Zn2+ as a chelate via carboxylate and phenolate oxygen, forming six-membered rings. All the crystallographic data indicate that this structure is identical to the UTSA-74 structure reported earlier.5
 | ||
| Fig. 2 The coordination of dobdc4– to the Zn2+ ions and the coordination geometries of the Zn2+ ions in UTSA-74 (left), HIMS-74 (middle) and MOF-7447 (right). The hydrogen atoms and the guest solvent molecules were omitted for clarity. Only the oxygen atom is represented for the disordered coordinated solvent molecules. | ||
Using a polarising optical microscope, the analysis of the materials obtained in DMF/EtOH/H2O solvent mixtures revealed UTSA-74 crystals as well as chiral alkaloid modulators as aggregates in solution. Therefore, the isolated solid materials were washed thoroughly with the synthesis solvent mixture to fully remove the alkaloids and then all materials were dried under ambient conditions. Notably, the PXRD patterns of these samples matched perfectly the simulated pattern of MOF-74 topology (Fig. 3). As indicated by PXRD, the conversion of UTSA-74 to MOF-74 is quantitative and it is most likely caused by the atmospheric humidity due to the prolonged air exposure during the drying process. Indeed, the water-induced transformation of UTSA-74 to MOF-74 was also reported by Ameloot et al.48
 | ||
| Fig. 3 PXRD patterns of the dried materials obtained in DMF (top) and NMP (bottom), and the simulated PXRD patterns for MOF-7447 and HIMS-74. | ||
The formation of UTSA-74 in our work is likely facilitated by the modulating effect of the alkaloids or the DMF/H2O/EtOH solvent mixture used or perhaps a combination of both. To further test the role of the alkaloid during the crystallisation process of UTSA-74, we performed the synthesis in the absence of the alkaloid. In this case, however, no solid material was obtained. This result clearly demonstrates that the CN and CD alkaloids play a key role in the formation of the UTSA structure. Furthermore, using XYZ spatial descriptors, we calculated the length of the alkaloids as 11.8 × 9.0 × 7.2 Å3 for CD and 11.8 × 9.3 × 7.3 Å3 for CN. This indicates diffusion restrictions for the alkaloids within the 8.81 Å pores of UTSA-7449 (Fig. S1†), thereby explaining the absence of chirality in the isolated molecular structures.
Our results show that the unstable UTSA-74 phase forms prior to that of MOF-74 in the presence of Cinchona alkaloid modulators. Therefore, subsequent studies aimed at understanding the role of the solvent mixture in stabilising the structure. We hypothesised that the solvent mixture may also act as a modulator in our synthetic procedure, a phenomenon also frequently reported.15,50,51 Consequently, we replaced DMF by the structurally very different N-methylpyrrolidone (NMP), which is known to lead to internal shearing stress and formation of a chiral framework, in the case of MOF-5.52 We envisioned that the dobdc4– linker could exhibit a twisting behaviour upon interaction with the NMP molecules as observed for the benzene-1,4-dicarboxylate linker (bdc2−) in MOF-5. However, replacing DMF with NMP and using a molar ratio for Zn2+:dobdc4–:alkaloid of 3:1:3 led to a new structure, namely, [Zn2(dobdc)(NMP)]·⅓NMP (hereafter denoted as HIMS-74).
HIMS-74 crystallises in the monoclinic P21/a space group. Its asymmetric unit contains two crystallographically independent Zn2+ ions. One Zn2+ ion is tetracoordinated (Fig. 2, middle), being surrounded by two phenolate oxygens and two carboxylate oxygens, forming a distorted tetrahedral geometry. The Zn–O bond lengths vary between 1.926(3) and 1.970(3) Å. The second Zn2+ centre is pentacoordinated, with the coordination sphere defined by two carboxylate oxygens, two phenolate oxygens and the oxygen of the coordinated NMP solvent molecule. The NMP is oriented towards the channels of the framework and its position imposes a distorted square pyramidal geometry. In this case, the Zn–O bond length varies from 1.926(3) to 2.092(3) Å. The dobdc4– linkers are hexadentate coordinated, bridging the two independent Zn2+ ions via the phenoxo group (Fig. 2, middle), whilst the carboxylate groups are bridging Zn2+ ions of the same type.
