
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMono- and bimetallic amidinate samarium complexes – synthesis, structure, and hydroamination catalysis†
Neda
Kazeminejad
,
Luca
Münzfeld
,
Michael T.
Gamer
and
Peter W.
Roesky
 *
*
Institut für Anorganische Chemie, Karlsruher Institut für Technologie (KIT), Engesserstr. 15, Geb. 30.45, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 9th May 2019
Abstract
In order to investigate the difference between mono- and bimetallic systems in the catalytic hydroamination/cyclization reaction two mono- and bimetallic amidinate samarium catalysts, featuring comparable coordination environments, were synthesized. Both systems comprise two {N(SiMe3)2}− leaving groups to minimize the steric influence of the corresponding amidinate ligand. The bimetallic system is based on a bis(amidinate) 4,6-dibenzofuran derivative, while N,N′-bis(2,6-diisopropylphenyl)benzamidinate was employed as ligand for the monometallic catalyst. For the hydroamination/cyclization reaction five different substrates were investigated. Additionally, kinetic studies were carried out to gain deeper understanding of the mechanism.
Introduction
Amidinates and the closely related guanidinates are a very well established class of ligands, which have been widely used in coordination chemistry.1–5 In general, amidinates [RC(NR′)2]− are monoanionic nitrogen-donor ligands, which can be easily accessed by different synthetic routes. Moreover, the steric and electronic properties of amidinates can be tuned by adapting the substituents R and R′.In lanthanide chemistry, amidinates were introduced about three decades ago by Edelmann et al.6–8 Ever since they have emerged as versatile ligands for the synthesis of both di- and trivalent lanthanide complexes.7–13 Some of these complexes have also been used for different applications such as homogeneous catalysis, or as precursors for atomic layer deposition (ALD) and metalorganic chemical vapor deposition (MOCVD).7,8,11,14–30
Nevertheless bis(amidinates), which were introduced into rare-earth chemistry by Hessen, Teuben et al. about two decades ago, are less common.31 Such ligand systems basically contain two amidinate functions, that are linked by a flexible or rigid organic spacer. Initially Teuben used these ligands for the synthesis of monometallic compounds, whereas nowadays linked bis(amidinates) are additionally applied in the synthesis of bimetallic complexes. Trifonov, Shen and others employed bis(amidinates) linked by e.g. 1,3-diaminopropane,32–35o-, m- and p-phenylene,36–40 pyridinediyl,39,40 1,4-cyclohexene,36 naphthalene41–44 or propyl45 for the synthesis of mono- and bimetallic rare-earth complexes.
Aside from f-element chemistry Hagadorn et al. established dibenzofuran and phenanthrene linked bis(amindinates) for aluminium, titanium, and zirconium based complexes.46–49
Some of the bimetallic complexes ligated by linked bis(amidinates) have been employed as catalysts.34,36,42,45 In general, bimetallic catalysts with both metals in well-defined distance to each other can feature cooperative substrate activation, a highly interesting property for the application in efficient catalytic transformations.50–53 On the other hand, it is very challenging to obtain similar bimetallic catalysts together with their monometallic reference systems to study these effects in detail.
Recently, we introduced bis(amidinate) ligands linked by rigid dibenzofuran and phenanthrene backbones into lanthanide chemistry for the first time. The design of this ligand scaffold was inspired by the work of Hagadorn et al. mentioned above.46–49 Depending on the ligand scaffold and the lanthanide source, either monometallic complexes or bi- and tetra-metallic macrocycles were obtained.54 The mono- and bi-metallic systems (A and B) shown in Scheme 1 were used as catalysts in the intramolecular hydroamination reaction and their reactivity (mono vs. dinuclear) was compared. Depending on the nuclearity of the catalyst a difference in kinetics was observed.
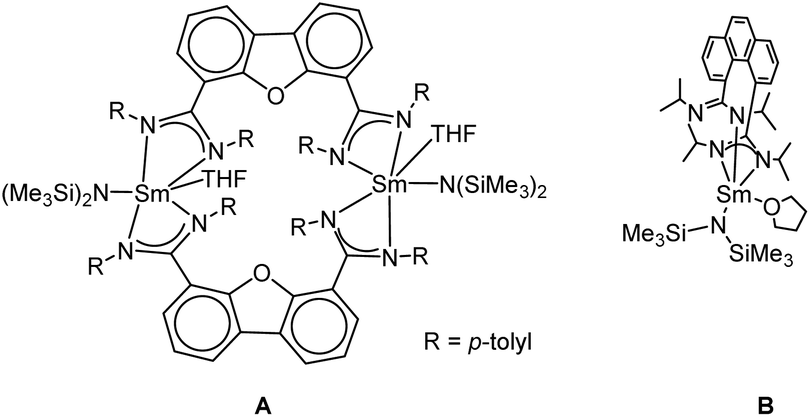 | ||
| Scheme 1 Recently reported mono and bimetallic catalyst for the intramolecular hydroamination reaction.54 | ||
Both systems shown in Scheme 1 feature one, respectively two samarium atoms ligated by two amidinate units and one {N(SiMe3)2}− leaving group.54 Herein, we now report on related but more open systems, in which the metal is ligated by one amidinate unit only. Thus, two {N(SiMe3)2}− leaving groups can be bound to the central metal ion. For a reasonable comparison in the intramolecular hydroamination catalysis, we attempted to design the ligand framework of the mono- and the linked bis(amidinates) as closely related as possible.