The dinuclear Zn2(O)2(CO2)3 secondary building units (SBUs) consist of two edge-sharing polyhedra which are linked to each other via the carboxylate oxygen donors of the dobdc4– linkers to form a 3D network with 1D channels (Fig. 4). Notably, the molecular structure of HIMS-74 is built up from dinuclear SBUs as also observed in the case of UTSA-74 and therefore their 3D molecular structures are very similar (Fig. 4). The main difference is that the SBUs of HIMS-74 consist of a tetrahedron and a square pyramid whilst the UTSA-74 SBUs are composed of a tetrahedron and an octahedron. This reflects the critical role of NMP in stabilising the square pyramidal geometry for the Zn2+ ion as compared to DMF, which facilitates the stabilisation of the octahedral geometry.
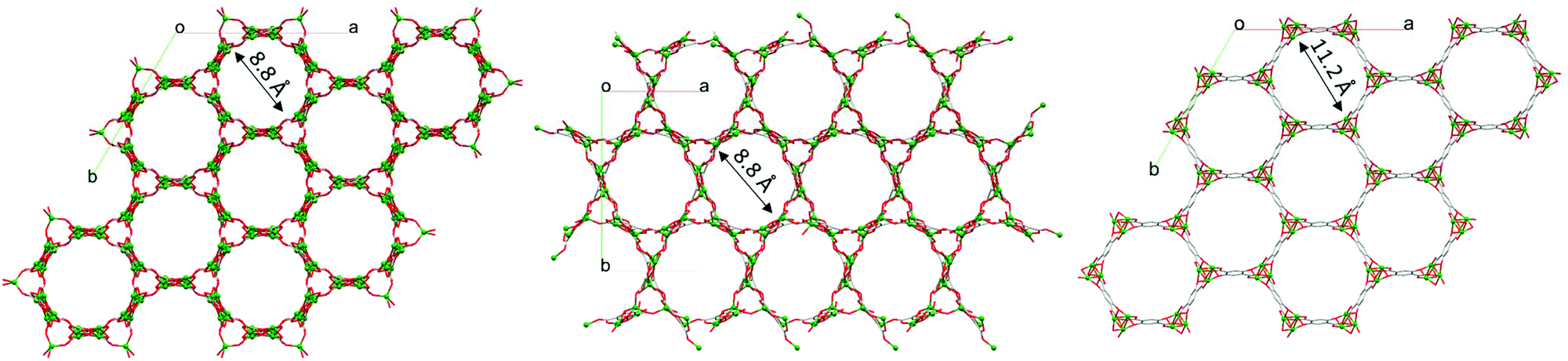 | ||
| Fig. 4 View of the 3D structures of UTSA-74 (left), HIMS-74 (middle) and MOF-74 (right) along the c axis. Hydrogen atoms, and guest and coordinated solvent molecules are omitted for clarity. | ||
The PXRD patterns (Fig. 3, bottom) of the crystalline solids isolated from NMP/H2O/EtOH solvent mixture show an excellent matching with the calculated pattern using the single-crystal XRD data of HIMS-74. To understand the role of the alkaloids in the synthesis of HIMS-74, we also performed the synthesis in their absence. Interestingly, the solid materials obtained in these reactions proved to be ZnO nanoparticles, as indicated by PXRD (Fig. S2†). Moreover, performing the synthesis using one equivalent of alkaloids does not facilitate the formation of the HIMS-74 structure. Therefore, we believe that both the alkaloids and the solvent mixture play key roles in the crystallization process of HIMS-74.
Our studies indicate that some weak interactions between the alkaloids and the SBUs of UTSA-74 and HIMS-74 may favour the formation of dinuclear SBUs as compared to the helical rods present in MOF-74. However, further investigations are necessary to determine the nature of these types of interactions. No induction of chirality using Cinchona alkaloids as modulators was observed. This suggests that the interaction of the alkaloid with the porous framework is weak at best. Under hydrothermal conditions, the alkaloid (pKa1 = 4.1 and pKa2 = 8.2 for CN and pKa1 = 4.0 and pKa2 = 8.2 for CD)53 is easily deprotonated and therefore can coordinate via its N and O donor atoms (amine and hydroxyl groups) to the Zn2+ ions.46 Thus, it is likely that the precursors of the HIMS-74 SBUs are doubly bridged chiral dinuclear units (see Fig. 5 for details) that prevent the formation of helical rods as observed for MOF-74. However, the increase in the basicity in the reaction mixture due to NMP decomposition causes the full deprotonation of the H4dobdc linker (pKa1 = 12.20 and pKa2 = 26.67).54 The hexadentate dobdc4– replaces the Cinchona alkaloid and forms HIMS-74 achiral SBUs (Fig. 6).