Results and discussion
Ligands
Both systems presented in here are literature known. For the synthesis of the bimetallic complexes a bis(amidinate) ligand based on a dibenzofuran backbone, iPrLDBFH2 (Scheme 2), was applied. Following a procedure reported by Hagadorn et al.,49 dibenzofuran was deprotonated with a slight excess of n-butyllithium to give 4,6-dilithiodibenzofuran. Further reaction with di-isopropylcarbodiimid (DIC) and hydrolyzation resulted in the proligand iPrLDBFH2 (Scheme 2). The proligand was characterized by NMR and IR spectroscopy. The data is in agreement with the literature.49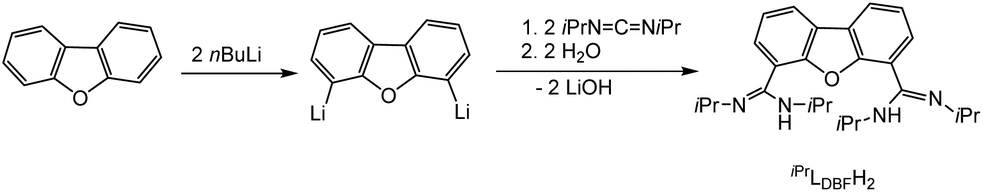 | ||
| Scheme 2 Synthesis of iPrLDBFH2.49 | ||
For the monometallic complex the proligand N,N′-bis(2,6-diisopropylphenyl)benzamidine (DippLPhH) was prepared analogue to the literature in a one-pot reaction via the so-called carbodiimide route.55 Phenyl lithium was reacted with bis(2,6-diisopropylphenyl)carbodiimide in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio to give the corresponding lithium salt Li[DippLPh]. After hydrolysis, the amidine DippLPhH was obtained (Scheme 3). The NMR data correspond to those in the literature.55
:1 molar ratio to give the corresponding lithium salt Li[DippLPh]. After hydrolysis, the amidine DippLPhH was obtained (Scheme 3). The NMR data correspond to those in the literature.55
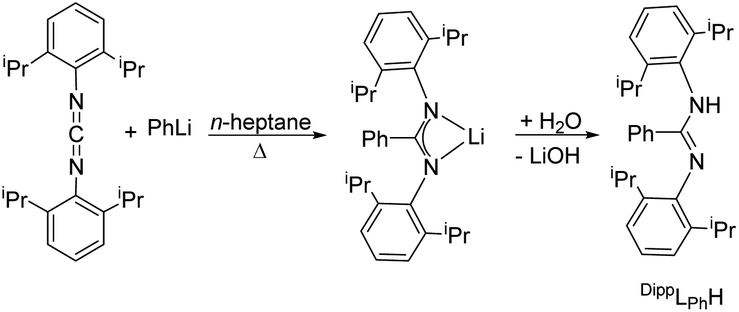 | ||
| Scheme 3 Synthesis of DippLPhH.55 | ||
Metal complexes
The synthesis of the desired bimetallic complex [Sm2(iPrLDBF){N(SiMe3)2}4] (1) was achieved by an amine elimination reaction. The neutral proligand iPrLDBFH2 was treated in refluxing toluene with the homoleptic amido complex [Sm{N(SiMe3)2}3] in a 1:1 stoichiometric ratio for three days (Scheme 4). After workup, compound 1 was crystallised from hot toluene to give single crystals suitable for X-Ray diffraction.
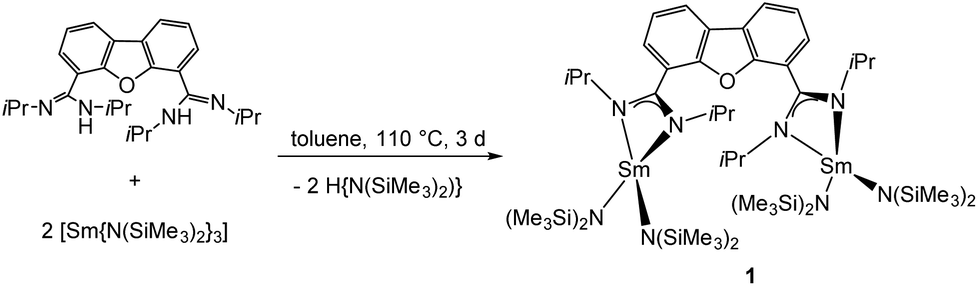 | ||
| Scheme 4 Synthesis of 1. | ||
Compound 1 crystallizes in the orthorhombic space group I222, with four molecules in the unit cell (Fig. 1). The crystals showed signs of slight twinning but no applicable twin law could be found for the final refinement. As expected, a bimetallic complex, in which two {Sm{N(SiMe3)2}2} fragments coordinate to each amidinate function, was obtained. A crystallographic C2 axis along the center of the furan ring is observed. The amidinate function coordinates in a slightly asymmetric fashion in a κ2-(N,N′) mode to the metal atoms (Sm–N1 2.37(2) Å and Sm–N2 2.42(2) Å). The observed bond distances are somewhat shorter than those in A (2.448 Å).54 In total the samarium atoms are four-fold coordinated in a distorted tetrahedral fashion by two nitrogen atoms of the amidinate function and two nitrogen atoms from the {N(SiMe3)2}− groups. The metal-to-metal distance in 1 is 8.84 Å and thus 0.6 Å longer than the metal-to-metal distance in A.
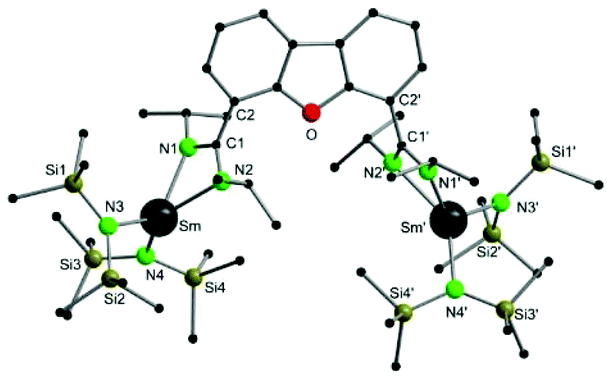 | ||
| Fig. 1 Molecular structure of 1 in the solid-state. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å], angles [°]: Sm–Sm′ 8.884, Sm–N1 2.37(2), Sm–N2 2.42 (2), Sm–N3 2.282(14), Sm–N4 2.30 (2), Si1–N3 1.73(2), Si2–N3 1.70(2), Si3–N4 1.68(2), Si4–N4 1.71(2), N1–C1 1.35(2), N2–C1 1.29(2); N1–Sm–N2 55.4(6), N2–C1–N1 115.0(2), N3–Sm–Si4 136.0(5). | ||
Compound 1 was also characterized by 1H NMR, 13C{1H} NMR, IR spectroscopy and elemental analysis. The NMR spectra were measured at room temperature in THF for a better resolution. However, due to the paramagnetic nature of the samarium ions some line broadening is observed (Fig. S3 and S4†).
For accessing a mononuclear complex ligated by only one amidinate ligand, a ligand with a high steric demand of the substituent on the nitrogen atom is needed. Otherwise, product mixtures with a metal to ligand ratio ranging from 1:1 up to 1:3, are obtained, which are difficult to separate.
The desired complex [Sm(DippLPh){N(SiMe3)2}2] (2) was obtained in a similar synthetic protocol as described above for 1. Reaction of DippLPhH with a slight excess of [Sm{N(SiMe3)2}3] in refluxing THF resulted in an amine elimination reaction, which gave the monometallic complex 2 in good yields. Single crystals were obtained after recrystallization from hot THF (Scheme 5).
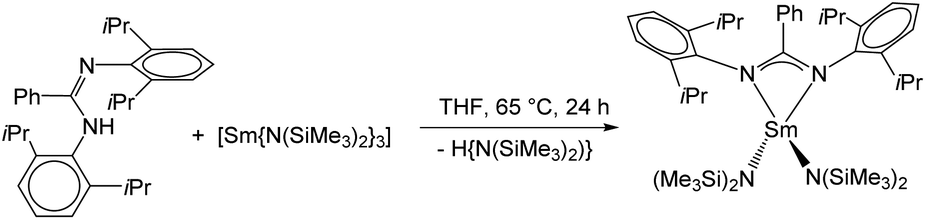 | ||
| Scheme 5 Synthesis of 2. | ||
Compound 2 was fully characterized by common analytic techniques and its solid-state structure was established by single crystal X-ray diffraction (Fig. 2). As described above for compound 1, significant line broadening and shifting of resonances is observed due to the paramagnetic character of the samarium ion (Fig. S5†).
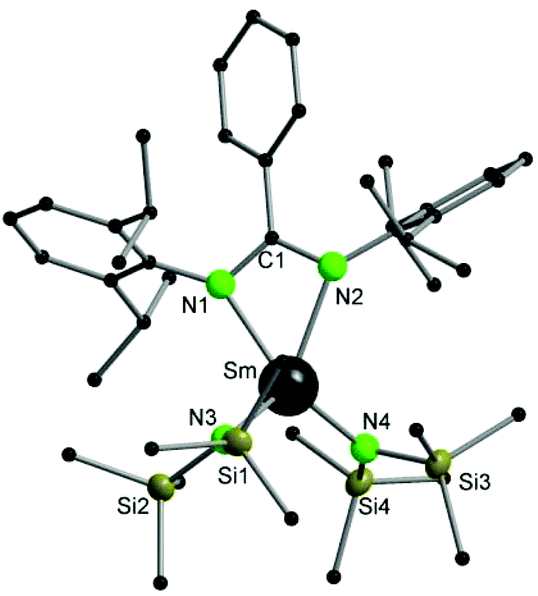 | ||
| Fig. 2 Molecular structure of 2 in the solid-state. Hydrogen atoms and solvent molecules are omitted for clarity. Selected bond lengths [Å], angles [°]: Sm–N1 2.421(3), Sm–N2 2.415(3), Sm–N3 2.306(3), Sm–N4 2.297(3), Si1–N3 1.727(3), Si2–N3 1.705(4), Si3–N4 1.715(3), Si4–N4 1.728(4), N1–C1 1.330(5), N2–C1 1.354(5); N1–Sm–N2 55.56(11), N3–Sm–N4 121.49(13), N1–C1–N2 114.2(4). | ||
Compound 2 crystallizes in the monoclinic space group P21/n with four molecules of 2 and four molecules of THF in the unit cell. The amidinate group coordinates to the metal in a syn-conformation with a κ2-(N,N′) mode, whereas the Sm–N1/2 bond distances are 2.421(3) Å and 2.415(3) Å, respectively. As observed in 1, the samarium atom is four fold coordinated in a distorted tetrahedral fashion by two nitrogen atoms of the amidinate function and two nitrogen atoms from the {N(SiMe3)2}− groups. The Sm–N bond distances to the {N(SiMe3)2}− groups are Sm–N3 2.306(3) Å and Sm–N4 2.297(3) Å.
Due to the similar binding modes of the ligands and coordination polyhedrons observed in compounds 1 and 2 as well as utilization of the same leaving groups ({N(SiMe3)2}−), we consider compounds 1 and 2 as a suitable couple to compare mono- and bimetallic complexes in hydroamination catalysis.
Catalytic hydroamination cyclization reaction
Hydroamination is the addition of ammonia or an organic amine nitrogen–hydrogen bond to a carbon–carbon double or triple bond in one step. The advantage of this synthetic route is the straightforward access to amines without any side-products. In contrast, most of the classical amine synthesis require multistep reactions and are accompanied by the formation of side-products. Although the catalytic hydroamination reaction is thermodynamically feasible under normal conditions, the high activation barrier hampers its use in synthesis. Since the pioneering work of Marks et al.56–60 in the early 1990ies a large number of homogeneous catalysts for the hydroamination reaction have been established. About two decades ago Livinghouse et al. and our group could show that homoleptic lanthanide amides [Ln{N(SiMe3)2}3] are active as catalysts in the catalytic hydroamination reaction but co-ligands are beneficial in many cases.61,62 The progress in this area over the last decade has been reviewed extensively.56,63–89 On the other hand bimetallic catalysis in hydroamination reactions was not investigated in detail.In our previous contribution, we reported on the hydroamination cyclization catalyzed by compounds A and B.54 In the intramolecular hydroamination reactions, both of the complexes give excellent yields. It was demonstrated that the monometallic complex B shows faster conversion and different kinetics than the bimetallic system A, e.g. for the formation of the five-membered rings Ib and IIb (Table 1), zero-order kinetics with respect to the substrate concentration were observed for the bimetallic catalyst A, while first order kinetics with respect to the substrate concentration were determined for the monometallic catalyst B.