 | ||
| Fig. 5 Possible coordination of the Cinchona alkaloid to the Zn2+ ions which precedes the formation of HIMS-74 SBUs. | ||
 | ||
| Fig. 6 Role of the synthetic conditions in the formation of MOF-74 isomers. | ||
Phase transformations
As discussed above, the UTSA-74 isomer is not stable and converts to MOF-74 under ambient conditions or in the presence of water. In UTSA-74, there are two types of coordination polyhedra for Zn2+ ions, tetrahedron and octahedron, whilst both Zn2+ ions have an octahedral geometry in MOF-74. The SBUs of UTSA-74 consist of Zn2(O)2(CO2)4 units connected by chains of dobdc4– linkers.48 MOF-74 contains helical chains of edge-sharing ZnO6 octahedra that are interconnected via dobdc4– linkers.47 Ameloot et al.48 attributed the formation of the discrete SBUs in UTSA-74 to a stabilising effect of the DMSO solvent. However, Chen et al.5 synthesised UTSA-74 in DMF/H2O, a similar solvent mixture used for preparing MOF-74.47 Our studies show that both the solvent mixture (DMF/H2O/EtOH) and the Cinchona alkaloids control the formation of UTSA-74. All these results indicate that the formation of the UTSA-74 framework is very complex and depends on many more factors, including the anions present in the reaction mixture (e.g. CH3COO−)48 as well as the temperature and reaction time (e.g. 72 hours).48As proposed by Ameloot et al.,48 it is important to consider the potential formation of UTSA-74 in the nucleation and growth process of MOF-74. This implies to consider a sequential stepwise model, known as Ostwald's Rule of Stages,55 rather than a classical nucleation and growth model. A careful selection of the reaction parameters and an in-depth morphological analysis of the single crystals formed are required to understand such a complex phase transition. The Zn-UTSA-74 single crystals can be easily distinguished from those of Zn-MOF-74 by analysing their morphology using polarised light microscopy. Initial studies reported that the Zn-UTSA-74 crystallises as not only yellow rod crystals5 but also hexagonal48 crystals were observed. In situ PXRD and SEM analysis of the Zn-UTSA-74 to Zn-MOF-74 phase transitions have shown that the nucleation and growth of the Zn-MOF-74 occurs at the surface of hexagonally shaped UTSA-74 crystals.48 This indicates that the MOF-74 phase is thermodynamically more stable. The conversion of the Zn2+ dimers of UTSA-74 to the one-dimensional infinite Zn2+ chains in MOF-74 increases the number of Zn2+-linker bonds from four to five, giving a hydrolytically more stable coordination environment.48
HIMS-74 is a highly stable structure under ambient conditions and it does not undergo any structural changes. Attempts to replace the strongly coordinated NMP molecules have revealed that exposing HIMS-74 to polar organic solvents (e.g. CH3OH) leads to a loss in crystallinity and partial or complete transition to UTSA-74 is likely to occur (Fig. S3†). SEM analysis taken after immersion of HIMS-74 in CH3OH (6 days) indicates that the morphology is no longer homogeneous and a wide distribution of crystal sizes is obtained (Fig. S4†). TGA-DSC analysis on the recovered sample also showed that no NMP solvent was present (Fig. S5†); however, N2 adsorption studies indicated the lack of porosity in this material (Fig. S6†). The integrity of the HIMS-74 framework is retained in acetone (Fig. S3†); however, NMP is not replaced in the sample as shown in the TGA-DSC analysis (Fig. S5†). To further test the stability of HIMS-74 in water, the solid crystalline material was immersed in water for 20 h. PXRD and SEM studies indicate that HIMS-74 is converted to MOF-74 (Fig. S7 and S8†). These results show that HIMS-74 also undergoes a water-mediated isomerization to MOF-74 and possibly UTSA-74, but the HIMS-74 to MOF-74 phase transition is faster than that of HIMS-74 to UTSA-74 (Fig. S7†).