| Ent. | Substrate | Yield | Kat. | T [°C] | t [min] | Conv. [%] |
|---|---|---|---|---|---|---|
| Conditions: Complex 1: (10 mg, 2 mol%); complex 2: (10 mg, 4 mol%), C6D6, calculated by 1H NMR spectroscopy, using ferrocene as the internal standard. | ||||||
| 1 |
|
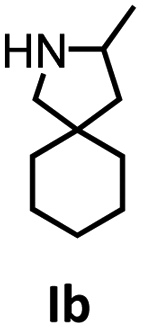
|
1 | 25 | 18 | Quant. |
| 2 | 2 | 25 | 8 | Quant. | ||
| 3 |
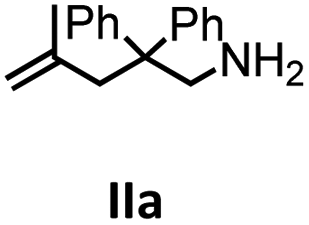
|
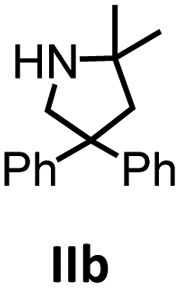
|
1 | 25 | 34 | Quant. |
| 4 | 2 | 25 | 5 | Quant. | ||
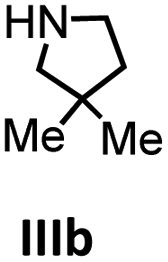
| 5 |
|
|
1 | 35 | 137 | Quant. |
| 6 | 2 | 35 | 49 | Quant. | ||
| 7 |
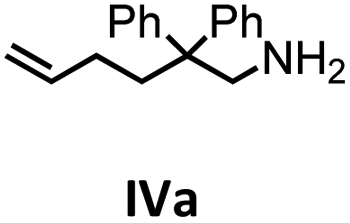
|
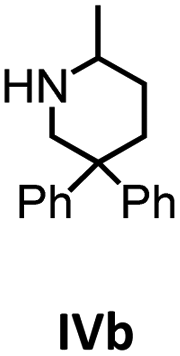
|
1 | 50 | 104 | 98 |
| 8 | 1 | 60 | 43 | 98 | ||
| 9 | 2 | 50 | 15 | 98 | ||
| 10 | 2 | 60 | 10 | Quant. | ||
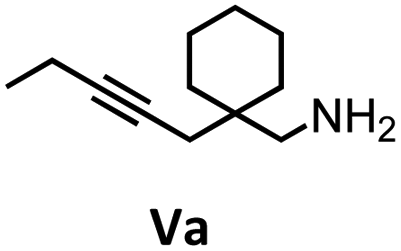
| 11 |
|
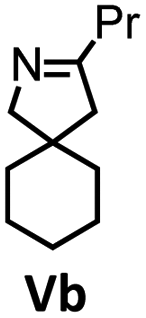
|
1 | 25 | 36 | Quant. |
| 12 | 2 | 25 | 19 | Quant. | ||
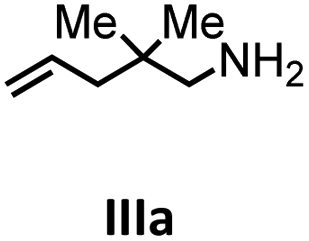
Herein, we compare now the more open bi- and monometallic catalysts 1 and 2, which both have two {N(SiMe3)2}− leaving groups. The catalytic hydroamination experiments were carried out under rigorous anaerobic conditions in C6D6 at different temperatures with catalyst loadings of 2 mol% for the bimetallic system 1 and 4 mol% for the monometallic catalyst 2, in order to have the same substrate to metal ratio. The conversion was followed by 1H NMR spectroscopy using ferrocene as an internal standard. To investigate the substrate dependency five different substrates were employed, of which four are amino alkenes (Table 1, Ia–IVa) and one is an amino alkyne (Table 1, Va).
All substrates were converted in (almost) quantitative yields, as shown in Table 1. The differences in reactivity of the substrates can be explained by the Thorpe–Ingold effect, which means that bulky substituents at the β-position to the amino group favor the cyclization.90–95
In general, compared to the bis(amidinate) complexes A and B, compounds 1 and 2 show faster conversions for all substrates depicted in Table 1. Obviously the more open coordination sphere of the mono(amidinate) complexes 1 and 2 is beneficial. In addition it was shown by Rodríguez et al. that a lutetium catalyst with two leaving groups can bind two cyclized substrate molecules simultaneously.96 In general, the monometallic catalyst 2 shows faster conversions than the bimetallic system 1. We do not have a straightforward explanation for this observation. However, since four substrate molecules can bind to the samarium ions in 1, there may be a diffusion limit to reach this concentration. In this case separating the metals into two catalytic active molecules may be an advantage.
To get some understanding of the mechanism, kinetic studies of the hydroamination cyclization of the aminoalkenes IIIa and IVa by using 1 and 2 as catalyst were carried out. All reactions were monitored in situ by 1H NMR spectroscopy at different temperatures. The full data is shown in the ESI.† Substrate IIIa was reacted at 35 °C and 60 °C by using 1 as catalyst. Since 2 shows a higher catalytic activity, the reactions were monitored at 15 °C, 25 °C, and 35 °C. In each case, a linear dependency between ln[substrate] vs. time was observed indicating that the reaction is first order in substrate concentration (see Fig. 3 and S9–S13†).