Unlike UTSA-74, HIMS-74 is stable upon separation from the mother liquor and drying in air, indicating that NMP has a stabilizing effect due to its strong coordination to the Zn2+ ions. Moreover, the close orientation of the coordinated NMP molecules towards the aromatic linkers in the single-crystal XRD analysis indicates a bonding via CH⋯π interactions (ca. 3.2 Å). One non-coordinated NMP molecule is also in close contact with the coordinated NMP and it interacts via moderate hydrogen bonding through the carbonyl O atoms, with a CH⋯O bond distance of 2.6–2.8 Å. Earlier studies52,56 also showed that the molecular size and the chemical nature of NMP give rise to structural transformation, e.g. chiral transformation in MOF-5.
MOF-74 is intensely studied for gas storage,57,58 molecular separations59 and catalysis applications.60,61 Notably, MOF-74 also shows a very high CO2 uptake due to the presence of three different CO2 adsorption sites.62 The first one is based on the electrostatic interaction between the open metal site and CO2, the second one is related to the van der Waals interaction of the CO2 with the host framework, and the third one is only related to the filling of the MOF channels. Although UTSA-74 has the same number of open metal sites per SBU as MOF-74, a smaller CO2 uptake was observed for UTSA-74 as compared to MOF-74.5 This was ascribed to the loose packing of the CO2 molecules in UTSA-74 (7.5 Å O⋯O) as compared to MOF-74 (3.6 Å O⋯O)5 and also to the slightly smaller pore size. The available open sites in HIMS-74 indicate two adsorption sites per SBU unit, similar to UTSA-74, and therefore similar CO2 uptake is expected. Nevertheless, we have not been able to verify it experimentally because the adsorption studies indicated that HIMS-74 is not porous (Fig. S9 and S10†). This is due to the unsuccessful removal of the coordinated NMP molecules without loss of sample crystallinity (Fig S11†) and it clearly demonstrates the key role of NMP in stabilizing the HIMS-74 framework.
Conclusions
This work demonstrates that the designed synthesis of MOFs remains a complex process and that the starting materials, synthesis solvents and the modulators play critical role in the stabilisation of various MOF structures. Specifically, we showed that using Cinchona alkaloids as modulators as well as the choice of solvent have direct implications for the crystallisation of different MOF-74 isomers. In addition to the previously known MOF-74 and UTSA-74 frameworks, a new HIMS-74 structure is formed when using NMP instead of DMF as coordinating solvent molecules. Unlike UTSA-74, the HIMS-74 isomer is more stable under ambient conditions and it undergoes a solvent-mediated isomerisation to MOF-74 only by immersing it in water. A comparative study of the adsorption properties of the three isomers was not possible because HIMS-74 is not stable upon solvent removal. This work highlights a new strategy to control framework formation when using Zn2+ nodes and dobdc4– linkers. It can also be applied to other MOF systems, thus affording the facile synthesis of structural isomers.Experimental
Materials and methods
All chemicals were purchased from commercial suppliers and used without further purification. The bulk synthesis of UTSA-74 used for the phase transition studies was performed following a reported procedure.48 The MOF-74 sample for SEM studies was prepared using a procedure from the literature.63:H2O:EtOH (20:1:1 v:v:v, 15 mL) were placed in a 20 mL Teflon screw-capped Duran™ Pyrex tube. The mixture was sonicated and then placed in a preheated oven and kept for 48 h at 120 °C. The reaction mixture was then cooled down to room temperature and the solid crystalline material was filtered and washed three times with the synthesis solvent. IR (KBr, cm−1): 3434 (m, b), 2959 (w), 2927 (w), 1656 (s, s), 1559 (s, s), 1511 (w), 1450 (m, s), 1410 (s, s), 1363 (w), 1307 (w), 1239 (m, s), 1196 (m, s), 1114 (w, s), 1063 (w), 910 (w), 879 (m, s), 815 (m, s), 676 (w), 635 (w), 578 (w, s).
:H2O:EtOH (20:1:1 v:v:v, 15 mL) were placed in a 20 mL Teflon screw-capped Duran™ Pyrex tube. The mixture was sonicated and then placed in a preheated oven and kept for 48 h at 120 °C. The reaction mixture was then cooled down to room temperature and the solid crystalline material was filtered and washed three times with NMP and one time with acetone. IR (KBr, cm−1): 3436 (m, b), 2958 (w), 2927 (w), 2873 (w), 1662 (m, s), 1646 (m, s), 1535 (s, s), 1469 (m, s), 1434 (m, s), 1410 (m, s), 1383 (m, b), 1304 (w, b), 1256 (m, s), 1234 (m, s), 1214 (m, s), 1119 (w, b), 1031 (w), 990 (w), 891 (m, s), 825 (m, s), 802 (m, s), 670 (w), 610 (m, s). C, H, N analysis (%): calcd for [Zn2(dobdc)(NMP)]·⅓NMP (Zn2C15.33H15.33N1.33O7.33) C 38.54, H 3.09, N 4.08; found C 38.72, H 3.54, N 4.14.
Physical characterisation
Powder X-ray diffraction (PXRD) patterns were recorded on a Rigaku MiniFlex II X-Ray diffractometer using Cu Kα radiation (λ = 1.5406 Å). The X-ray tube was operated at 30 kV and 15 mA. Measurements were carried out in an angle (2θ) range of 3–60° with a turning speed of 2° min−1. PDF card no. 2300112 was used as a reference for the PRXD of ZnO. Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed in the 35–800 °C range at 5 K min−1 using a NETZSCH Jupiter® STA 449F3 instrument. The measurements were carried out under a flow of argon (20 mL min−1) and protective argon (20 mL min−1). N2 and CO2 adsorption isotherms were measured on a Thermo Scientific Surfer instrument at 77 K and 273 K, respectively. The as-synthesised HIMS-74 was pre-treated in a vacuum with 1° min−1 heating from room temperature to 150 °C and a hold of 2 h. The methanol exchanged HIMS-74 sample was heated under vacuum from room temperature to 80 °C with a ramp of 2° min−1 and a hold of 1 h, then heating to 120 °C with 4° min−1 and 1 h hold, and lastly to 220 °C with 2° min−1 and a final hold of 12 h. Scanning electron micrographs were obtained using a field emission scanning electron microscope FEI-Verios 460. The samples were previously sputtered with a 20 mm thick Au layer using a Leica EM ACE600 Double sputter coater.Single-crystal X-ray diffraction (SCXRD) data of HIMS-74 and UTSA-74 analogue were collected at 100(2) K in the BL13-XALOC beamline64 at the ALBA synchrotron, on a single-axis goniometer with a Pilatus 6 M detector using a monochromatic X-ray beam (λ = 0.72656 Å). The data frames were integrated and scaled using the XDS software package.65 Absorption correction was not applied. The structure was solved by direct methods and subsequently refined by correction of F2 against all reflections, using SHELXT2013 and SHELXL2013 within the WinGX package.66–68 Both structures contain some disordered molecules. Attempts to adequately model the disordered molecules were unsatisfactory; therefore, the PLATON/SQUEEZE routine was applied to mask out the disordered electron density.69
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Research Priority Area Sustainable Chemistry of the University of Amsterdam, http://suschem.uva.nl, and was funded by a TOP-PUNT grant from the NWO. It was also supported by Spanish MINECO (projects PN RTI2018-095622-B-I00), the Catalan AGAUR (project 2014 SGR 80), the ERC under the EU-FP7 (ERC-Co 615954), and the CERCA Program/Generalitat de Catalunya. ICN2 was supported by the Severo Ochoa program from Spanish MINECO (Grant No. SEV-2017-0706). The authors would like to thank Thierry Slot for his assistance in SEM measurements.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Z. Bao, L. Yu, Q. Ren, X. Lu and S. Deng, J. Colloid Interface Sci., 2011, 353, 549–556 CrossRef CAS PubMed.
- A. K. Adhikari and K.-S. Lin, Chem. Eng. J., 2016, 284, 1348–1360 CrossRef CAS.
- K. Sillar, A. Hofmann and J. Sauer, J. Am. Chem. Soc., 2009, 131, 4143–4150 CrossRef CAS PubMed.