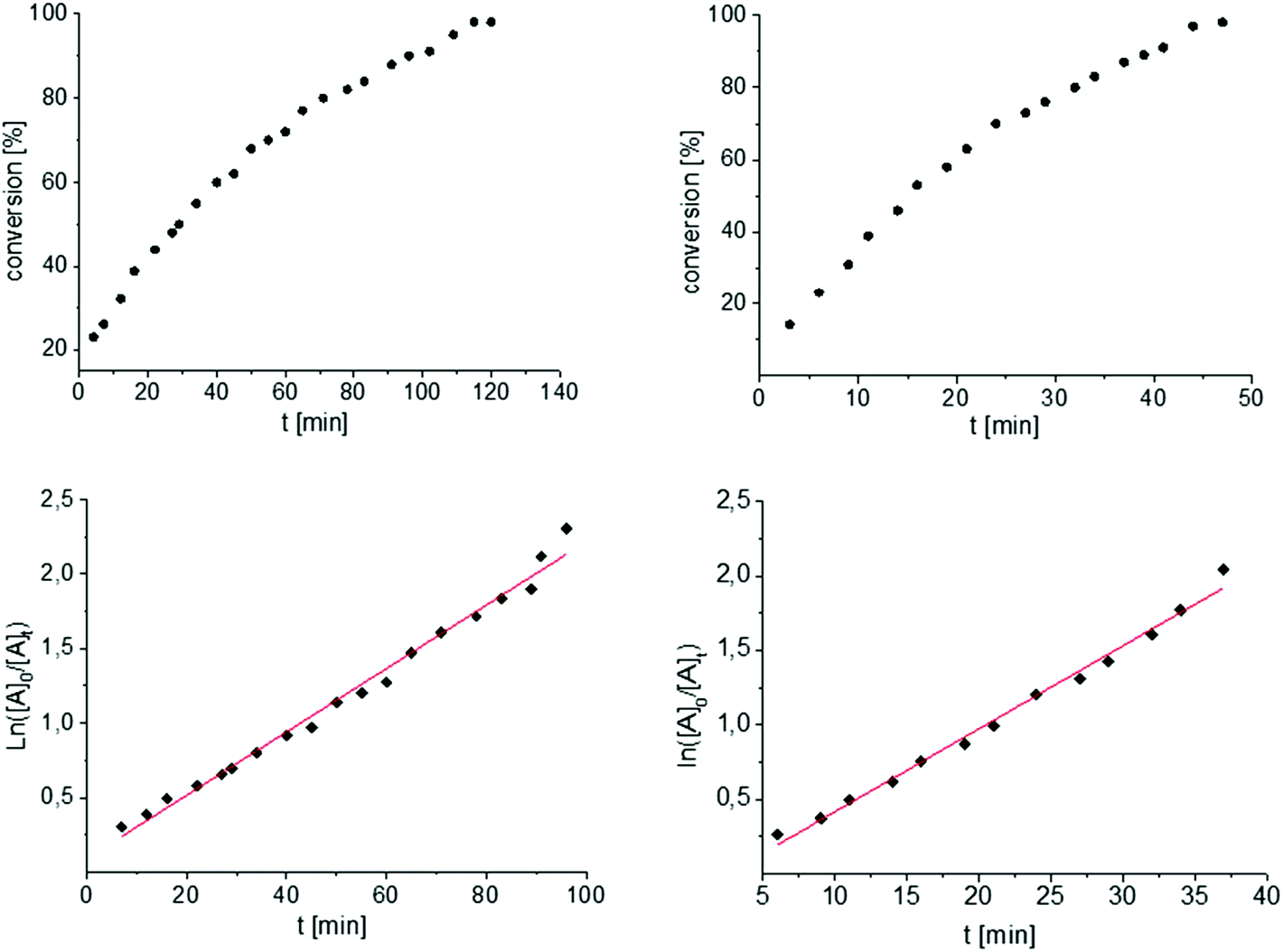 | ||
| Fig. 3 Conversion of IIIa with 1 (left) and 2 (right) at 35 °C. First order kinetics in respect to the substrate concentration for both reactions were observed (Table 1, entries 5 and 6, see also Fig. S9 and S13†). For the determination of the kinetic the final phase (from about 90% conversion) was cut off. | ||
In contrast to the previously reported catalysts A and B neither different kinetics for the same substrate nor any temperature dependency of the kinetic order were observed by comparing a mono vs. a bimetallic system.
In addition, the cyclization leading to a six-membered ring was investigated in detail. Substrate IVa was reacted at 40, 50, and 60 °C by using 1 as catalyst and at 30, 35, and 50 °C with the faster system 2 as catalyst (Fig. 4 and S14–S29†). In each case, a zero order kinetics in substrate concentration was determined.