- F. Luo, C. Yan, L. Dang, R. Krishna, W. Zhou, H. Wu, X. Dong, Y. Han, T.-L. Hu, M. O'Keeffe, L. Wang, M. Luo, R.-B. Lin and B. Chen, J. Am. Chem. Soc., 2016, 138, 5678–5684 CrossRef CAS PubMed.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- X.-Z. Song, Y.-L. Meng, Z. Tan, L. Qiao, T. Huang and X.-F. Wang, Inorg. Chem., 2017, 56, 13646–13650 CrossRef CAS PubMed.
- N. Bhardwaj, S. K. Bhardwaj, J. Mehta, K.-H. Kim and A. Deep, ACS Appl. Mater. Interfaces, 2017, 9, 33589–33598 CrossRef CAS.
- P. Freund, I. Senkovska and S. Kaskel, ACS Appl. Mater. Interfaces, 2017, 9, 43782–43789 CrossRef CAS PubMed.
- A. W. Stubbs, L. Braglia, E. Borfecchia, R. J. Meyer, Y. Román-Leshkov, C. Lamberti and M. Dincă, ACS Catal., 2018, 8, 596–601 CrossRef CAS.
- P. Deria, D. A. Gómez-Gualdrón, I. Hod, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14449–14457 CrossRef CAS PubMed.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- L. Liu, T.-Y. Zhou and S. G. Telfer, J. Am. Chem. Soc., 2017, 139, 13936–13943 CrossRef CAS PubMed.
- R. Seetharaj, P. V. Vandana, P. Arya and S. Mathew, Arabian J. Chem., 2016, 12, 295–315 CrossRef.
- Z. Hu, I. Castano, S. Wang, Y. Wang, Y. Peng, Y. Qian, C. Chi, X. Wang and D. Zhao, Cryst. Growth Des., 2016, 16, 2295–2301 CrossRef CAS.
- A. M. Bumstead, D. B. Cordes, D. M. Dawson, K. K. Chakarova, M. Y. Mihaylov, C. L. Hobday, T. Düren, K. I. Hadjiivanov, A. M. Z. Slawin, S. E. Ashbrook, R. R. R. Prasad and P. A. Wright, Chem. – Eur. J., 2018, 24, 6115–6126 CrossRef CAS PubMed.
- E. Bagherzadeh, S. M. Zebarjad and H. R. M. Hosseini, Eur. J. Inorg. Chem., 2018, 2018, 1909–1915 CrossRef CAS.
- V. Bon, I. Senkovska, I. A. Baburin and S. Kaskel, Cryst. Growth Des., 2013, 13, 1231–1237 CrossRef CAS.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., 2009, 121, 4833–4837 CrossRef.
- A. Schaate, P. Roy, T. Preuße, S. J. Lohmeier, A. Godt and P. Behrens, Chem. – Eur. J., 2011, 17, 9320–9325 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- R. J. Marshall, C. L. Hobday, C. F. Murphie, S. L. Griffin, C. A. Morrison, S. A. Moggach and R. S. Forgan, J. Mater. Chem. A, 2016, 4, 6955–6963 RSC.
- O. V. Gutov, S. Molina, E. C. Escudero-Adán and A. Shafir, Chem. – Eur. J., 2016, 22, 13582–13587 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- S. Diring, S. Furukawa, Y. Takashima, T. Tsuruoka and S. Kitagawa, Chem. Mater., 2010, 22, 4531–4538 CrossRef CAS.
- G. Zahn, P. Zerner, J. Lippke, F. L. Kempf, S. Lilienthal, C. A. Schröder, A. M. Schneider and P. Behrens, CrystEngComm, 2014, 16, 9198–9207 RSC.
- V. Bon, I. Senkovska, M. S. Weiss and S. Kaskel, CrystEngComm, 2013, 15, 9572–9577 RSC.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC.
- J. M. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Chem. Mater., 2015, 27, 2286–2289 CrossRef CAS.
- O. V. Gutov, M. G. Hevia, E. C. Escudero-Adán and A. Shafir, Inorg. Chem., 2015, 54, 8396–8400 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans., 2016, 45, 4090–4099 RSC.
- M. H. Rosnes, M. Opitz, M. Frontzek, W. Lohstroh, J. P. Embs, P. A. Georgiev and P. D. C. Dietzel, J. Mater. Chem. A, 2015, 3, 4827–4839 RSC.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2009, 131, 4995–5000 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- J. Liu, A. I. Benin, A. M. B. Furtado, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2011, 27, 11451–11456 CrossRef CAS PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS.