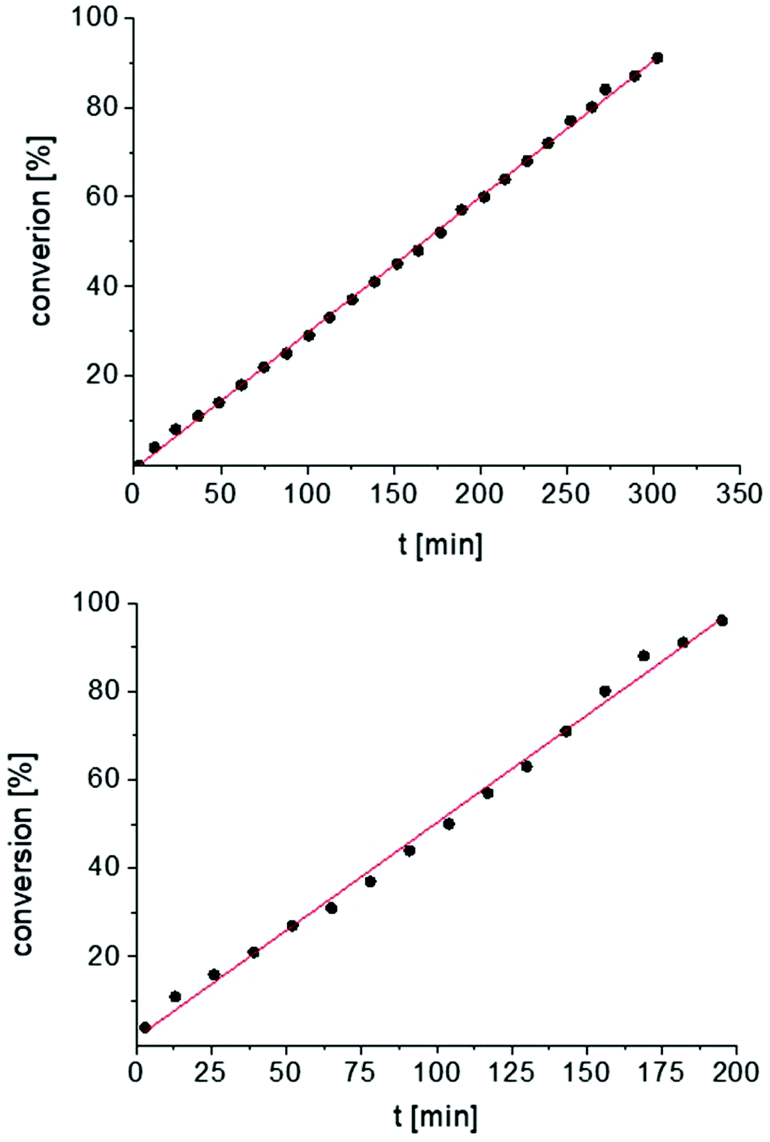 | ||
| Fig. 4 Conversion of IVa with 1 at 40 °C (top) and 2 at 30 °C (bottom). Zero order kinetics with respect to the substrate concentration for both reactions were observed (see also Fig. S14 and S17†). | ||
In contrast to catalysts A and B, compounds 1 and 2 always show the same kinetic order for the same substrate. This may be an effect of the more remote metal centers in the bimetallic catalyst 1, which means that both catalytic centers operate independently of each other.
Summary
In summary, we have prepared a mono and a bimetallic complex with one amidinate ligand and two {N(SiMe3)2}− leaving groups. Both systems show similar kinetics for the conversions of aminoalkenes to five and six membered rings. Although both systems have a similar coordination sphere, the monometallic system is the more efficient catalyst. Since it is known that both leaving groups of the metal can be substituted by two substrates, up to four substrate molecules can bind to the bimetallic system. We suggest that the lower performance of the bimetallic catalyst may be caused by diffusion control of the substrate coordination.Experimental97
General procedures
All air- and water-sensitive materials were prepared under an argon or a nitrogen atmosphere on a Schlenk line or in a glovebox. THF was distilled from potassium metal under nitrogen before use. Toluene, n-heptane and n-pentane were dried using an MBraun solvent purification system (SPS-800). Deuterated solvents were purchased from Eurisotop (99 atom% D) and were dried and stored under vacuum with Na/K alloy. All other chemicals were purchased and used without further purification. NMR spectra were recorded on a Bruker Avance II 300 MHz or Avance III 400 MHz NMR spectrometer. Elemental analyses were carried out on an Elementar Vario Micro Cube. IR spectra were performed on a Bruker TENSOR 37 spectrometer via the attenuated total reflection method (ATR). [Sm{N(SiMe3)2}3] was prepared using a modified procedure of Bradley et al.98 We found, that using 2.90 equivalents of K{N(SiMe3)2} instead of 3 equivalents Li{N(SiMe3)2} gave good yields without the formation of ‘ate’ complexes or the need for additional purification.54iPrLDBFH2 was prepared according to a literature procedure.49X-Ray crystallographic studies of 1–2
A suitable crystal was covered in mineral oil (Aldrich) and mounted on a glass fiber or a mylar loop. The crystal was transferred directly to the cold stream of a STOE IPDS 2 diffractometer.All structures were solved by using the program SHELXS/T102,103 using Olex2.104 The remaining non-hydrogen atoms were located from successive difference Fourier map calculations. The refinements were carried out by using full-matrix least-squares techniques on F2 by using the program SHELXL.102,103 In each case, the locations of the largest peaks in the final difference Fourier map calculations, as well as the magnitude of the residual electron densities, were of no chemical significance. Positional parameters, hydrogen atom parameters, thermal parameters, bond distances and angles have been deposited as ESI.†
973 reflections measured, 9097 unique (Rint = 0.0945) which were used in all calculations. The final wR2 was 0.2681 (all data) and R1 was 0.0984 (I > 2(I)).
921 reflections measured, 10159 unique (Rint = 0.0431) which were used in all calculations. The final wR2 was 0.0836 (all data) and R1 was 0.0386 (I > 2(I)).
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as a supplementary publication no. CCDC-1904969–1904970.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the DFG-funded transregional collaborative research center SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” project B3 is gratefully acknowledged.Notes and references
- P. C. Junk and M. L. Cole, Chem. Commun., 2007, 1579–1590 RSC.
- F. T. Edelmann, in Adv. Organomet. Chem, ed. F. H. Anthony and J. F. Mark, Academic Press, 2008, vol. 57, pp. 183–352 Search PubMed.
- A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS.
- F. T. Edelmann, in Adv. Organomet. Chem, ed. F. H. Anthony and J. F. Mark, Academic Press, 2013, vol. 61, pp. 55–374 Search PubMed.
- F. T. Edelmann, in Adv. Organomet. Chem, ed. F. H. Anthony and J. F. Mark, Academic Press, 2008, vol. 57, pp. 183–352 Search PubMed.
- M. Wedler, F. Knösel, U. Pieper, D. Stalke, F. T. Edelmann and H.-D. Amberger, Chem. Ber., 1992, 125, 2171–2181 CrossRef CAS.
- F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC.
- F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- F. T. Edelmann, Angew. Chem., Int. Ed. Engl., 1995, 34, 2466–2488 CrossRef CAS.
- F. T. Edelmann, Top. Curr. Chem., 1996, 179, 113–148 CrossRef CAS.
- F. T. Edelmann, Struct. Bonding, 2010, 137, 109–164 CrossRef CAS.
- F. T. Edelmann, J. Alloys Compd., 1994, 207–208, 182–188 CrossRef CAS.
- J. R. Hagadorn and J. Arnold, Organometallics, 1996, 15, 984–991 CrossRef CAS.
- B. S. Lim, A. Rahtu and R. G. Gordon, Nat. Mater., 2003, 2, 749–754 CrossRef CAS PubMed.
- A. P. Milanov, R. A. Fischer and A. Devi, Inorg. Chem., 2008, 47, 11405–11416 CrossRef CAS PubMed.
- J. Paivasaari, C. L. Dezelah, IV, D. Back, H. M. El-Kaderi, M. J. Heeg, M. Putkonen, L. Niinisto and C. H. Winter, J. Mater. Chem., 2005, 15, 4224–4233 RSC.
- S. Bambirra, M. W. Bouwkamp, A. Meetsma and B. Hessen, J. Am. Chem. Soc., 2004, 126, 9182–9183 CrossRef CAS PubMed.
- V. Y. Rad'kov, G. G. Skvortsov, D. M. Lyubov, A. V. Cherkasov, G. K. Fukin, A. S. Shavyrin, D. Cui and A. A. Trifonov, Eur. J. Inorg. Chem., 2012, 2289–2297 CrossRef.
- L. Zhang, M. Nishiura, M. Yuki, Y. Luo and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 2642–2645 CrossRef CAS PubMed.
- S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2003, 522–523 RSC.
- Y. Luo, Y. Yao, Q. Shen, J. Sun and L. Weng, J. Organomet. Chem., 2002, 662, 144–149 CrossRef CAS.
- C. Villiers, P. Thuéry and M. Ephritikhine, Eur. J. Inorg. Chem., 2004, 4624–4632 CrossRef CAS.
- E. A. Bijpost, R. Duchateau and J. H. Teuben, J. Mol. Catal. A: Chem., 1995, 95, 121–128 CrossRef CAS.
- A. A. Trifonov, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes I: State-of-the-art and Perspectives, ed. J. Campora and G. Giambastiani, Springer Netherlands, Dordrecht, 2011, pp. 119–152 Search PubMed.
- S. Bambirra, H. Tsurugi, D. van Leusen and B. Hessen, Dalton Trans., 2006, 1157–1161 RSC.
- S. Bambirra, M. W. Bouwkamp, A. Meetsma and B. Hessen, J. Am. Chem. Soc., 2004, 126, 9182–9183 CrossRef CAS PubMed.
- S. Ge, A. Meetsma and B. Hessen, Organometallics, 2008, 27, 3131–3135 CrossRef CAS.
- A. A. Trifonov, Rare-Earth Metal Complexes Supported by Nitrogen-Containing Ligands in Olefin Polymerization, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes I. ed. J. Campora and G. Giambastiani, Catalysis by Metal Complexes, Springer Netherlands, 2011, vol. 34, pp. 119–152 Search PubMed.
- I. V. Basalov, O. S. Yurova, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2016, 55, 1236–1244 CrossRef CAS PubMed.
- L. Zhang, M. Nishiura, M. Yuki, Y. Luo and Z. Hou, Angew. Chem., 2008, 120, 2682–2685 CrossRef.
- S. Bambirra, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 2001, 20, 782–785 CrossRef CAS.
- J. Wang, H. Sun, Y. Yao, Y. Zhang and Q. Shen, Polyhedron, 2008, 27, 1977–1982 CrossRef CAS.
- J. Wang, J. Li, F. Xu and Q. Shen, Adv. Synth. Catal., 2009, 351, 1363–1370 CrossRef CAS.
- W. Li, M. Xue, F. Xu, J. Tu, Y. Zhang and Q. Shen, Dalton Trans., 2012, 41, 8252–8260 RSC.
- J. Tu, W. Li, M. Xue, Y. Zhang and Q. Shen, Dalton Trans., 2013, 42, 5890–5901 RSC.
- J. Yang, P. Xu and Y. Luo, Chin. J. Chem., 2010, 28, 457–462 CrossRef CAS.
- A. O. Tolpygin, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Organometallics, 2012, 31, 5405–5413 CrossRef CAS.
- G. G. Skvortsov, G. K. Fukin, S. Y. Ketkov, A. V. Cherkasov, K. A. Lyssenko and A. A. Trifonov, Eur. J. Inorg. Chem., 2013, 4173–4183 CrossRef CAS.
- A. O. Tolpygin, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2014, 53, 1537–1543 CrossRef CAS PubMed.
- G. G. Skvortsov, A. O. Tolpygin, G. K. Fukin, J. Long, J. Larionova, A. V. Cherkasov and A. A. Trifonov, Eur. J. Inorg. Chem., 2017, 4275–4284 CrossRef CAS.
- M. V. Yakovenko, A. V. Cherkasov, G. K. Fukin, D. Cui and A. A. Trifonov, Eur. J. Inorg. Chem., 2010, 3290–3298 CrossRef CAS.
- M. V. Yakovenko, A. A. Trifonov, E. Kirillov, T. Roisnel and J.-F. Carpentier, Inorg. Chim. Acta, 2012, 383, 137–142 CrossRef CAS.
- C. Wang, X. Zhang, M. Xue, Y. Zhang and Q. Shen, Dalton Trans., 2013, 42, 7009–7018 RSC.
- M. V. Yakovenko, N. Y. Udilova, T. A. Glukhova, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, New J. Chem., 2015, 39, 1083–1093 RSC.
- X. Yu, M. Li, J. Hong, X. Zhou and L. Zhang, Chem. – Eur. J., 2019, 25, 2569–2576 CAS.
- J. R. Hagadorn, Chem. Commun., 2001, 2144–2145 RSC.