- G. Srinivas, J. Ford, W. Zhou and T. Yildirim, Int. J. Hydrogen Energy, 2012, 37, 3633–3638 CrossRef CAS.
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Int. J. Hydrogen Energy, 2011, 36, 10834–10844 CrossRef CAS.
- W. Yan, Z. Guo, H. Xu, Y. Lou, J. Chen and Q. Li, Mater. Chem. Front., 2017, 1, 1324–1330 RSC.
- J. Zhang, S. Chen, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2008, 130, 12882–12883 CrossRef CAS PubMed.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- T. Kaczorowski, I. Justyniak, T. Lipińska, J. Lipkowski and J. Lewiński, J. Am. Chem. Soc., 2009, 131, 5393–5395 CrossRef CAS.
- J. Lewiński, T. Kaczorowski, I. Justyniak and D. Prochowicz, Chem. Commun., 2010, 47, 950–952 RSC.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- B. Bueken, H. Reinsch, N. Heidenreich, A. Vandekerkhove, F. Vermoortele, C. E. A. Kirschhock, N. Stock, D. D. Vos and R. Ameloot, CrystEngComm, 2017, 19, 4152–4156 RSC.
- D. Dubbeldam, S. Calero and T. J. H. Vlugt, Mol. Simul., 2018, 44, 653–676 CrossRef CAS.
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
- X. Zhou, P. Liu, W.-H. Huang, M. Kang, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2013, 15, 8125–8132 RSC.
- J. D. Evans and F.-X. Coudert, J. Am. Chem. Soc., 2016, 138, 6131–6134 CrossRef CAS PubMed.
- Clarke's Analysis of Drugs and Poisons, ed. A. C. Moffat, M. D. Osselton, B. Widdop and J. Watts, Pharmaceutical Press, London, UK, 4th edn, 2011 Search PubMed.
- P. Lu, Y. Wu, H. Kang, H. Wei, H. Liu and M. Fang, J. Mater. Chem. A, 2014, 2, 16250–16267 RSC.
- M. J. Van Vleet, T. Weng, X. Li and J. R. Schmidt, Chem. Rev., 2018, 118, 3681–3721 CrossRef CAS.
- S.-Y. Zhang, D. Li, D. Guo, H. Zhang, W. Shi, P. Cheng, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2015, 137, 15406–15409 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- Y. Liu, H. Kabbour, C. M. Brown, D. A. Neumann and C. C. Ahn, Langmuir, 2008, 24, 4772–4777 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- G. Calleja, R. Sanz, G. Orcajo, D. Briones, P. Leo and F. Martínez, Catal. Today, 2014, 227, 130–137 CrossRef CAS.
- D. Ruano, M. Díaz-García, A. Alfayate and M. Sánchez-Sánchez, ChemCatChem, 2015, 7, 674–681 CrossRef CAS.
- W. L. Queen, M. R. Hudson, E. D. Bloch, J. A. Mason, M. I. Gonzalez, J. S. Lee, D. Gygi, J. D. Howe, K. Lee, T. A. Darwish, M. James, V. K. Peterson, S. J. Teat, B. Smit, J. B. Neaton, J. R. Long and C. M. Brown, Chem. Sci., 2014, 5, 4569–4581 RSC.
- L. Garzón-Tovar, A. Carné-Sánchez, C. Carbonell, I. Imaz and D. Maspoch, J. Mater. Chem. A, 2015, 3, 20819–20826 RSC.
- J. Juanhuix, F. Gil-Ortiz, G. Cuní, C. Colldelram, J. Nicolás, J. Lidón, E. Boter, C. Ruget, S. Ferrer and J. Benach, J. Synchrotron Radiat., 2014, 21, 679–689 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- G. M. Sheldrick, Z. Dauter, K. S. Wilson, H. Hope and L. C. Sieker, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1993, 49, 18–23 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Pore size distributions, TGA/DSC, PXRD, SEM, adsorption isotherms, crystallographic table. CCDC 1884101 and 1884102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01572h |
| This journal is © The Royal Society of Chemistry 2019 |