- J. R. Hagadorn and M. J. McNevin, Organometallics, 2003, 22, 609–611 CrossRef CAS.
- J. R. Hagadorn, M. J. McNevin, G. Wiedenfeld and R. Shoemaker, Organometallics, 2003, 22, 4818–4824 CrossRef CAS.
- B. Clare, N. Sarker, R. Shoemaker and J. R. Hagadorn, Inorg. Chem., 2004, 43, 1159–1166 CrossRef CAS PubMed.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- M. Trincado and H. Grützmacher, in Cooperative Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 67–110 Search PubMed.
- J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- N. Kazeminejad, D. Munzel, M. T. Gamer and P. W. Roesky, Chem. Commun., 2017, 53, 1060–1063 RSC.
- J. Hong, L. Zhang, K. Wang, Z. Chen, L. Wu and X. Zhou, Organometallics, 2013, 32, 7312–7322 CrossRef CAS.
- S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS PubMed.
- M. R. Gagne, L. Brard, V. P. Conticello, M. A. Giardello, C. L. Stern and T. J. Marks, Organometallics, 1992, 11, 2003–2005 CrossRef CAS.
- M. R. Gagne, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275–294 CrossRef CAS.
- M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241–10254 CrossRef CAS.
- L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- Y. K. Kim, T. Livinghouse and J. E. Bercaw, Tetrahedron Lett., 2001, 42, 2933–2935 CrossRef CAS.
- M. R. Bürgstein, H. Berberich and P. W. Roesky, Chem. – Eur. J., 2001, 7, 3078–3085 CrossRef.
- P. W. Roesky and T. E. Müller, Angew. Chem., Int. Ed., 2003, 42, 2708–2710 CrossRef CAS PubMed.
- K. C. Hultzsch, Org. Biomol. Chem., 2005, 3, 1819–1824 RSC.
- X. Han and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2006, 45, 1747–1749 CrossRef CAS PubMed.
- I. Aillaud, J. Collin, J. Hannedouche and E. Schulz, Dalton Trans., 2007, 5105–5118 RSC.
- J.-J. Brunet, N.-C. Chu and M. Rodriguez-Zubiri, Eur. J. Inorg. Chem., 2007, 4711–4722 CrossRef CAS.
- A. V. Lee and L. L. Schafer, Eur. J. Inorg. Chem., 2007, 2245–2255 CrossRef.
- R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC.
- T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550–567 RSC.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- R. Dorta, in Iridium Complexes in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 145–172 Search PubMed.
- G. Zi, Dalton Trans., 2009, 9101–9109 RSC.
- A. N. Duncan and R. A. Widenhoefer, Synlett, 2010, 419–422 CAS.
- K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026–18029 CrossRef CAS.
- T. Li, J. Jenter and P. W. Roesky, Struct. Bonding, 2010, 137, 165–228 CrossRef CAS.
- A. L. Reznichenko and K. C. Hultzsch, in Chiral Amine Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 341–375 Search PubMed.
- J. G. Taylor, L. A. Adrio and K. K. Hii, Dalton Trans., 2010, 39, 1171–1175 RSC.
- Z. Guofu, J. Organomet. Chem., 2011, 696, 68–75 CrossRef.
- J. Jenter, A. Lühl, P. W. Roesky and S. Blechert, J. Organomet. Chem., 2011, 696, 406–418 CrossRef CAS.
- T. Li, S. Schulz and P. W. Roesky, Chem. Soc. Rev., 2012, 41, 3759–3771 RSC.
- J. Hannedouche and E. Schulz, Chem. – Eur. J., 2013, 19, 4972–4985 CrossRef CAS PubMed.
- N. Nishina and Y. Yamamoto, Top. Organomet. Chem., 2013, 43, 115–143 CrossRef CAS.
- A. Reznichenko and K. Hultzsch, Top. Organomet. Chem., 2013, 43, 51–114 CrossRef CAS.
- R. Manzano, T. Wurm, F. Rominger and A. S. K. Hashmi, Chem. – Eur. J., 2014, 20, 6844–6848 CrossRef CAS PubMed.
- W. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941–2955 RSC.
- A. Arcadi, Top. Heterocycl. Chem., 2016, 46, 53–85 CAS.
- T. Li, J. Wiecko and P. W. Roesky, in Zinc-catalyzed hydroamination reactions, ed. S. Enthaler and X. Wu, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 83–118 Search PubMed.
- A. L. Reznichenko and K. C. Hultzsch, Org. React., 2016, 88, 1–554 CAS.
- M. R. Gagne and T. J. Marks, J. Am. Chem. Soc., 1989, 111, 4108–4109 CrossRef CAS.
- Y. Li, P.-F. Fu and T. J. Marks, Organometallics, 1994, 13, 439–440 CrossRef CAS.
- Y. Li and T. J. Marks, J. Am. Chem. Soc., 1996, 118, 9295–9306 CrossRef.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- A. L. Reznichenko, F. Hampel and K. C. Hultzsch, Chem. – Eur. J., 2009, 15, 12819–12827 CrossRef CAS PubMed.
- M. R. Gagne, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275–294 CrossRef CAS.
- A. Otero, A. Lara-Sánchez, C. Nájera, J. Fernández-Baeza, I. Márquez-Segovia, J. A. Castro-Osma, J. Martínez, L. F. Sánchez-Barba and A. M. Rodríguez, Organometallics, 2012, 31, 2244–2255 CrossRef CAS.
- N. Kazeminejad, Dissertation, Karlsruhe, Institute of Technology, 2017.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC.
- M. Rastätter, A. Zulys and P. W. Roesky, Chem. – Eur. J., 2007, 13, 3606–3616 CrossRef PubMed.
- M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670–9685 CrossRef CAS PubMed.
- J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584–12605 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and IR spectra and crystallographic studies. CCDC 1904969–1904970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01418g |
| This journal is © The Royal Society of Chemistry 2019 |